
Counter Anion Effects on the Formation and Structural Transformations of Mo(vi)-Hydrazone Coordination Assemblies: Salts, Solvates, Co-Crystals, and Neutral Complexes




Abstract
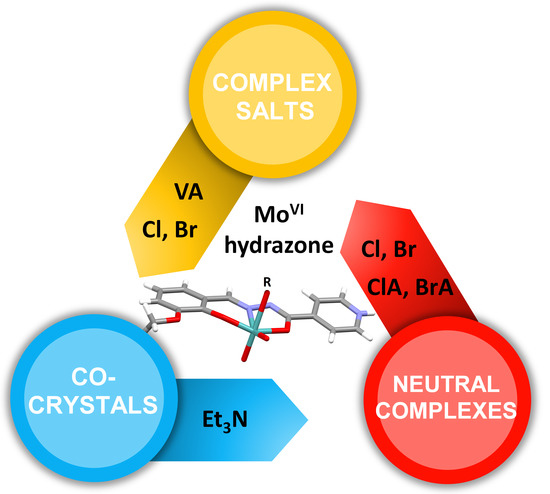
:
1. Introduction
2. Materials and Methods
2.1. Synthesis
2.1.1. Synthesis of [MoO2(HL)(MeOH)]Cl·MeOH ([1H]Cl·MeOH)
2.1.2. Synthesis of [MoO2(HL)(MeOH)]Br ([1H]Br)
2.1.3. Synthesis of [MoO2(HL)(MeOH)](ClA)0.5·2MeOH ([1H](ClA)0.5·2MeOH)
2.1.4. Synthesis of [MoO2(HL)(MeOH)](BrA)0.5·2MeOH ([1H](BrA)0.5·2MeOH)
2.1.5. Synthesis of [MoO2(HL)(MeOH)]Cl·0.5VA ([1H]Cl·0.5VA)
2.1.6. Synthesis of [MoO2(HL)(MeOH)]Br·0.5VA ([1H]Br·0.5VA)
2.1.7. Synthesis of [MoO2(HL)Cl]·MeOH (2Cl·MeOH)
2.1.8. Synthesis of [MoO2(HL)Cl] (2Cl)
2.1.9. Synthesis of [MoO2(HL)Br]·0.5MeCN (2Br·0.5MeCN)
2.1.10. Synthesis of [MoO2(L)(MeOH)] (1)
2.1.11. Synthesis of [MoO2(L)]n (2)
2.2. Single Crystal and Powder X-ray Diffraction
2.3. Thermal and Spectroscopic Measurements
2.4. In Vitro Cytotoxic Activity
3. Results and Discussion
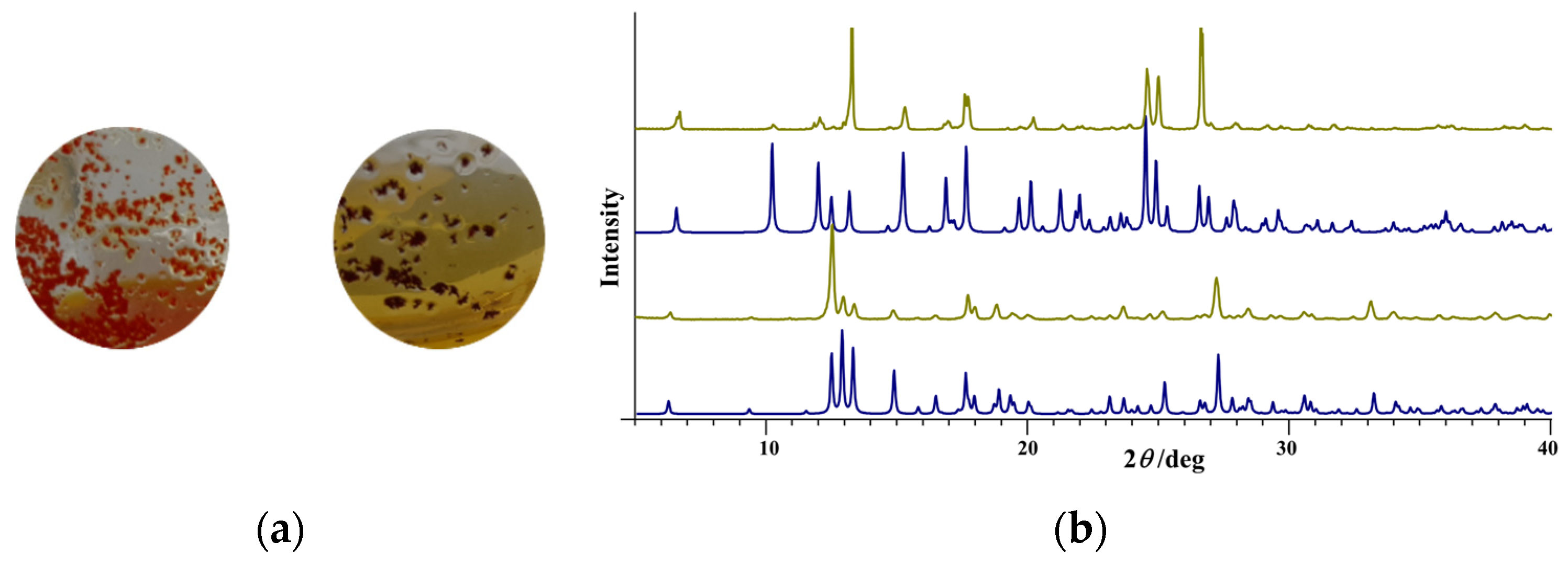
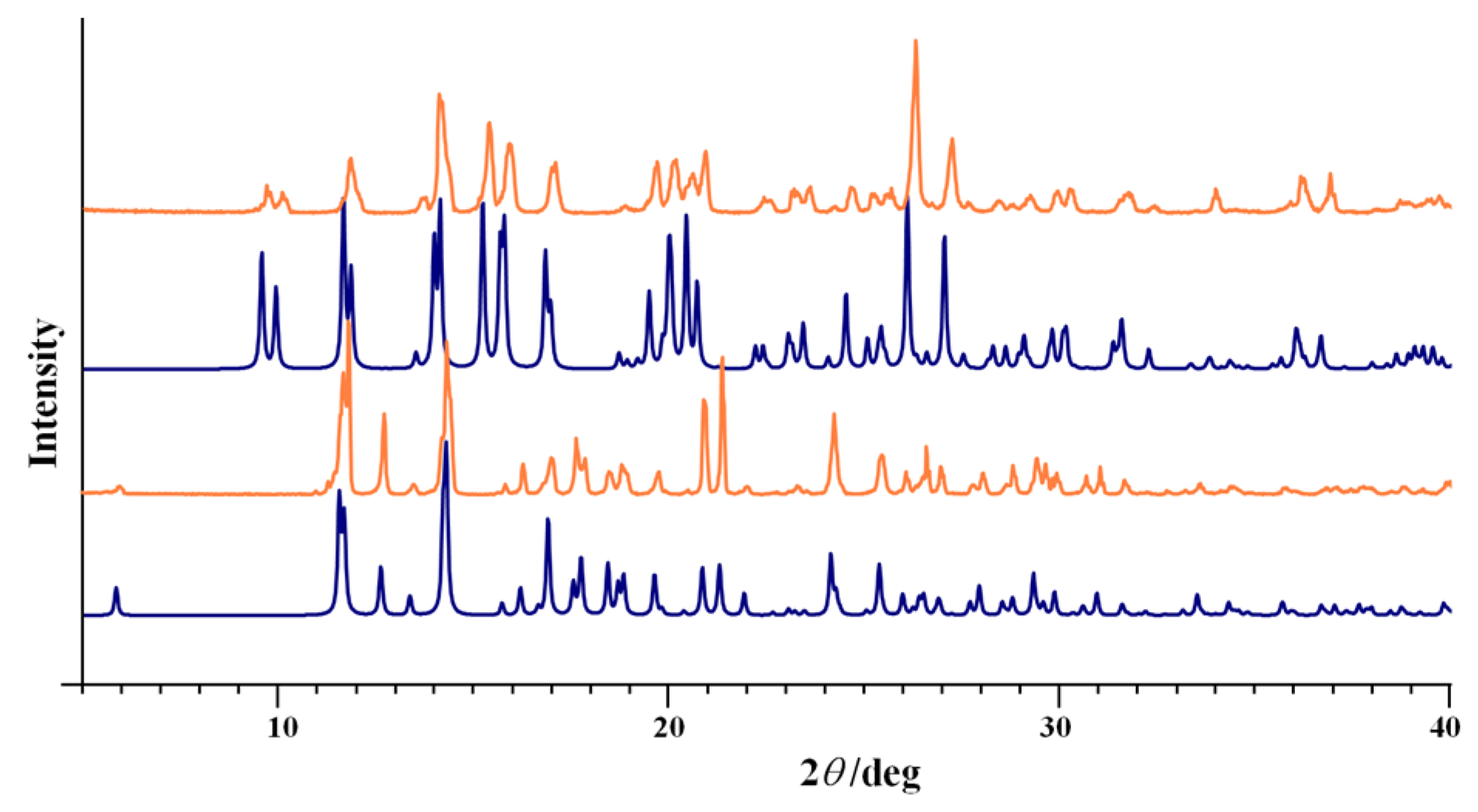
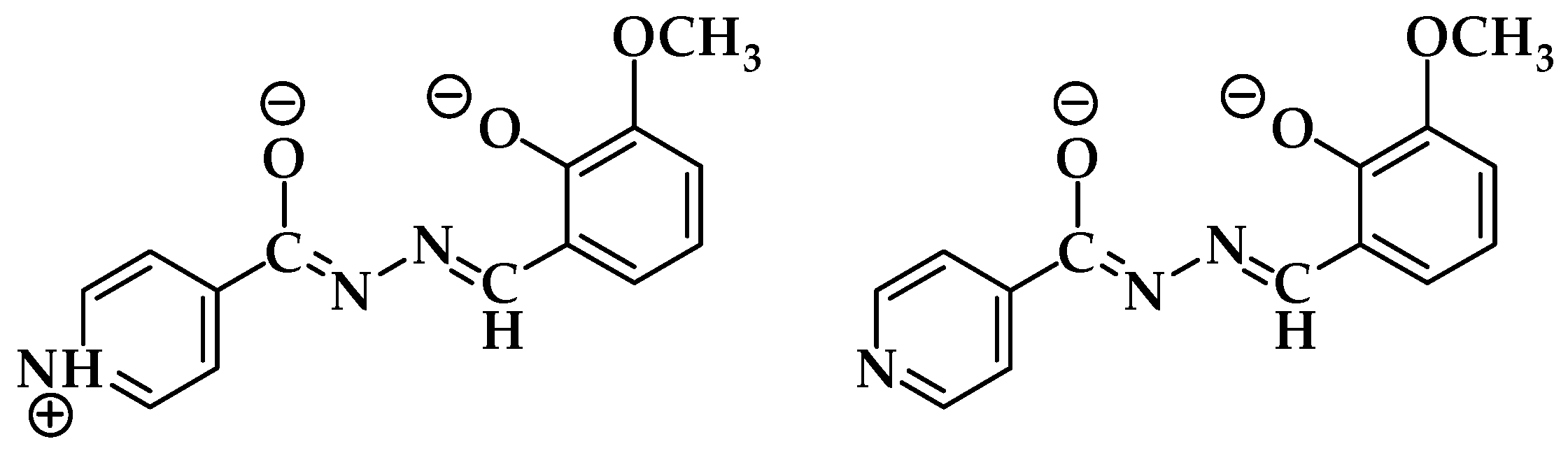
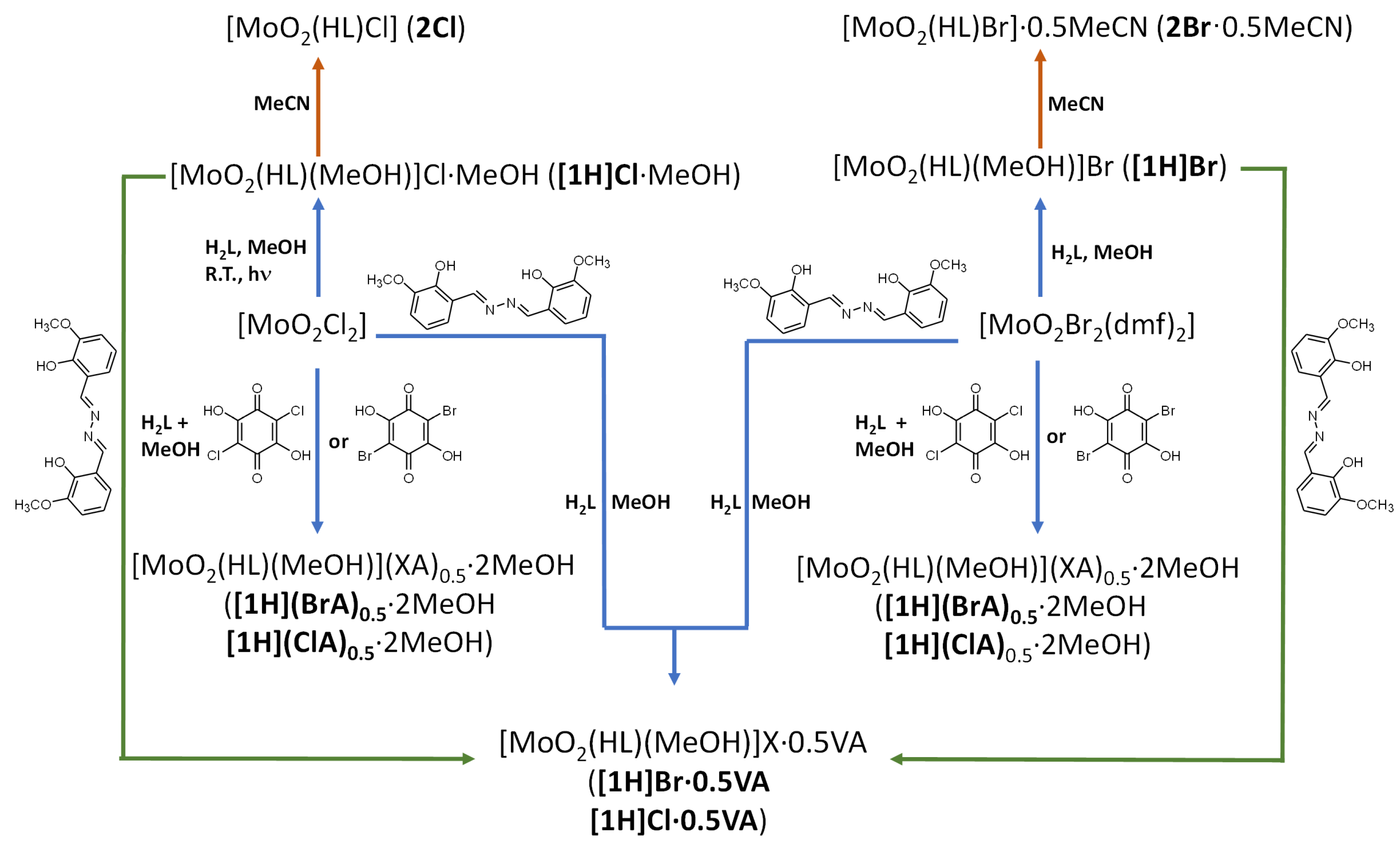
3.1. Complex Salts, Solvates and Co-Crystals—Synthesis of [1H]X, [1H](XA)0.5·2MeOH, and [1H]X·0.5VA (X = Cl or Br, XA = ClA or BrA)
3.2. Counter Anion Effects and Transformation of Complex Assemblies—Synthesis of 2Cl and 2Br·0.5MeCN
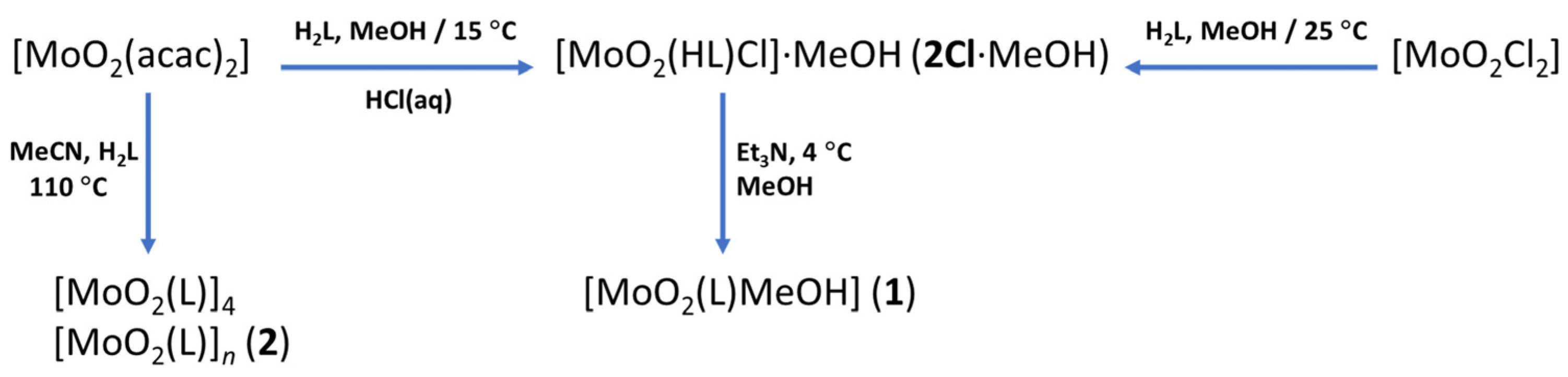
3.3. Deprotonated Mo-Hydrazone Assemblies—Synthesis of 1 and 2
3.4. Crystalographic Studies
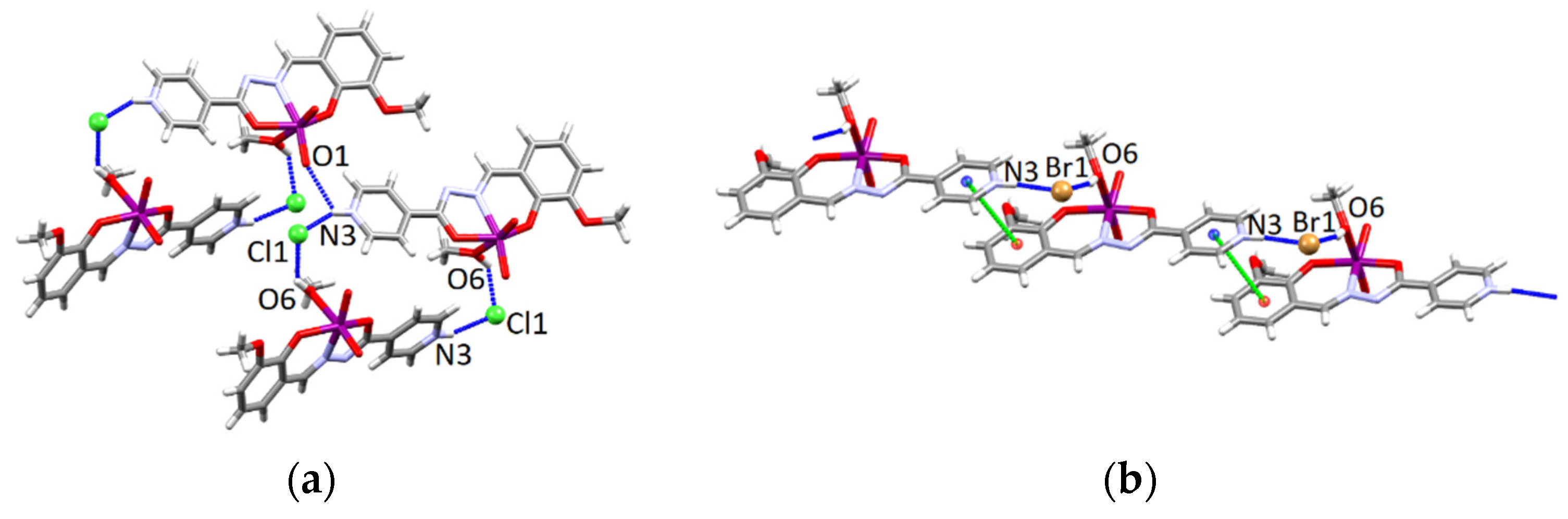
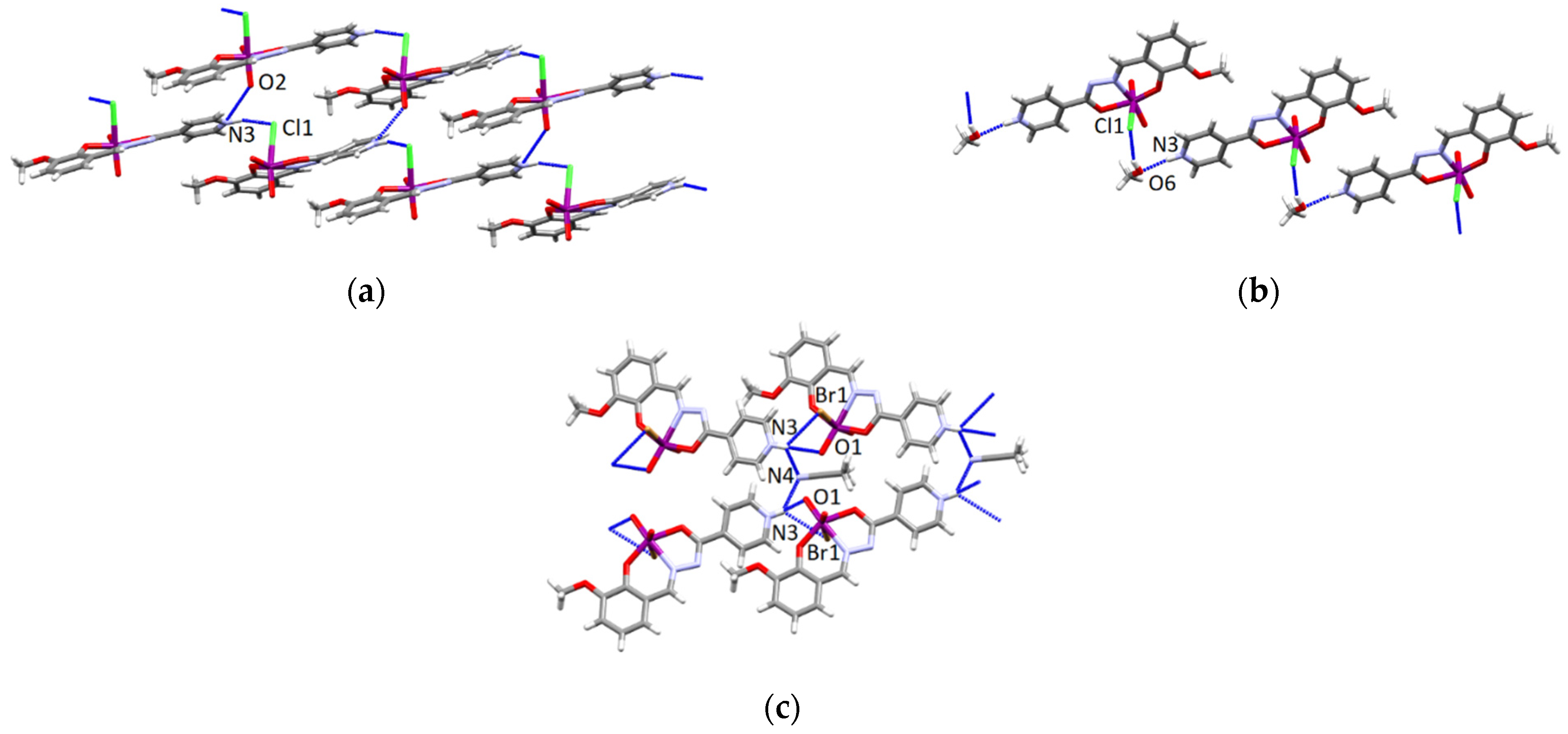
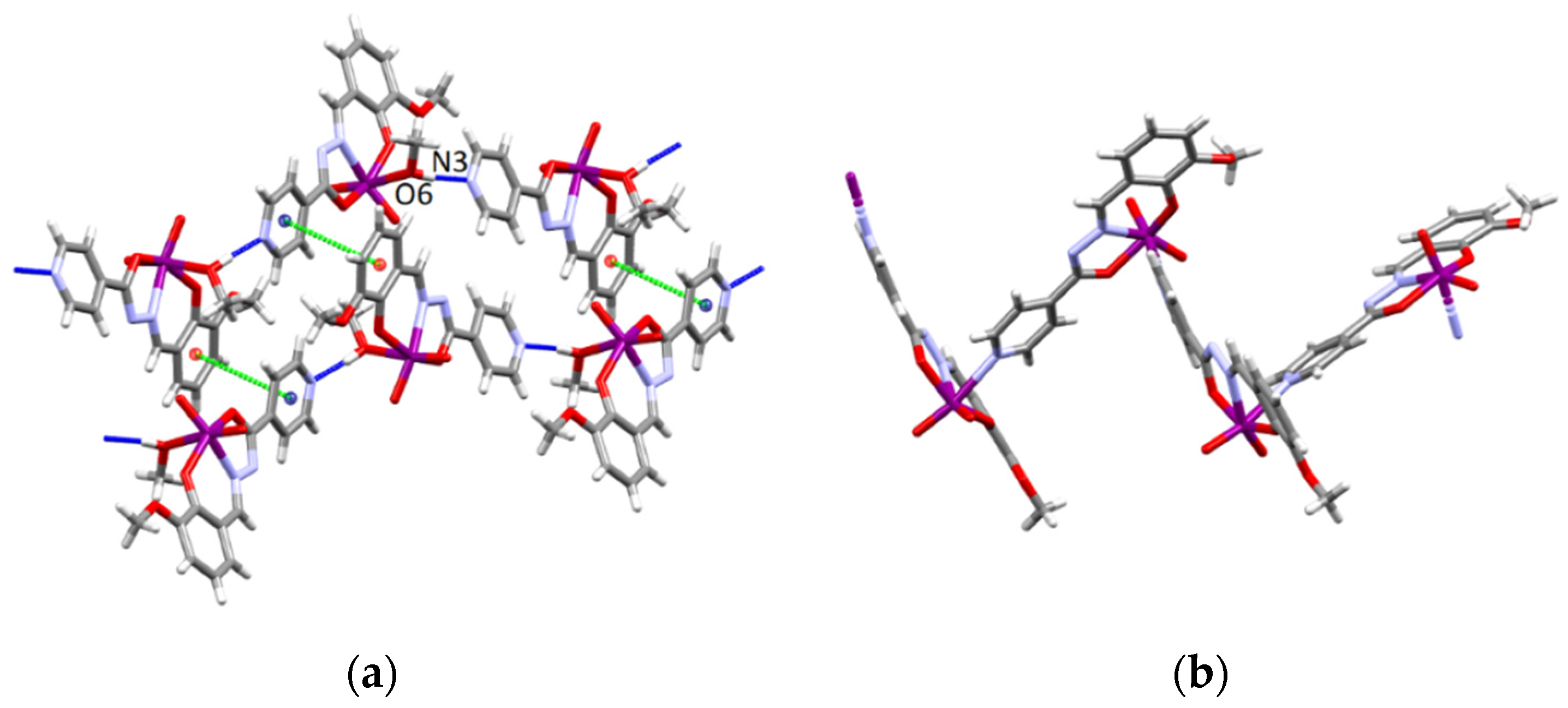
3.4.1. Structures of the Complex Salts, Solvates and Co-Crystals [1H]X, [1H](XA)0.5·2MeOH, and [1H]X·0.5VA (X = Cl or Br, XA = ClA or BrA)
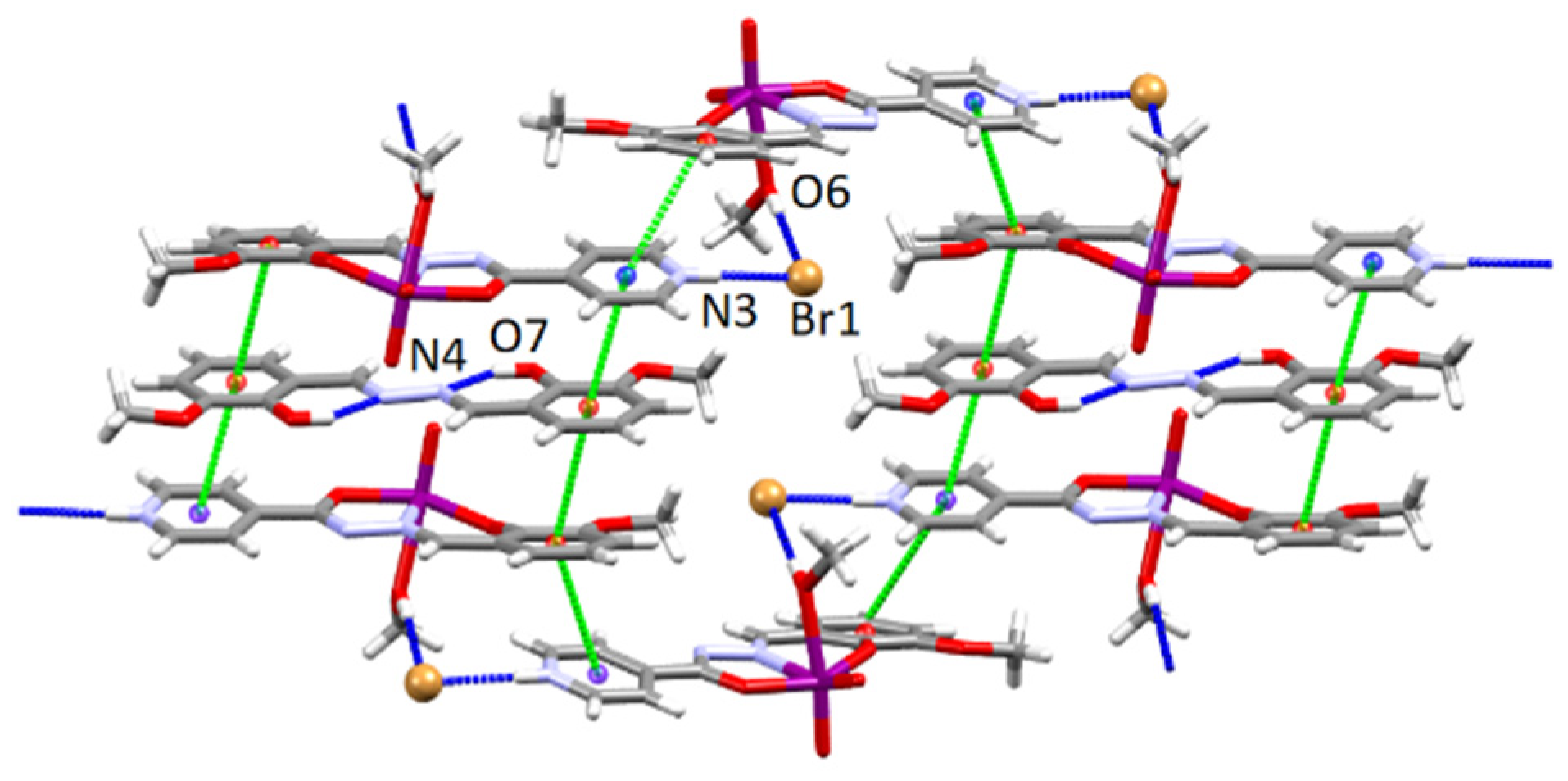
3.4.2. Structures of the Neutral Complexes 2Cl, 2Cl·MeOH and 2Br·0.5MeCN
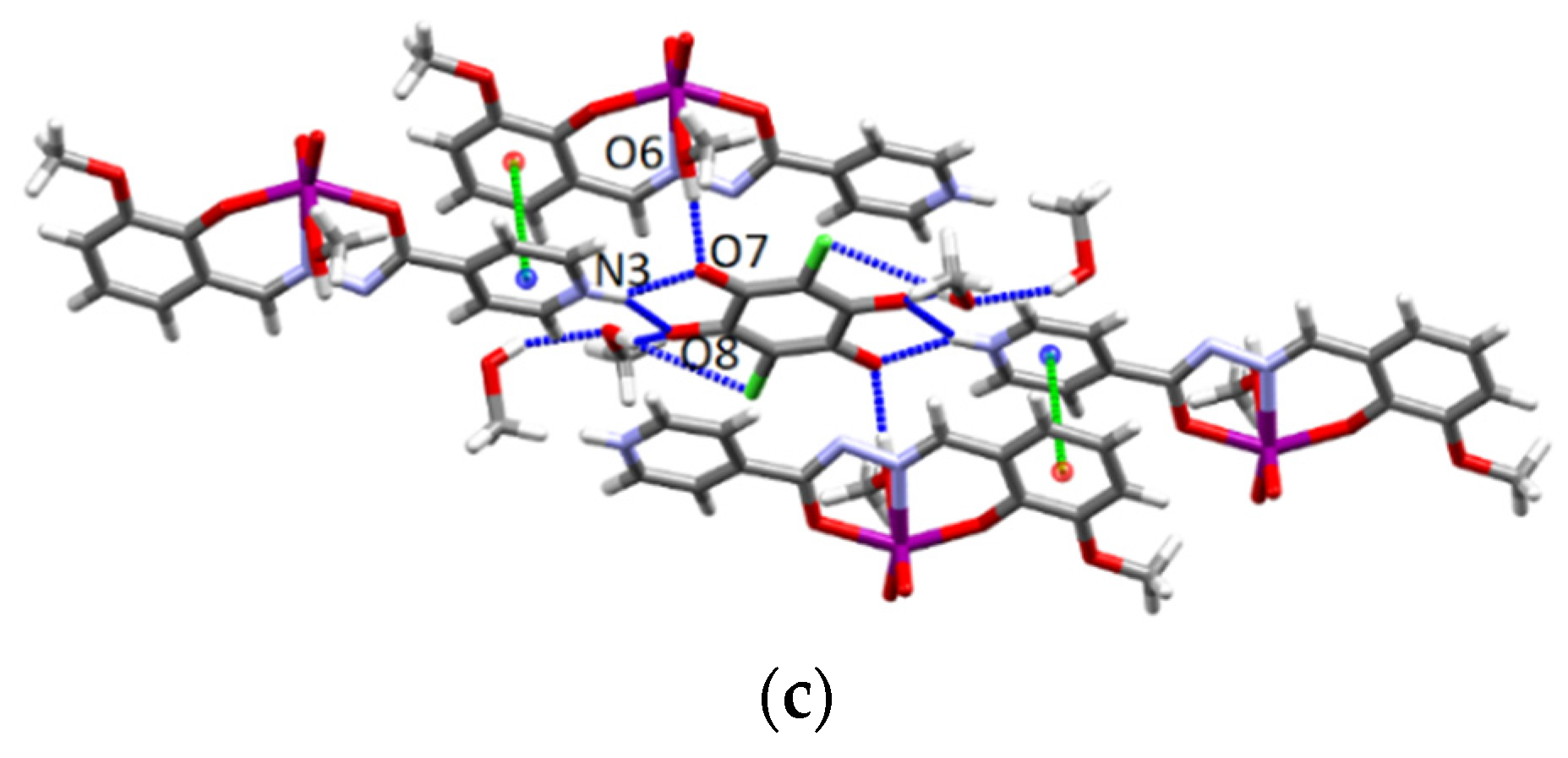
3.4.3. Structures of the Deprotonated Mo-Hydrazone Assemblies 1 and 2
3.5. Spectroscopic Studies
3.6. Therogravimetric Studies
