3.1. Microstructures and Chemical Compositions of the Two As–Cast Alloys
The as–cast microstructures of both alloys are illustrated in
Figure 1, with a ×250 SEM/BSE micrographs for both of them (general view) and a ×4 enlargement in the bottom right corner of the ×250 images for observing the morphologies of the carbides in better conditions (×1000). The two alloys are obviously only double–phased. They contain first a dendritic matrix with a 25 µm fineness (approximate secondary dendrite arms spacing). The second phase is HfC, as specified by EDS spot analysis on the coarsest carbides found in the microstructures. In the Co-32.5Cr-0.25C-3.72Hf alloy, the HfC carbides are present only as one of the two phases of a eutectic compound located in the interdendritic areas (the second phase in this eutectic is the matrix itself). The morphology of these eutectic HfC carbides is Chinese script. This means that the HfC are locally made of interconnected elongated parts closely mixed with the peripherical parts of the dendrites. This type of morphology results from the re–distribution of the elements between the HfC part and the cobalt solid solution part of the eutectic compound, during their coupled growths). The HfC carbides are present in great quantity, and they form an interdendritic network almost continuously. In the Co-32.5Cr-0.50C-7.44Hf alloy, the eutectic script–like HfC are also present, with a fraction that is higher than in the first alloy, as an interconnected interdendritic carbide network. Other carbides are also present, with a coarse morphology, dispersed in the microstructure as polygonal carbides. Their morphology and distribution in the alloy suggest that these HfC are of a pre–eutectic nature (i.e., the first solid phase to crystallize at the beginning of solidification). Despite the presence of supplementary chromium in a significant quantity (32.5 wt.%, i.e., 30% Cr more by comparison with 25 wt.% Cr), one can remark that no chromium carbides appeared. As for the previous alloys with 25 wt.% Cr [
18,
19], the Hf atoms still monopolize carbon, for forming exclusively HfC, again. EDS spot analyses performed on the coarsest white carbides (to avoid penetration of the interaction peer in the subjacent matrix) show high concentrations of Hf and C (
