
Control of Molecular Packing in Crystal and Electron Communication of Two Ferrocenyl Moieties across Chiral Isomannide or Isosorbide Bridge

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
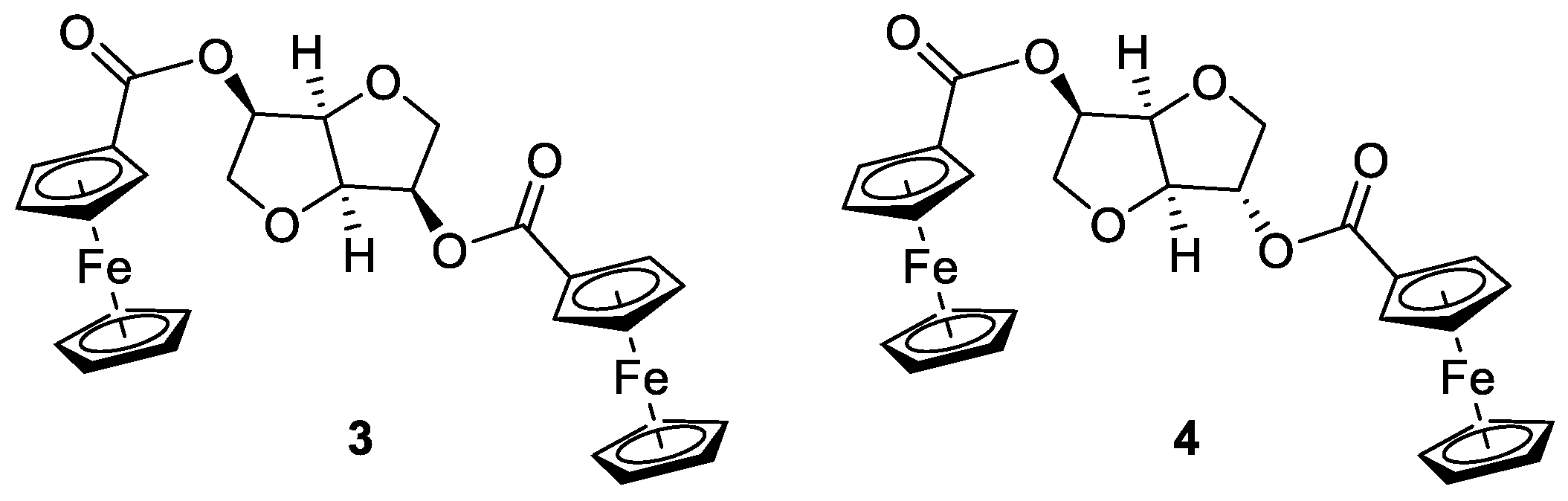
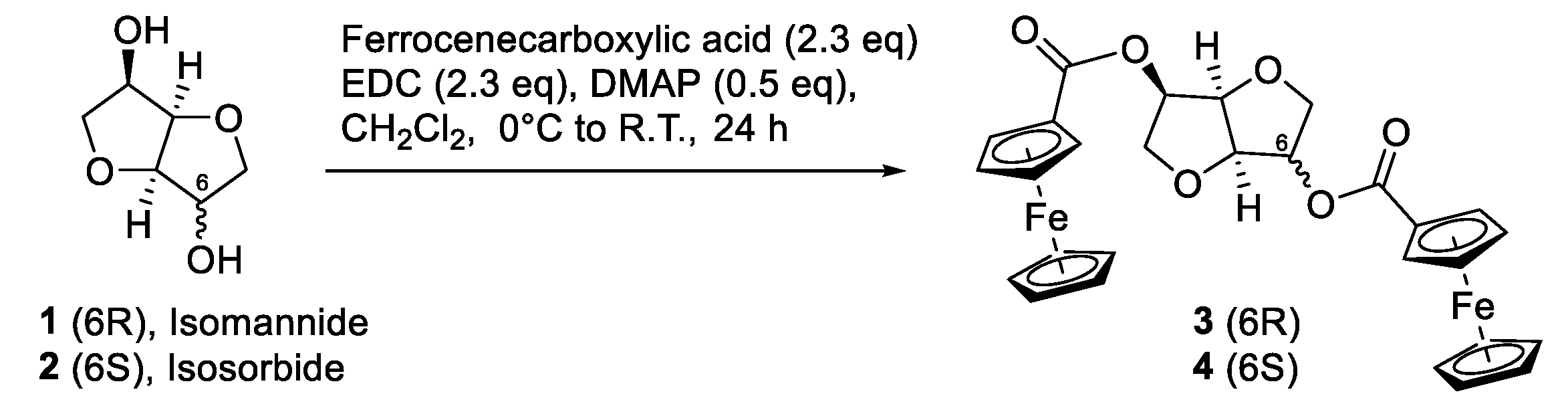