3.7. Cytotoxicity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Su, X.; Aprahamian, I. Hydrazone-based switches, metallo-assemblies and sensors. Chem. Soc. Rev. 2014, 43, 1963–1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehn, J.-M. From supramolecular chemistry towards constitutional dynamic chemistry and adaptive chemistry. Chem. Soc. Rev. 2007, 36, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Ray, A.; Sen, S.; Mitra, S.; Hughes, D.L.; Butcher, R.J.; Batten, S.; Turner, D. Pseudohalide-induced structural variations in hydrazone-based metal complexes: Syntheses, electrochemical studies and structural aspects. Inorg. Chim. Acta 2008, 361, 2692–2700. [Google Scholar] [CrossRef]
- Jayanthi, E.; Kalaiselvi, S.; Padma, V.V.; Bhuvanesh, N.S.P.; Dharmaraj, N. Solvent assisted formation of ruthenium(iii) and ruthenium(ii) hydrazone complexes in one-pot with potential in vitro cytotoxicity and enhanced LDH, NO and ROS release. Dalton Trans. 2016, 45, 1693–1707. [Google Scholar] [CrossRef]
- Li, Y.; Yang, Z.; Zhou, M.; Li, Y.; He, J.; Wang, X.; Lin, Z. Ni(ii) and Co(ii) complexes of an asymmetrical aroylhydrazone: Synthesis, molecular structures, DNA binding, protein interaction, radical scavenging and cytotoxic activity. RSC Adv. 2017, 7, 41527–41539. [Google Scholar] [CrossRef] [Green Version]
- Ay, B.; Şahin, O.; Demir, B.S.; Saygideger, Y.; López-De-Luzuriaga, J.M.; Mahmoudi, G.; Safin, D.A. Antitumor effects of novel nickel–hydrazone complexes in lung cancer cells. New J. Chem. 2020, 44, 9064–9072. [Google Scholar] [CrossRef]
- Kobayashi, A.; Yamamoto, D.; Horiki, H.; Sawaguchi, K.; Matsumoto, T.; Nakajima, K.; Chang, H.-C.; Kato, M. Photoinduced Dimerization Reaction Coupled with Oxygenation of a Platinum(ii)–Hydrazone Complex. Inorg. Chem. 2014, 53, 2573–2581. [Google Scholar] [CrossRef]
- Bebić, N.; Topić, E.; Mandarić, M.; Hrenar, T.; Vrdoljak, V. Extending the structural landscape of Mo(vi) hydrazonato inorganic–organic POM-hybrids: An experimental and computational study. CrystEngComm 2021, 23, 6349–6358. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Prugovečki, B.; Matković-Čalogović, D.; Dreos, R.; Siega, P.; Tavagnacco, C. Zigzag Chain, Square Tetranuclear, and Polyoxometalate-Based Inorganic−Organic Hybrid Compounds—Molybdenum vs. Tungsten. Cryst. Growth Des. 2010, 10, 1373–1382. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Prugovečki, B.; Matković-Čalogović, D.; Pisk, J.; Dreos, R.; Siega, P. Supramolecular Hexagon and Chain Coordination Polymer Containing the MoO22+ Core: Structural Transformation in the Solid State. Cryst. Growth Des. 2011, 11, 1244–1252. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Prugovečki, B.; Matković-Čalogović, D.; Hrenar, T.; Dreos, R.; Siega, P. Three Polymorphic Forms of a Monomeric Mo(vi) Complex: Building Blocks for Two Metal–Organic Supramolecular Isomers. Intermolecular Interactions and Ligand Substituent Effects. Cryst. Growth Des. 2013, 13, 3773–3784. [Google Scholar] [CrossRef]
- Maurya, M.R.; Rana, L.; Avecilla, F. Catalytic oxidation of internal and terminal alkenes by oxidoperoxidomolybdenum(vi) and dioxidomolybdenum(VI) complexes. Inorg. Chim. Acta 2015, 429, 138–147. [Google Scholar] [CrossRef]
- Sinha, S.; Chakraborty, M.; Pramanik, N.R.; Raychaudhuri, T.K.; Mondal, T.K.; Sarkar, D.; Drew, M.G.; Ghosh, S.; Mandal, S.S. Dimer formation by symbiotic donor–acceptor interaction between two molecules of a specially designed dioxomolybdenum(vi) complex containing both donor and acceptor centers—A structural, spectroscopic and DFT study. Polyhedron 2013, 55, 192–200. [Google Scholar] [CrossRef]
- Bafti, A.; Razum, M.; Topić, E.; Agustin, D.; Pisk, J.; Vrdoljak, V. Implication of oxidant activation on olefin epoxidation catalysed by Molybdenum catalysts with aroylhydrazonato ligands: Experimental and theoretical studies. Mol. Catal. 2021, 512, 111764. [Google Scholar] [CrossRef]
- Mihalinec, J.; Pajski, M.; Guillo, P.; Mandarić, M.; Bebić, N.; Pisk, J.; Vrdoljak, V. Alcohol Oxidation Assisted by Molybdenum Hydrazonato Catalysts Employing Hydroperoxide Oxidants. Catalysts 2021, 11, 881. [Google Scholar] [CrossRef]
- Pisk, J.; Agustin, D.; Vrdoljak, V. Tetranuclear molybdenum(vi) hydrazonato epoxidation (pre)catalysts: Is water always the best choice? Catal. Commun. 2020, 142, 106027. [Google Scholar] [CrossRef]
- Biswal, D.; Pramanik, N.R.; Chakrabarti, S.; Drew, M.G.B.; Sarkar, B.; Maurya, M.R.; Mukherjee, S.K.; Chowdhury, P. New polymeric, dimeric and mononuclear dioxidomolybdenum(vi) complexes with an ONO donor ligand: Crystal structures, DFT calculations, catalytic performance and protein binding study of the ligand. New J. Chem. 2017, 41, 4116–4137. [Google Scholar] [CrossRef]