Figure 1, bottom). The obtained C content is slightly overestimated.
SEM/EDS allowed controlling the chemical composition of the whole alloys (
Table 1), except carbon for which this was not possible because of its too low atomic mass and its too low targeted weight content. The obtained contents in hafnium are not lower than introduced in the charges before melting, on the contrary. The obtained Hf contents are a little higher than targeted. This is a classical effect of the involvement of such heavy elements in numerous primary carbides. This is an effect that is also usually encountered with tantalum, for instance. The contents in Cr are well respected. Additionally, three EDS spot analyses were performed in the matrix of each alloy (
Table 2). Matrices are rich in chromium (content slightly higher than in the whole alloy) and they contain no hafnium. Hafnium is seemingly totally involved in carbides. This suggests a very high carbide–former behavior of this element.
3.2. Isothermal Oxidation
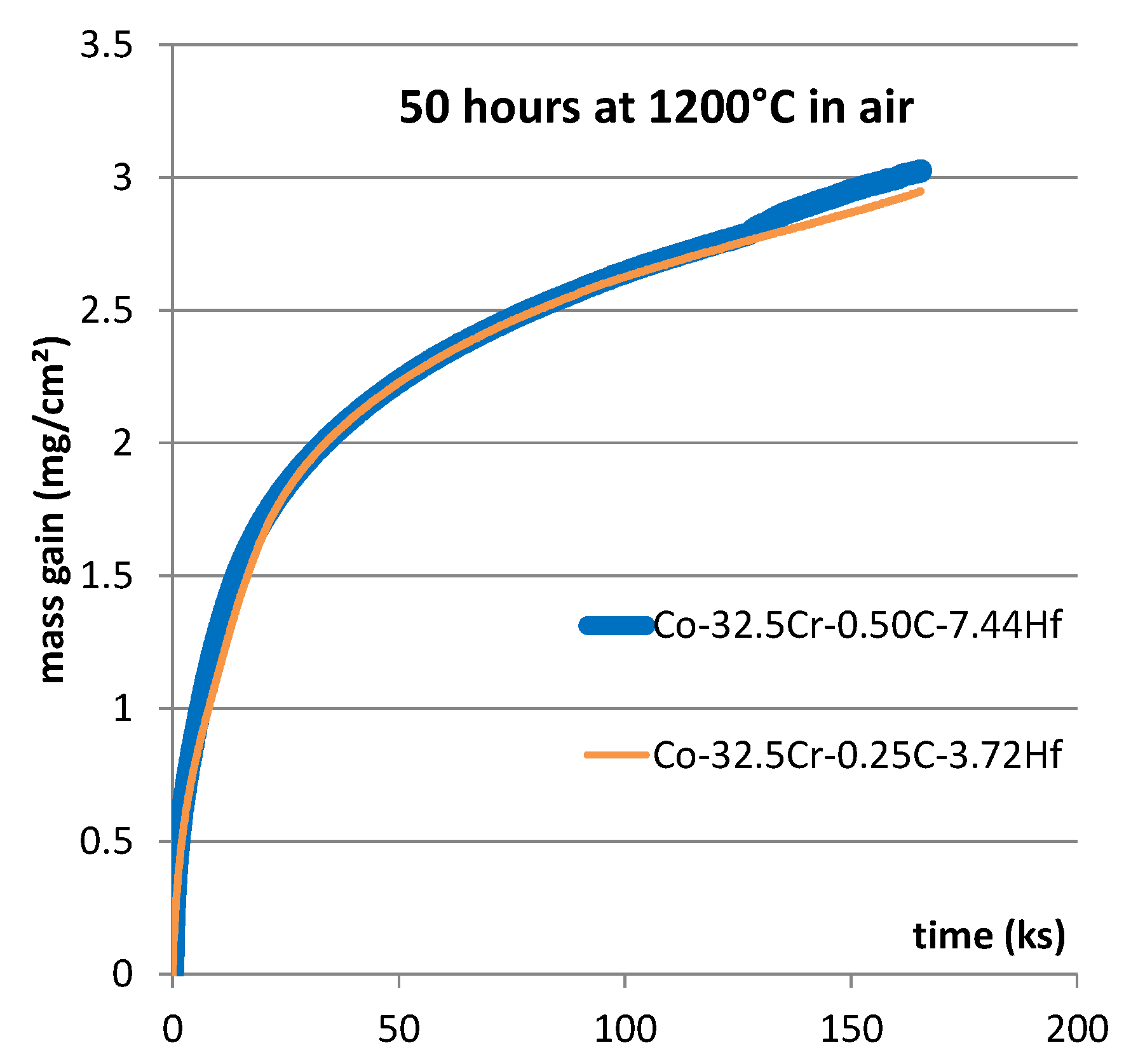
The mass gain curves of the two alloys are plotted together in
Figure 2. They are almost identical. Both of them start with a fast mass gain during 20 h approximately, followed by a strong slow down leading to a parabolic regime. Such a parabolic regime suggests that the whole surface of the sample is separated from the air by a continuous oxide scale. The growth of this scale is governed by the diffusion of the species involved in the oxidation process (Wagner’s law). In such a case, the best characteristic allowing to anticipate the oxidation rate for a longer time is the parabolic constant noted “Kp”. In the first time, the mass gain was plotted versus the square root of time to specify the value of Kp for each alloy at 1200 °C. Kp is usually deduced from the slope of the obtained straight line. However, the obtained {mass gain versus the square root of time}–curves are not linear and their slopes decrease with time. Considering on the first hand the high level of chromium content of both alloys, and on the second hand the high level of the test temperature, it appeared clear that chromia covered the samples and that this oxide was subjected to re–oxidation into gaseous CrO