2.1. General Procedure for the Synthesis of the Bis-Ferrocene Carboxylates 3 and 4 [30]
2.2. Electrochemical and Spectroelectrochemical Measurements
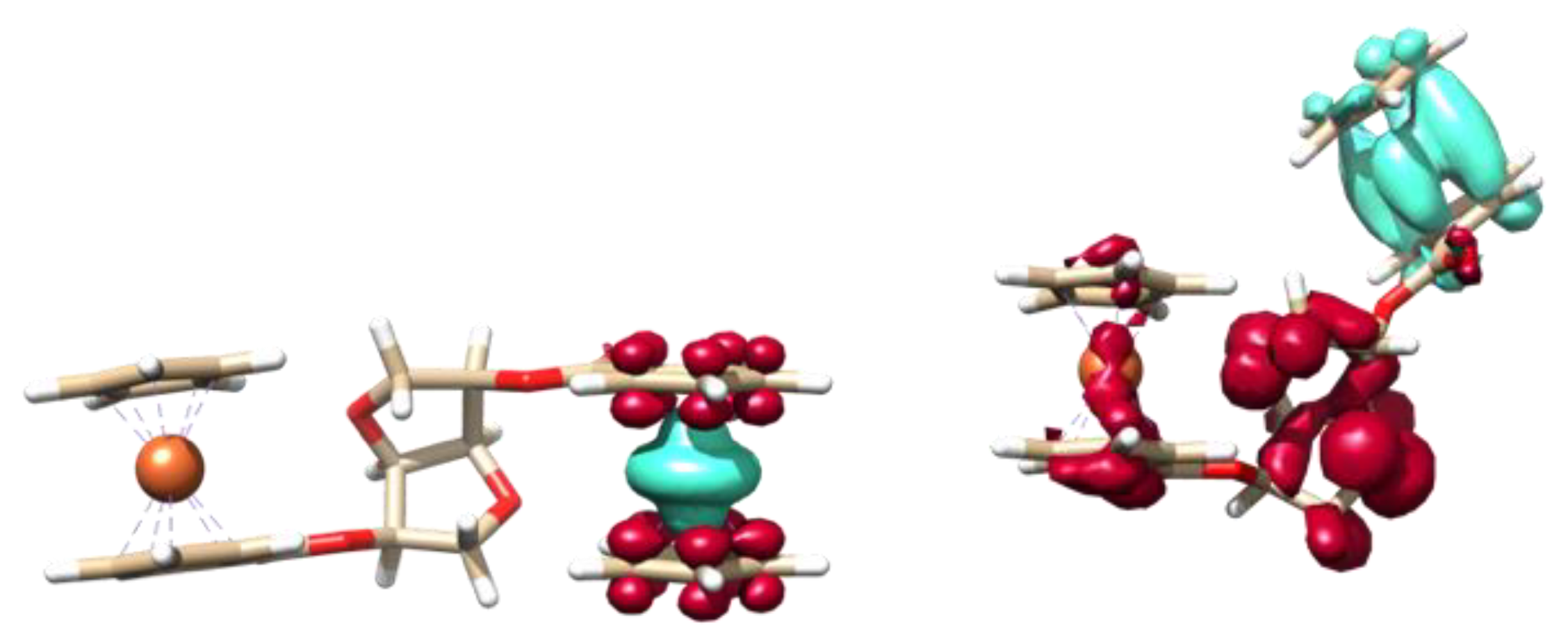
2.3. Computational Studies
3. Results and Discussion
3.1. Synthesis of Ferrocenyl Derivatives