- Kargar, H.; Fallah-Mehrjardi, M.; Behjatmanesh-Ardakani, R.; Munawar, K.S.; Ashfaq, M.; Tahir, M.N. Selective oxidation of benzyl alcohols to benzaldehydes catalyzed by dioxomolybdenum Schiff base complex: Synthesis, spectral characterization, crystal structure, theoretical and computational studies. Transit. Met. Chem. 2021, 46, 437–455. [Google Scholar] [CrossRef]
- Kargar, H.; Fallah-Mehrjardi, M.; Behjatmanesh-Ardakani, R.; Munawar, K.S.; Ashfaq, M.; Tahir, M.N. Synthesis, spectral characterization, SC-XRD, HSA, DFT and catalytic activity of a dioxidomolybdenum complex with aminosalicyl-hydrazone Schiff base ligand: An experimental and theoretical approach. Polyhedron 2021, 208, 115428. [Google Scholar] [CrossRef]
- Cvijanović, D.; Pisk, J.; Pavlović, G.; Šišak-Jung, D.; Matković-Čalogović, D.; Cindrić, M.; Agustin, D.; Vrdoljak, V. Discrete mononuclear and dinuclear compounds containing a MoO22+ core and 4-aminobenzhydrazone ligands: Synthesis, structure and organic-solvent-free epoxidation activity. New J. Chem. 2019, 43, 1791–1802. [Google Scholar] [CrossRef]
- Mazur, L.; Materek, I.; Bond, A.D.; Jones, W. Multicomponent Crystal Forms of a Biologically Active Hydrazone with Some Dicarboxylic Acids: Salts or Cocrystals? Cryst. Growth Des. 2019, 19, 2663–2678. [Google Scholar] [CrossRef]
- Zhuo, C.; Sun, Y.; Wang, F.; Zhang, J. In Situ Encapsulation of Organic Sulfates in Layered Structures of Zinc and Tris(4-(1H-Imidazol-1-yl)phenyl)amine. Cryst. Growth Des. 2020, 20, 4228–4231. [Google Scholar] [CrossRef]
- Braga, D.; Grepioni, F.; Shemchuk, O. Organic–inorganic ionic co-crystals: A new class of multipurpose compounds. CrystEngComm 2018, 20, 2212–2220. [Google Scholar] [CrossRef]
- Ngan, N.K.; Wong, R.C.S.; Lo, K.M.; Ng, S.W. [N′-(5-Chloro-2-oxidobenzyl-κO)-2,4-dihydroxybenzohydrazidato-κ2N′,O](methanol-κO)dioxidomolybdenum(VI)–4,4′-bipyridine (1/1). Acta Crystallogr. Sect. E Struct. Rep. Online 2011, 67, m747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.X.; Zhang, X.M.; Wang, X. Synthesis, characterization and crystal structure of cis-dioxomolybdenum (VI)-Schiff base complex [MoO2(O-C6H4CH–NN–COCH2NC5H5)CH3OH]Cl. Polyhedron 1994, 13, 441–444. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Prugovečki, B.; Pulić, I.; Cigler, M.; Sviben, D.; Vuković, J.P.; Novak, P.; Matković-Čalogović, D.; Cindrić, M. Dioxidomolybdenum(vi) complexes with isoniazid-related hydrazones: Solution-based, mechanochemical and UV-light assisted deprotonation. New J. Chem. 2015, 39, 7322–7332. [Google Scholar] [CrossRef] [Green Version]
- Mandarić, M.; Prugovečki, B.; Cvijanović, D.; Vuković, J.P.; Lovrić, J.; Skočibušić, M.; Odžak, R.; Cindrić, M.; Vrdoljak, V. Vapour- and solvent-mediated crystalline transformations in Mo(vi) hydrazone complexes controlled by noncovalent interactions. CrystEngComm 2019, 21, 6281–6292. [Google Scholar] [CrossRef]
- Kabir, K.; Tobita, H.; Matsuo, H.; Nagayoshi, K.; Yamada, K.; Adachi, K.; Sugiyama, Y.; Kitagawa, S.; Kawata, S. Crystal Engineering Using the Versatility of 2,5-Dichloro-3,6-dihydroxy-1,4-benzoquinone with Organic and Metal Complex Partners. Cryst. Growth Des. 2003, 3, 791–798. [Google Scholar] [CrossRef]
- Molčanov, K.; Kojić-Prodić, B.; Meden, A. Unique Electronic and Structural Properties of 1,4-Benzoquinones: Crystallochemistry of Alkali Chloranilate Hydrates. Croat. Chem. Acta 2009, 82, 387–396. Available online: https://hrcak.srce.hr/39656 (accessed on 9 April 2021).
- Ishida, H. Pyrrolidinium chloranilate. Acta Crystallogr. Sect. E Struct. Rep. Online 2004, 60, o974–o976. [Google Scholar] [CrossRef]
- Gotoh, K.; Asaji, T.; Ishida, H. Hydrogen bonding in two solid phases of phenazine–chloranilic acid (1/1) determined at 170 and 93 K. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2006, 63, o17–o20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kühl, O.; Goutal, S. Acid/Base versus Redox Reaction—The Case of Chloranilic Acid and Hydrazine: A Supramolecular Decision? Cryst. Growth Des. 2005, 5, 1875–1879. [Google Scholar] [CrossRef]
- Dworniczak, M. Anion radical formation in the reaction of chloranilic acid with n-methylpiperidine in acetonitrile. React. Kinet. Catal. Lett. 2003, 78, 65–72. [Google Scholar] [CrossRef]