3. Consequently, after having estimated the average slope and the value of Kp, both mass gain files were analyzed according to a method described in [
20]. By plotting versus the opposite value of the mass gain per surface unit area (noted “m”) the value of m multiplied by its derived value versus time dm/dt, the obtained graph may present a linear part. The equation of this linear part may lead to the value of a reliable Kp constant, as well as to the value of the constant Kv. The obtained value of Kp is reliable since it is not underestimated because of the loss of a part of chromia. The obtained Kv value represents the rate of linear loss of re–oxidized chromia.
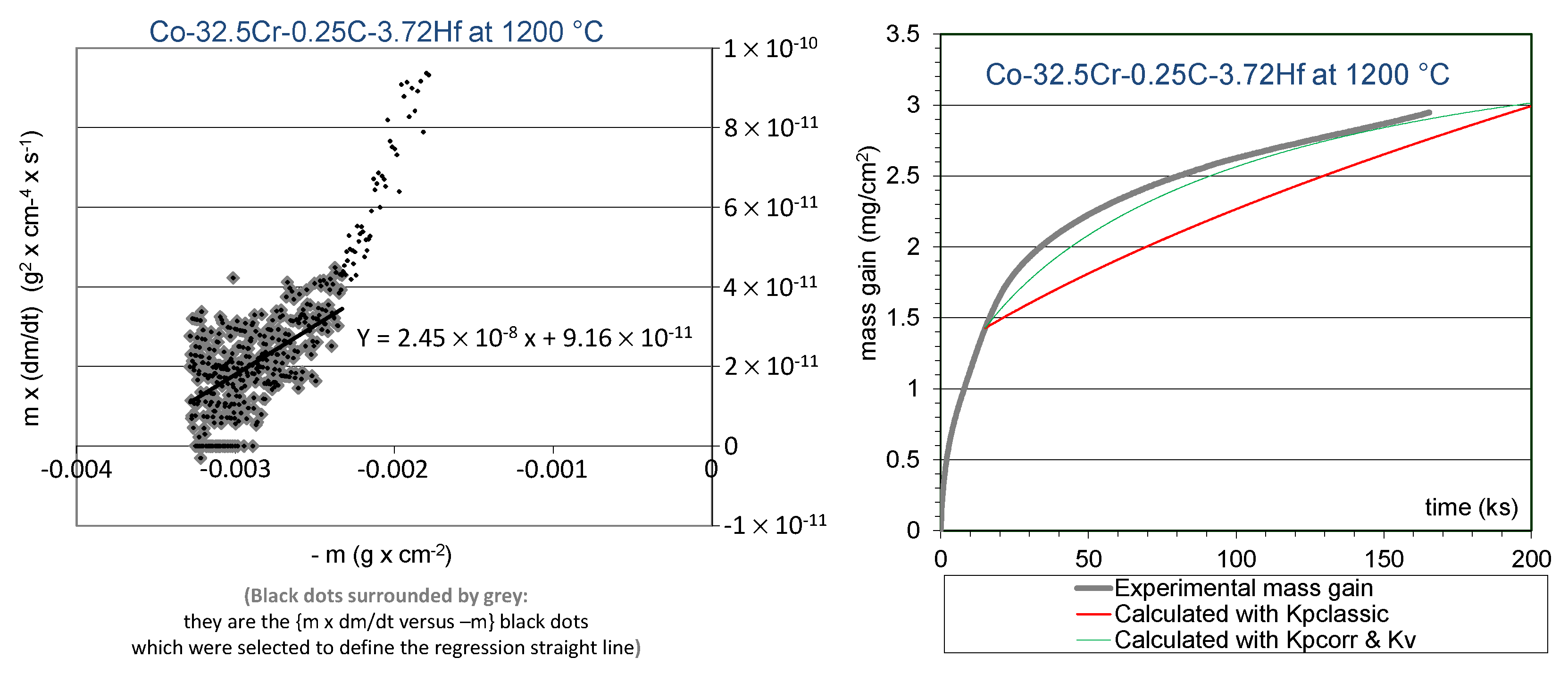
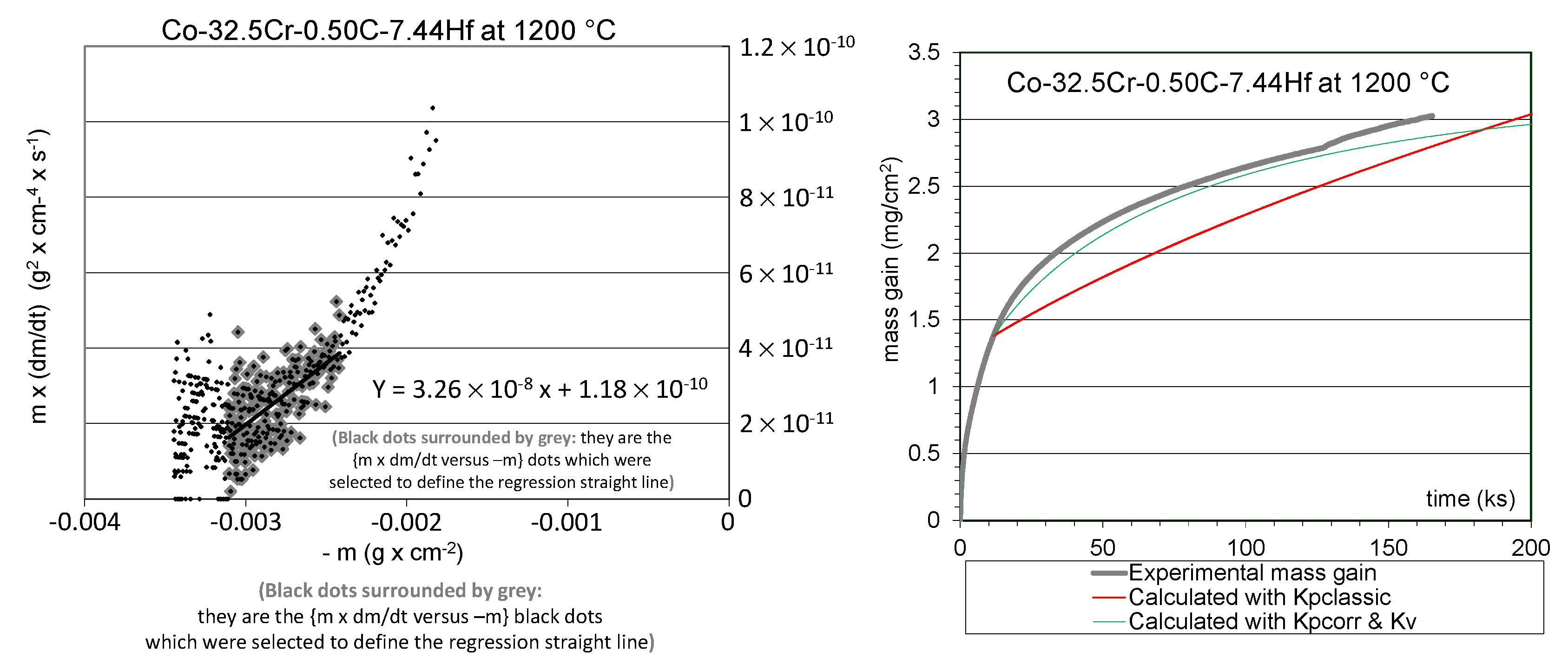
The {m × dm/dt versus -m}–plot led to the graphs showed in the left side of
Figure 3 for the Co-32.5Cr-0.25C-3.72Hf alloy and in the left side of
Figure 4 for the Co-32.5Cr-0.50C-7.44Hf alloy.
In both cases, only the points corresponding to the mass gain values beyond the first 20 h were considered. Indeed, this is over the [20 h; 46 h] time range that the chromia scale can be expected to be continuous. For the Co-32.5Cr-0.25C-3.72Hf alloy (
Figure 3, left), all points were kept for determining the equation of the regression straight line. For the Co-32.5Cr-0.50C-7.44Hf alloy (
Figure 4, left), the last points, recorded during the last ten hours before the isothermal stage’s end (on the left of the graph), were omitted since they correspond to a small jump in mass gain (visible in
Figure 2) due to a local detachment of the external scale.
The equations of the regression straight lines are given in
Figure 3 (left) and
Figure 4 (left). In each of them, the values of the ordinate at the origin are the parabolic constants Kp corrected from chromia volatilization, and the slopes are the volatilization constants Kv.
The values of all the kinetic constants determined by the exploitation of the curves shown in
Figure 2 and of the graphs shown in
Figure 3 and
Figure 4, are gathered in
Table 3. Model curves were plotted using these values obtained for the kinetic constants, to be compared to the experimental isothermal mass gain curves (
Figure 3, right;
Figure 4, right). The obtained values of Kp
corr volat and Kv
volat obviously represent the real mass gain kinetic much better than the Kp
classic values.