- Chang, M.; Kobayashi, A.; Nakajima, K.; Chang, H.-C.; Kato, M. Dimensionality Control of Vapochromic Hydrogen-Bonded Proton-Transfer Assemblies Composed of a Bis(hydrazone)iron(ii) Complex. Inorg. Chem. 2011, 50, 8308–8317. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Dosen, M.-A.; Chang, M.; Nakajima, K.; Noro, S.-I.; Kato, M. Synthesis of Metal−Hydrazone Complexes and Vapochromic Behavior of Their Hydrogen-Bonded Proton-Transfer Assemblies. J. Am. Chem. Soc. 2010, 132, 15286–15298. [Google Scholar] [CrossRef]
- Kawata, S.; Kitagawa, S.; Kumagai, H.; Kudo, C.; Kamesaki, H.; Ishiyama, T.; Suzuki, R.; Kondo, M.; Katada, M. Rational Design of a Novel Intercalation System. Layer-Gap Control of Crystalline Coordination Polymers, {[Cu(CA)(H2O)m](G)}n (m = 2, G = 2,5-Dimethylpyrazine and Phenazine; m = 1, G = 1,2,3,4,6,7,8,9-Octahydrophenazine). Inorg. Chem. 1996, 35, 4449–4461. [Google Scholar] [CrossRef]
- Jurić, M.; Molčanov, K.; Žilić, D.; Kojić-Prodić, B. From mononuclear to linear one-dimensional coordination species of copper(ii)–chloranilate: Design and characterization. RSC Adv. 2016, 6, 62785–62796. [Google Scholar] [CrossRef] [Green Version]
- Kabir, M.; Miyazaki, N.; Kawata, S.; Adachi, K.; Kumagai, H.; Inoue, K.; Kitagawa, S.; Iijima, K.; Katada, M. Novel layered structures constructed from iron–chloranilate compounds. Coord. Chem. Rev. 2000, 198, 157–169. [Google Scholar] [CrossRef]
- Maurya, M.R.; Rana, L.; Avecilla, F. Molybdenum complexes with a μ-O{MoO2}2 core: Their synthesis, crystal structure and application as catalysts for the oxidation of bicyclic alcohols using N-based additives. New J. Chem. 2017, 41, 724–734. [Google Scholar] [CrossRef]
- Gündüz, T. Some New Organic Complexes of Molybdenum. Commun. Fac. Sci. Univ. Ank. Ser. B Chem. Chem. Eng. 1964, 11, 021–031. [Google Scholar] [CrossRef]
- Gomes, L.R.; Low, J.N.; Correira, N.R.D.L.; Noguiera, T.C.; Pinheiro, A.C.; de Souza, M.V.; Wardell, J.L.; Wardell, S.M. Crystal structures and Hirshfeld surface analysis of four 1,4-bis(methoxyphenyl)-2,3-diazabuta-1,3-dienes: Comparisons of the intermolecular interactions in related compounds. Z. Krist. Cryst. Mater. 2019, 234, 59–71. [Google Scholar] [CrossRef]
- Ghosh, M.; Ta, S.; Matalobos, J.S.; Das, D. Azine based smart probe for optical recognition and enrichment of Mo(vi). Dalton Trans. 2018, 47, 11084–11090. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.J.J.; McDonald, J.W.; Newton, W.E. Synthesis of molybdenum(iv) and molybdenum(v) complexes using oxo abstraction by phosphines. Mechanistic implications. Inorg. Chem. 1976, 15, 2612–2615. [Google Scholar] [CrossRef]
- Arnáiz, F.J.; Aguado, R.; Pedrosa, M.R.; Mahía, J.; Maestro, M.A. Addition compounds of MoO2Br2 from MoO2Br2(H2O)2. Molecular structure of MoO2Br2{OP[N(CH3)2]3}2 and MoO2Br2{CH2[P(O)(C6H5)2]2}. Polyhedron 2002, 21, 1635–1642. [Google Scholar] [CrossRef]
- Graham, R.L.; Hepler, L.G. Heats of Formation of Molybdenum Oxychlorides. J. Phys. Chem. 1959, 63, 723–724. [Google Scholar] [CrossRef]
- Pisk, J.; Hrenar, T.; Rubčić, M.; Pavlović, G.; Damjanović, V.; Lovrić, J.; Cindrić, M.; Vrdoljak, V. Comparative studies on conventional and solvent-free synthesis toward hydrazones: Application of PXRD and chemometric data analysis in mechanochemical reaction monitoring. CrystEngComm 2018, 20, 1804–1817. [Google Scholar] [CrossRef]
- Kurteva, V.B.; Simeonov, S.; Stoilova-Disheva, M. Symmetrical Acyclic Aryl Aldazines with Antibacterial and Antifungal Activity. Pharmacol. Pharm. 2011, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- CrysAlisPro Software System, Version 1.171.38.41; Rigaku Oxford Diffraction: Oxford, UK, 2015.
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.J.; van de Streek, J.; Wood, P. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Degen, T.; Sadki, M.; Bron, E.; König, U.; Nénert, G. The High Score suite. Powder Diffr. 2014, 29, S13–S18. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Lei, Y.; Fu, C. Synthesis and X-Ray Structural Characterization of Dioxomolybdenum(vi) Complexes with 2’-(5-Chloro-2-hydroxybenzylidene)isonicotinohydrazide and 2′-(2-Hydroxy-3-methoxybenzylidene)isonicotinohydrazide. Synth. React. Inorg. Met. Nano-Metal Chem. 2011, 41, 704–709. [Google Scholar] [CrossRef]
- Alghool, S.; Slebodnick, C. Supramolecular structures of mononuclear and dinuclear dioxomolybdenum(vi) complexes via hydrogen bonds and π–π stacking, thermal studies and electrochemical measurements. Polyhedron 2014, 67, 11–18. [Google Scholar] [CrossRef]