In
Table 3 the first line contains the values of the linear constants K
l featuring in the {m = K
l × t}–equation of the straight line tangent to the mass gain curves in
Figure 2. These linear constants are the instantaneous rates of mass gain existing at the beginning of the isothermal stage. They are also called transient oxidation rates. The linear constant tends to be higher for the Co-32.5Cr-0.50C-7.44Hf alloy than for the other alloy. For each alloy, the K
l value obtained here is similar to the ones obtained for the previous alloys containing only 25 wt.% Cr (42 × 10
−5 mg/cm
2 for the Co-25Cr-0.25C-3.72Hf alloy and 80 × 10
−5 mg/cm
2 for the Co-25Cr-0.50C-7.44Hf [
19]). This tends to confirm that it is more the HfC carbides density than the chromium content that influences this first kinetic constant.
The second line (Kp classically determined, Kp
classic) and the third line (Kp corrected from volatilization of chromia, Kp
corr volat) in
Table 3 suggest that the Co-32.5Cr-0.50C-7.44Hf tends to oxidize a little faster than the other alloys in this study. However, the differences are not significant. In contrast, there are very great differences between the Kp
classic and Kp
corr volat values determined here, and the ones previously determined for the corresponding alloys containing only 25 wt.% Cr. For 32.5 wt.% Cr the Kp values are several ten times lower than for the previous alloys containing 7.5 wt.% Cr less (globally 600 to 800 × 10
−6 mg cm
−4 s
−1 for Kp
classic, and 1300 to 2500 × 10
−6 mg cm
−4 s
−1 for Kp
corr volat [
19]).
Differences can be also noted concerning the chromia volatilization constants, but they are much less spectacular. They are here slightly lower than the ones previously determined for the 25 wt.% Cr containing alloys: 250 to 330 × 10
−7 mg cm
−2 s
−1 here, against 470 to 530 × 10
−7 mg cm
−2 s
−1 for the 25wt.% Cr containing alloys [
19]. On the mass gain kinetic first, one can conclude that adding 7.5 wt.% Cr more to the earlier alloys considerably enhances their resistance to isothermal oxidation at 1200 °C.
3.3. Oxidation at Heating and Oxide Spallation at Cooling
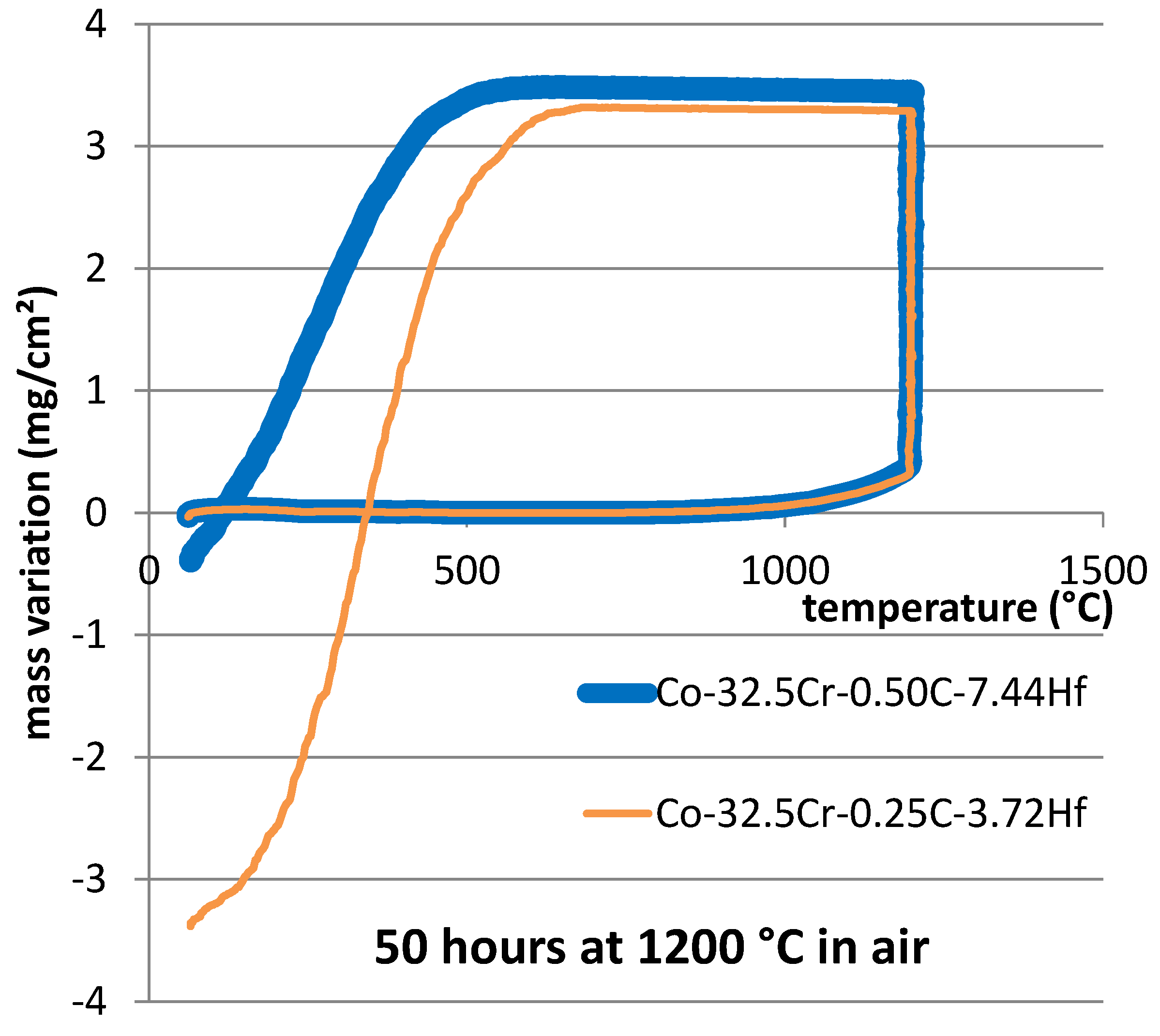
The whole files containing the recorded mass variation values were exploited too. After correction for air buoyancy variation, the two files were plotted versus temperature (
Figure 5).
One can see first that a small but significant part of the total mass gain was achieved during heating. Indeed, heating was performed in the flow of industrial air, and oxidation started to be detectable as soon as the temperature became high enough. The used thermo–balance detected the mass gain due to oxidation when the temperature reached about 740 °C. The values displayed in the first line of
Table 4 suggest that the Co-25Cr-0.50C-7.44Hf alloy was a little less reactive than the other alloy since the mass gain due to oxidation was detected a little later (at a temperature about 35 °C higher). There was no difference between the oxidation start temperatures at heating for the 25 wt.% Cr containing alloys: about 900 °C for both of them [
19]. This temperature is 150 °C higher than the oxidation start temperatures of the alloys studied here. This suggests that the 7.5 wt.% Cr addition increased the reactivity of the present alloys in comparison with the ones earlier studied. Probably due to that, the mass gains during the whole heating are, for the present alloys, 1.5 to 2 times higher than the ones of the 25 wt.% Cr containing alloys earlier studied (second line in
Table 4 and [
19]). In contrast, the mass gains achieved during the isothermal stage are here much lower than for the earlier alloys: 3.3–3.4 mg cm
−2 for 46 h at 1200 °C (third line in
Table 4) against 14–18 mg cm
−2 for the 25 wt.% Cr containing alloys [
19]. This is consistent with the differences in Kp constant values seen above. Consequently, the proportion of the mass gain during heating in the total mass gain achieved until the end of isothermal oxidation is here ten times higher than for the 25 wt.% Cr containing alloys: about 10% (fourth line in
Table 4) against 1–2 %.
The cooling taking place after the end of the isothermal stage usually induces more and more intense compressive stresses on the oxide scales externally formed around the samples. This generally provokes local ruptures of the scale and possibly the loss of oxide parts. These oxide losses are shown by mass losses then falls in the mass curves. Such phenomena occurred here (
Figure 5). This is more obvious for the Co-32.5Cr-0.25C-3.72Hf alloy than for the Co-32.5Cr-0.50C-7.44Hf alloy. The values of the parameters characterizing the oxide scale spallation phenomena were measured on the {m versus T}–curves (
Figure 5) and these values are reported in
Table 5. For the present oxidized samples, scale spallation started about 15 min later for the Co-32.5Cr-0.50C-7.44Hf alloy than for the other, and after 80 °C more cooling (first line in
Table 5). A similar difference was previously noticed for the 25 wt.% Cr containing alloys (about 650 °C for the Co-25Cr-0.25C-3.72Hf alloy and 570 °C for the Co-25Cr-0.50C-7.44Hf one [
19]. Oxide spallation occurred here sooner than for the 25 wt.% Cr containing alloys which started spalling off about 30 °C below. The total mass loss, achieved between the start of oxide spallation and the return to room temperature, appears as being lower for the Co-32.5Cr-0.50C-7.44Hf alloy than for the other alloy (bottom line in
Table 5). Such difference was also noticed for the 25 wt.% Cr containing alloys (about −50 g cm
−2 for the Co-25Cr-0.25C-3.72Hf alloy against −30 g cm
−2 for the Co-25Cr-0.50C-7.44Hf one). This allows pointing out the better resistance against oxide spallation when the Hf content is higher. The better isothermal oxidation resistance of the alloys of the present study allowed the 32.5 wt.% Cr containing alloys to lose less oxide masses (about ten times lower than for the 25 wt.% Cr containing ones [
19]).