- Bikas, R.; Lippolis, V.; Noshiranzadeh, N.; Farzaneh-Bonab, H.; Blake, A.J.; Siczek, M.; Hosseini-Monfared, H.; Lis, T. Electronic Effects of Aromatic Rings on the Catalytic Activity of Dioxidomolybdenum(vi)–Hydrazone Complexes. Eur. J. Inorg. Chem. 2016, 2017, 999–1006. [Google Scholar] [CrossRef] [Green Version]
- Randell, N.M.; Thompson, L.K.; Dawe, L.N. 6,6′-Dimethoxy-2,2′-{[(E,E)-hydrazine-1,2-diylidene]bis(methanylylidene)}diphenol methanol disolvate. Acta Crystallogr. Sect. E Struct. Rep. Online 2012, 68, o2711. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50/µmol L−1 | |
|---|---|---|
| HepG2 | THP-1 | |
| [1H]Cl | 98.33 | 2.77 |
| [1H](ClA)0.5 | >100 | 26.7 |
| [1H]Cl·0.5VA | >100 | 29.2 |
| 2Cl | >100 | 3.21 |
| 1 | >100 | 4.54 |
| staurosporine | 14.31 | 0.18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mandarić, M.; Prugovečki, B.; Kekez, I.; Musija, D.; Parlov Vuković, J.; Cindrić, M.; Vrdoljak, V. Counter Anion Effects on the Formation and Structural Transformations of Mo(vi)-Hydrazone Coordination Assemblies: Salts, Solvates, Co-Crystals, and Neutral Complexes. Crystals 2022, 12, 443. https://doi.org/10.3390/cryst12040443
Mandarić M, Prugovečki B, Kekez I, Musija D, Parlov Vuković J, Cindrić M, Vrdoljak V. Counter Anion Effects on the Formation and Structural Transformations of Mo(vi)-Hydrazone Coordination Assemblies: Salts, Solvates, Co-Crystals, and Neutral Complexes. Crystals. 2022; 12(4):443. https://doi.org/10.3390/cryst12040443
Chicago/Turabian StyleMandarić, Mirna, Biserka Prugovečki, Ivana Kekez, Danijela Musija, Jelena Parlov Vuković, Marina Cindrić, and Višnja Vrdoljak. 2022. "Counter Anion Effects on the Formation and Structural Transformations of Mo(vi)-Hydrazone Coordination Assemblies: Salts, Solvates, Co-Crystals, and Neutral Complexes" Crystals 12, no. 4: 443. https://doi.org/10.3390/cryst12040443
APA StyleMandarić, M., Prugovečki, B., Kekez, I., Musija, D., Parlov Vuković, J., Cindrić, M., & Vrdoljak, V. (2022). Counter Anion Effects on the Formation and Structural Transformations of Mo(vi)-Hydrazone Coordination Assemblies: Salts, Solvates, Co-Crystals, and Neutral Complexes. Crystals, 12(4), 443. https://doi.org/10.3390/cryst12040443