3.4. Post–Mortem Metallography Characterization of the Oxidized Samples
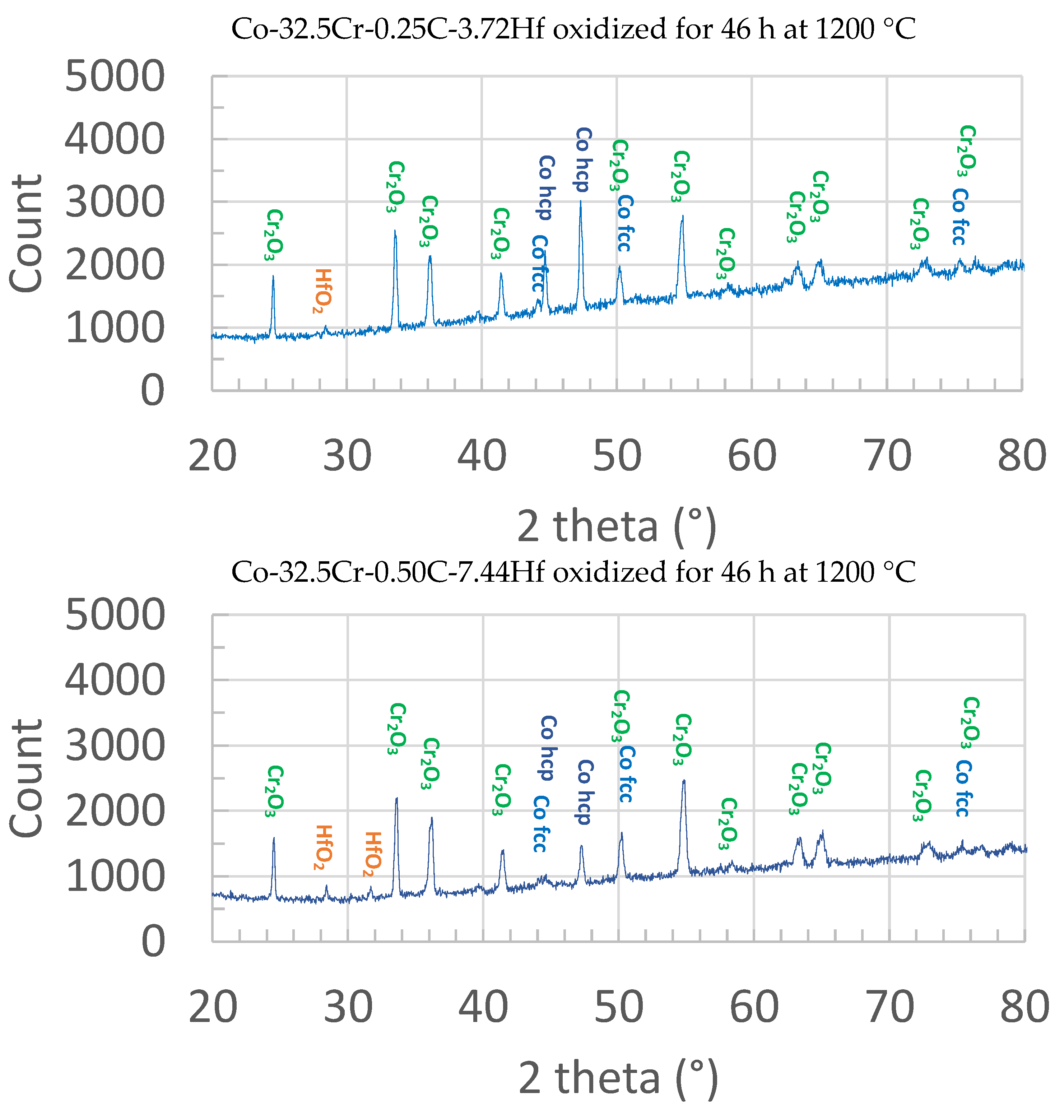
First, X-ray diffraction was performed on the oxidized surfaces of the oxidized samples. The obtained diffractograms are displayed in
Figure 6. One can see that, in both cases, the principal oxide present is clearly chromia. Very small peaks point out the presence of hafnium oxide. The other diffraction peaks correspond to the alloy, the matrix of which is double–phased (austenitic and hexagonal compact) due to partial allotropic transformation during the cooling.
The cross–sectional samples were prepared from the oxidized alloys by electrolytic coating, cutting and grinding/polishing. Despite the careful handling of the oxidized samples, parts of the oxide scales were unfortunately lost. These cross–sectional samples were examined all around. Low magnification SEM/BSE micrographs were taken where the oxide scale was still sufficiently present. These micrographs are provided in
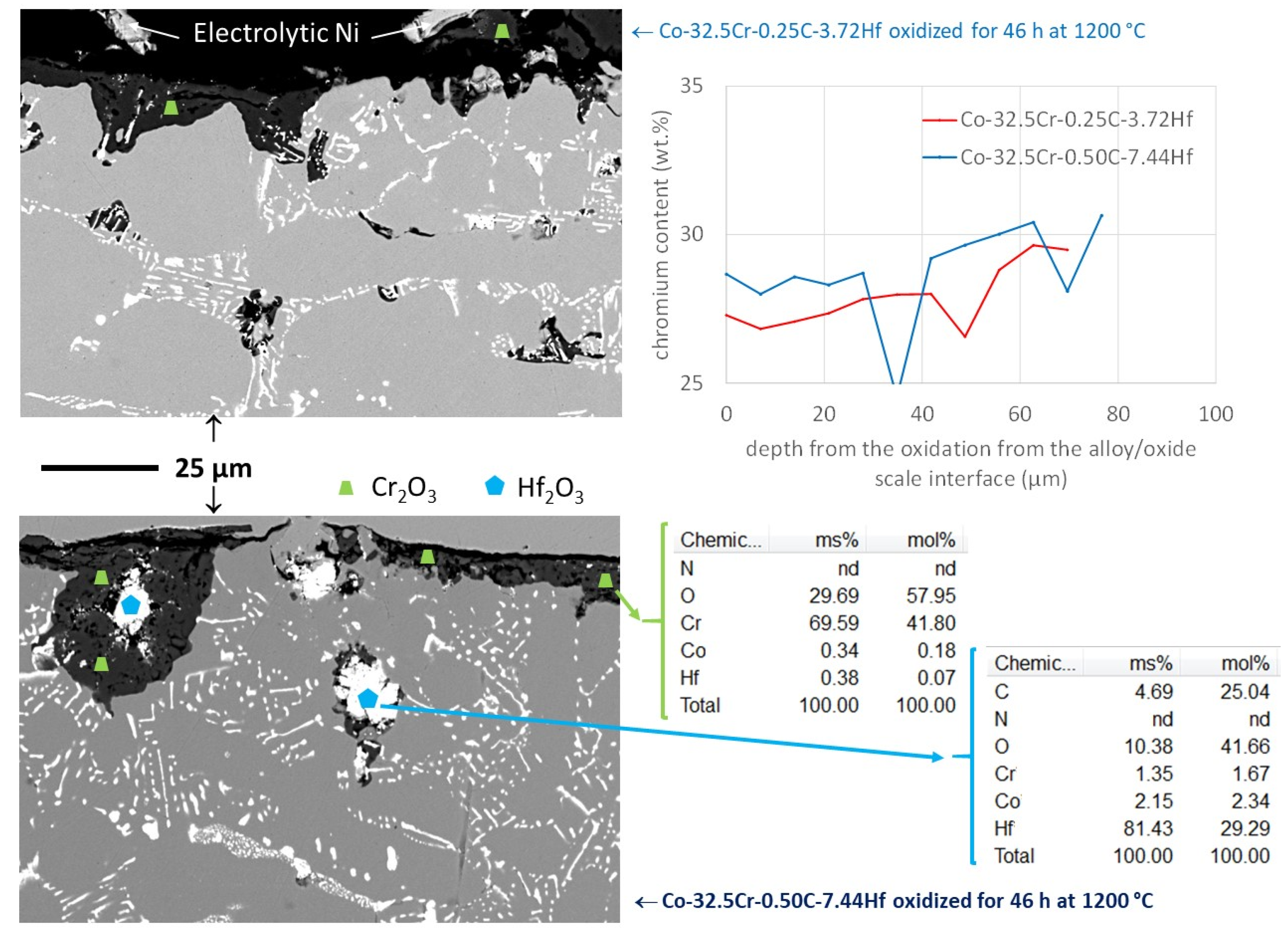
Figure 7 to benefit from a general view of the oxidized state. Obviously, oxidation acted both externally and internally (
Figure 8, left). Inward progression of oxidation was generalized, as this was already the case for the 25 wt.% Cr containing alloys studied previously [
19]. Chromia was present all around with the two types of location (external scale and internal oxides). Other oxides were also present as internal oxides. Several EDS spot analyses performed on these white internal oxides demonstrated that their stoichiometry was Hf
2O
3 (examples of EDS results inserted in
Figure 8) It seemed that the Hf
2O
3 oxide, mainly met in the subsurface of the Co-32.5Cr-0.50C-7.44Hf alloy, resulting from the oxidation of the compact pre–eutectic HfC present close to the oxidation front (
Figure 8, left). Unlike the 25 wt.% Cr containing alloys previously studied [
19], no cobalt oxides or CoCr
2O
4 spinel oxides were observed here. In addition, no local catastrophic oxidation started anywhere for the two alloys, unlike in the 25 wt.% Cr containing alloys which were affected by fast oxidation here and there before the end of the oxidation tests. Seemingly, the chromium content was high enough to maintain a chromia–forming behavior. Several EDS Cr profiles were acquired from the oxidation front toward the bulk. Two Cr profiles, one per alloy, are drawn together in
Figure 8 (left). The chromium content decreases from the content in the bulk over a depth equal to about 100 µm in both cases (
Figure 8, right). The minimal Cr content values were mostly about 27.5 wt.% Cr for both alloys, despite that lower contents (e.g., 24 wt.%) were met in some locations. Such Cr contents, significantly higher than the 12 to 15 wt.% Cr met for the 25 wt.% Cr containing alloys close to the oxidation front [
19], guaranteed the continuation of the chromia–forming behavior.
Concerning hafnium, as for the previous 25 wt.% Cr containing alloys [
19], the Hf content is extremely low (even inexistent) in the subsurface. The carbide network is almost not affected, even very close to the oxidation front where only a limited part of HfC was converted in hafnium oxide. This is totally different from TaC–strengthened cobalt alloys [
22] for which tantalum diffuses outward to be oxidized at the scale/alloy interface as CrTaO
4 and for which a TaC–free zone usually develops inward from the oxidation front. Seemingly Hf cannot enter in solid solution in the matrix and is, therefore, not able to quit carbides and diffuse.
Since the HfC network stayed in place for all the oxidation test duration, taking into account the very high density of this carbide network (in the Co-32.5Cr-0.50C-7.44Hf alloy but also in the other one), one may think that the diffusion of chromium itself may be made more difficult and slowed down. This was maybe a reason for why the 25 wt.% Cr containing alloys oxidized with the formation of chromia but also of other oxides [
19]. In the present case, with 7.5 wt.% Cr more, the Cr content initially present everywhere in the alloys and in the zones close to the sample surface, in particular, was high enough to allow the chromia–forming behavior to be less dependent on Cr diffusion from deeper in the alloys. However, because of the no disappearance of the carbide network, the diffusion barrier effect of the HfC may be a serious problem for oxidation in the long term. For 46 h at 1200 °C in air, the probable hindering to Cr diffusion caused by the HfC network was responsible for a significant inward oxygen diffusion. This oxygen diffusion from the oxidation front is revealed by the chromia islands formed internally.
3.6. Creep Behavior Control
Logically, enriching in chromium the creep–resistant 25 wt.% containing alloys previously studied [
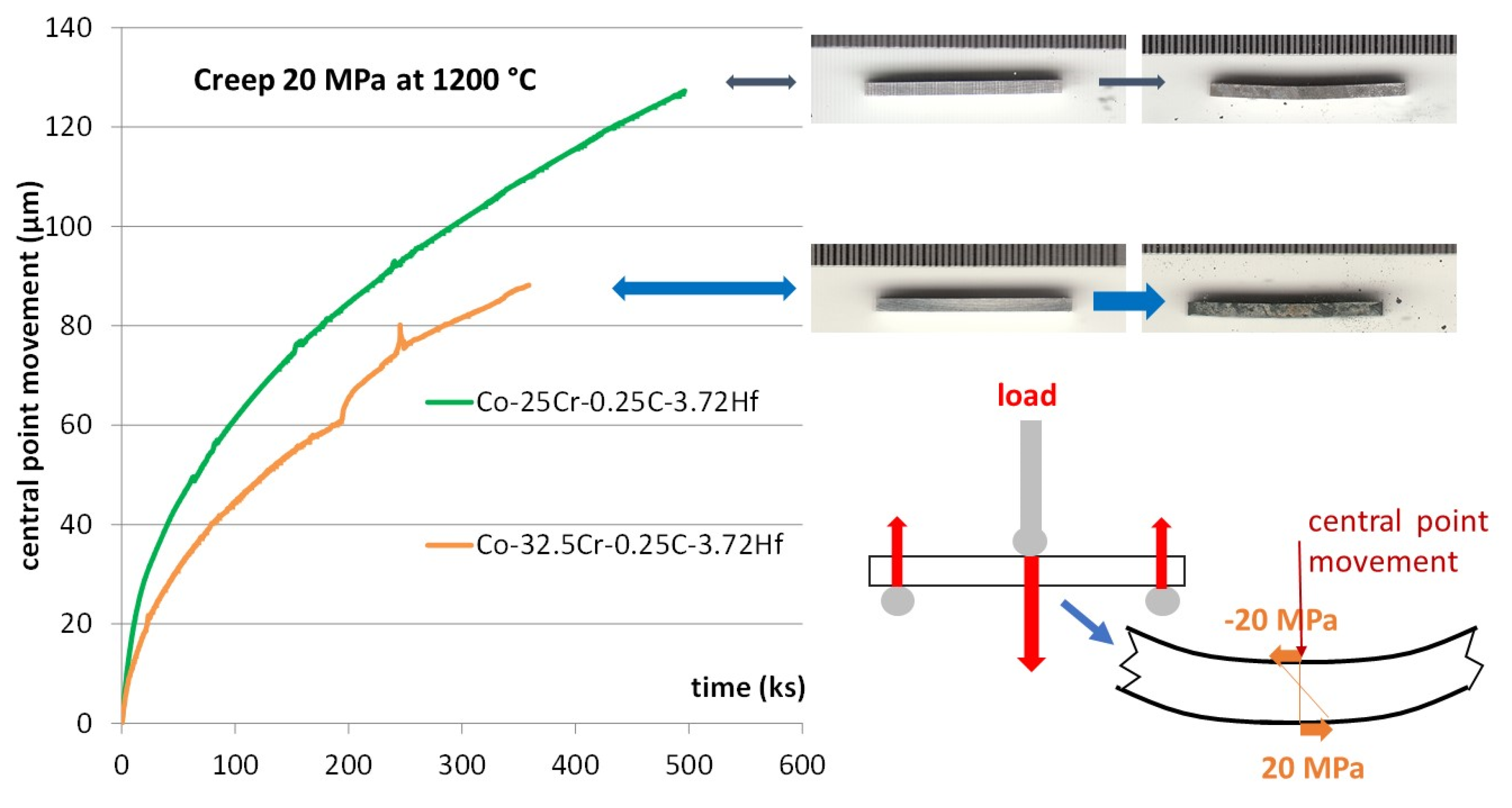
19] was expected to increase their resistance against high temperature oxidation (now verified and confirmed). However, this modification of the chemical composition of the alloy may also possibly influence the mechanical properties at high temperatures. It was just seen below that the stability of the carbides network was not affected by this Cr addition, neither in the as–cast condition nor after aging. However, the absence of any deleterious effect due to chromium addition needs to be checked by mechanically testing the alloys at high temperatures. A result of a flexural creep test at 1200 °C was earlier carried out on the Co-32.5Cr-0.25C-3.72Hf alloy and it demonstrated remarkable creep resistance. The same test was here performed, on the Cr–enriched corresponding alloy in the same conditions for comparison. The Co-32.5Cr-0.25C-3.72Hf alloy was thus tested here, at 1200 °C for a resulting maximal stress of 20 MPa, with the same apparatus and for the same protocol as earlier, during 100 h. The used device and the test protocol are noted by a scheme in the right–bottom corner of
Figure 10. Prior to heating to 1200 °C, a load (long red arrow downward oriented) is progressively applied—using the grey top rod—on the center of the top face of the parallelepiped sample, until inducing here 20 MPa for the horizontal compressive stress (and a 20 MPa horizontal tensile stress in the other side (according to the elastic theory). This load is equilibrated by the sum of the support’s reactions (short red arrows upward oriented). The sample deformation started, of course, before reaching 1200 °C, as soon as the temperature became high enough. The deformation re–initialized to 0 when the temperature reached 1200 °C. The creep deformation progression is characterized by the downward movement of the central point, counted with positive values.
The resulting deformation curve is plotted in the left part of
Figure 10, together with the one previously obtained for the 25 wt.% Cr containing alloy [
18]. The resistance is globally of the same level as the previous result, with a deformation rate a little lower but also with a little instability appearing at about t = 200,000 s. Of course, this needs to be confirmed by some repeated tests to take into account a possible lack of reproducibility.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}