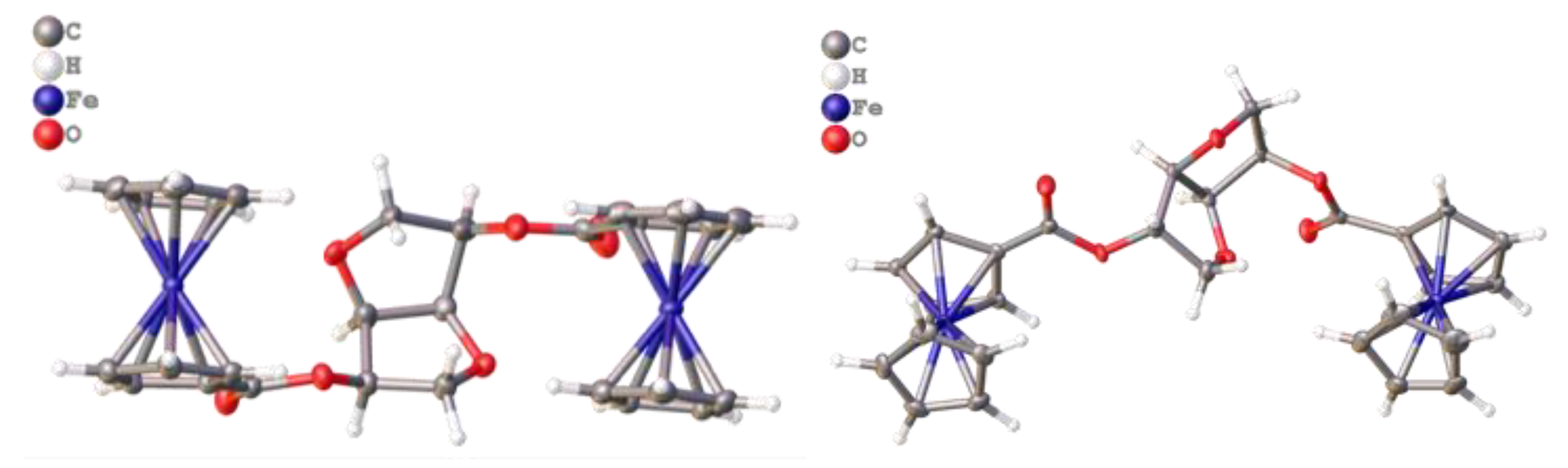
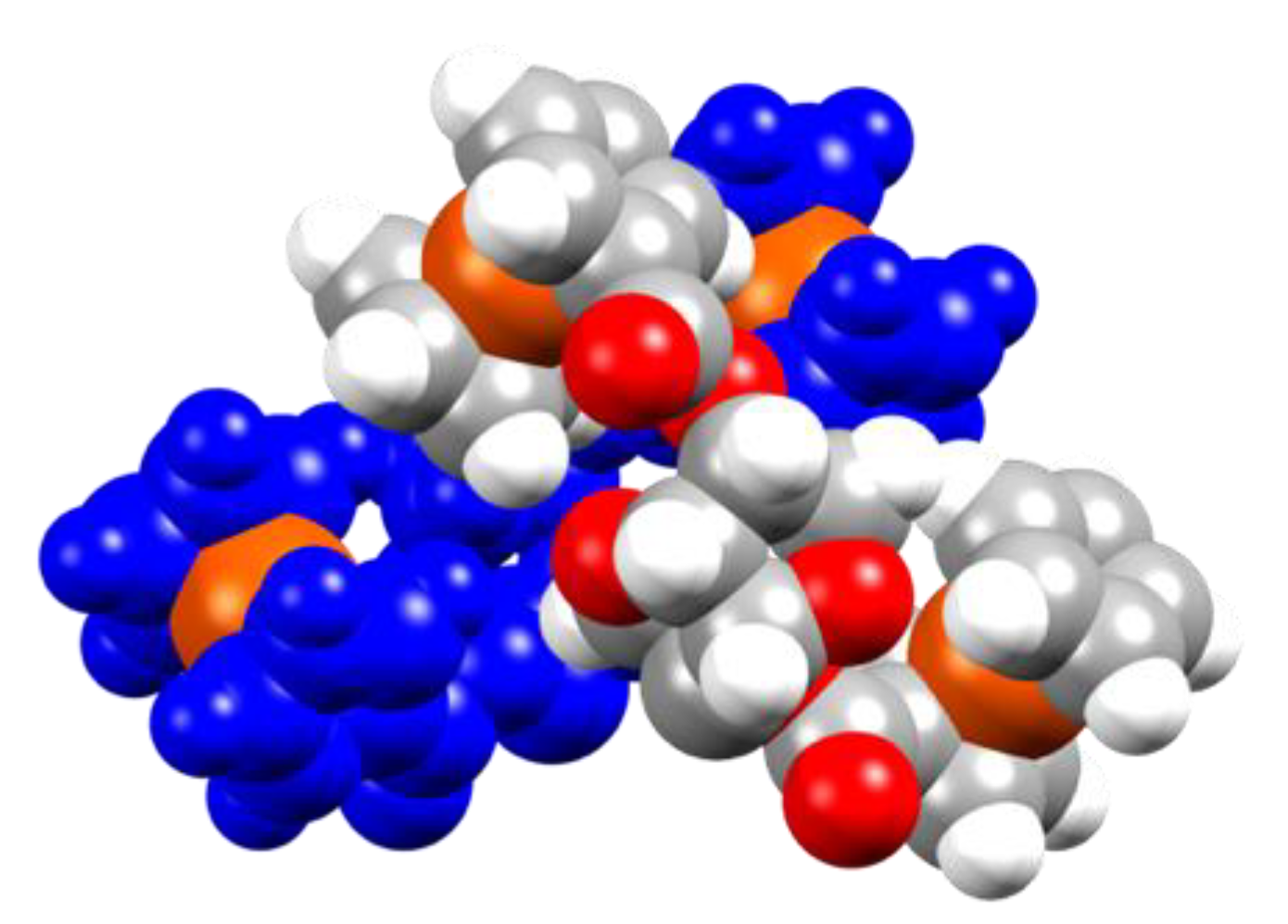
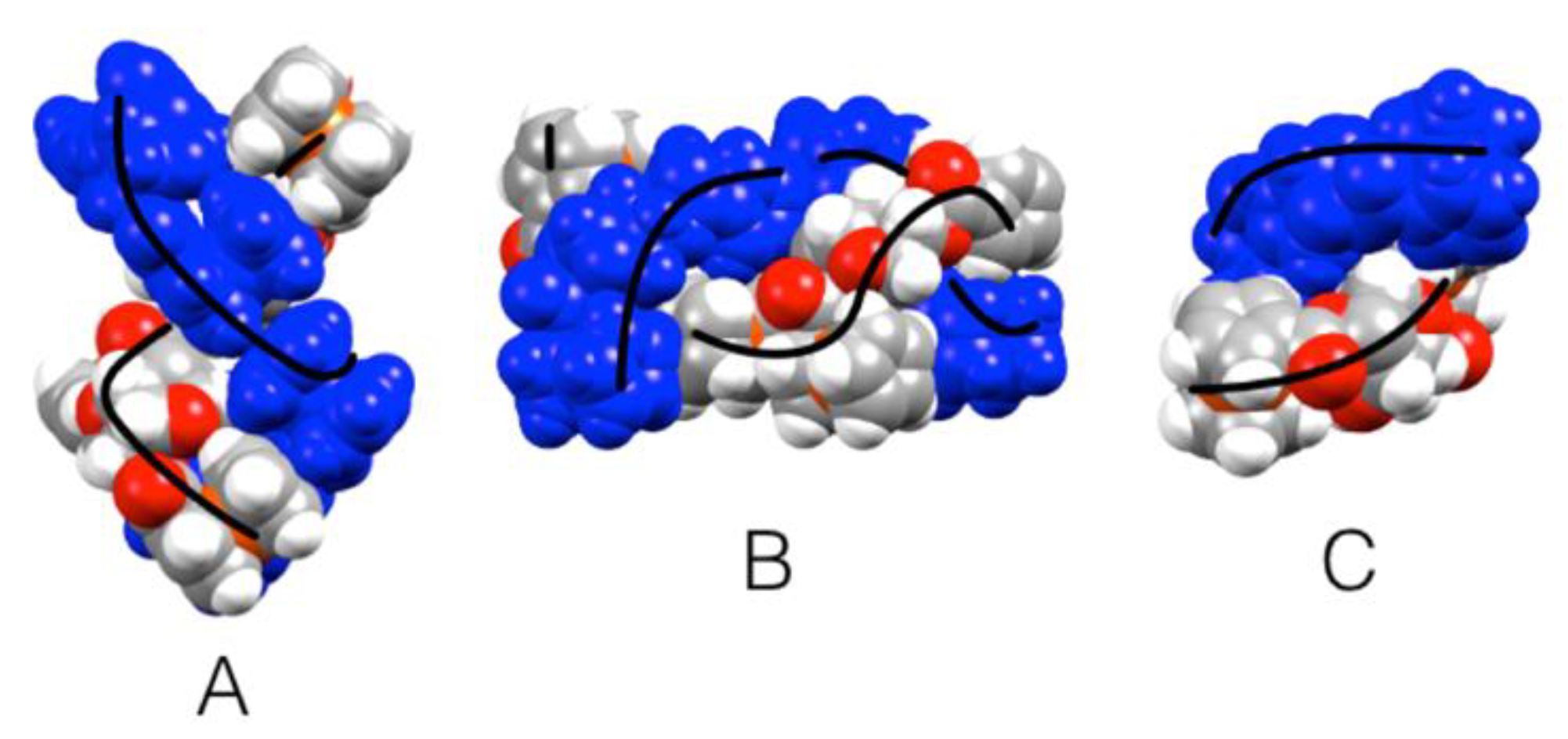
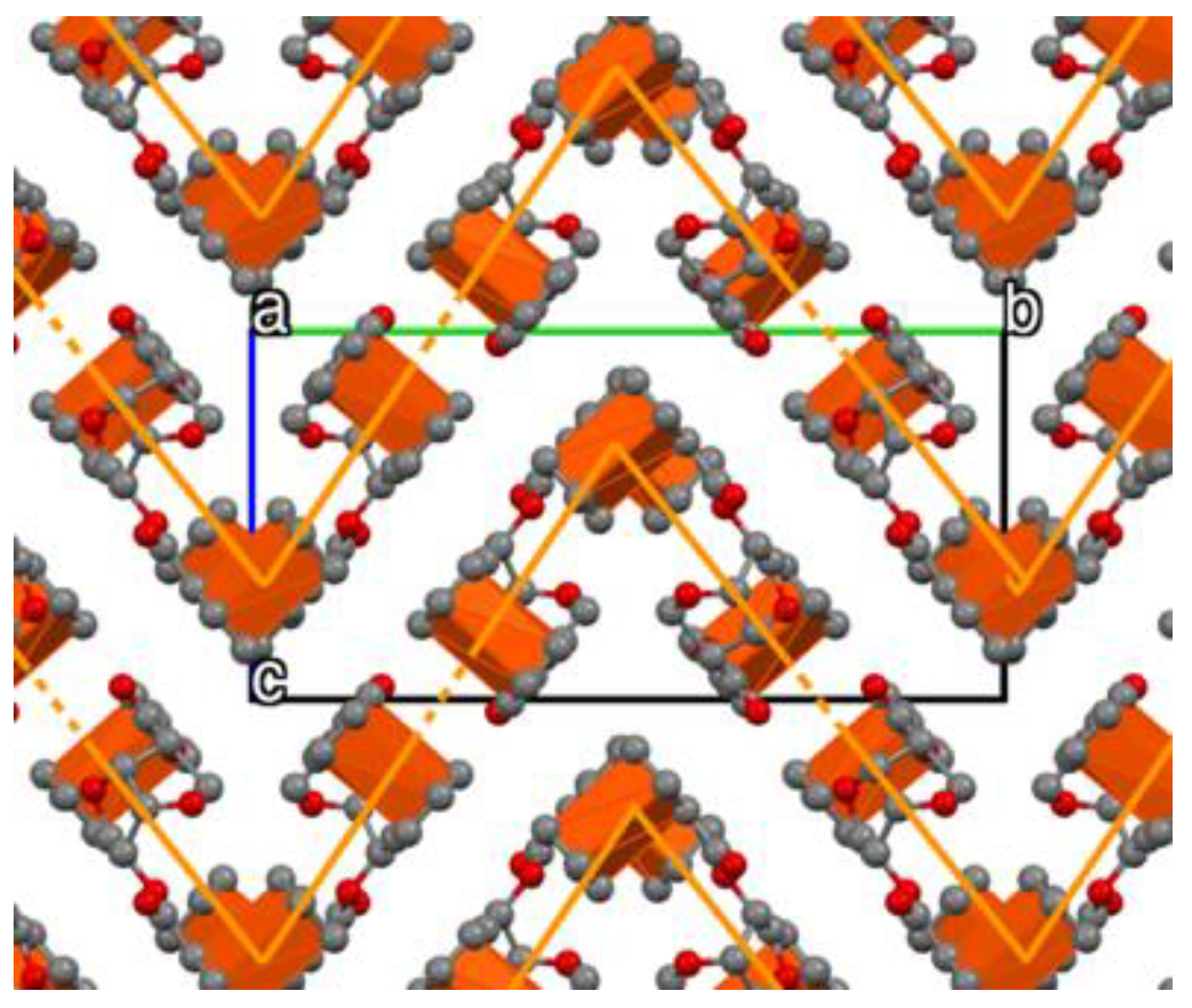
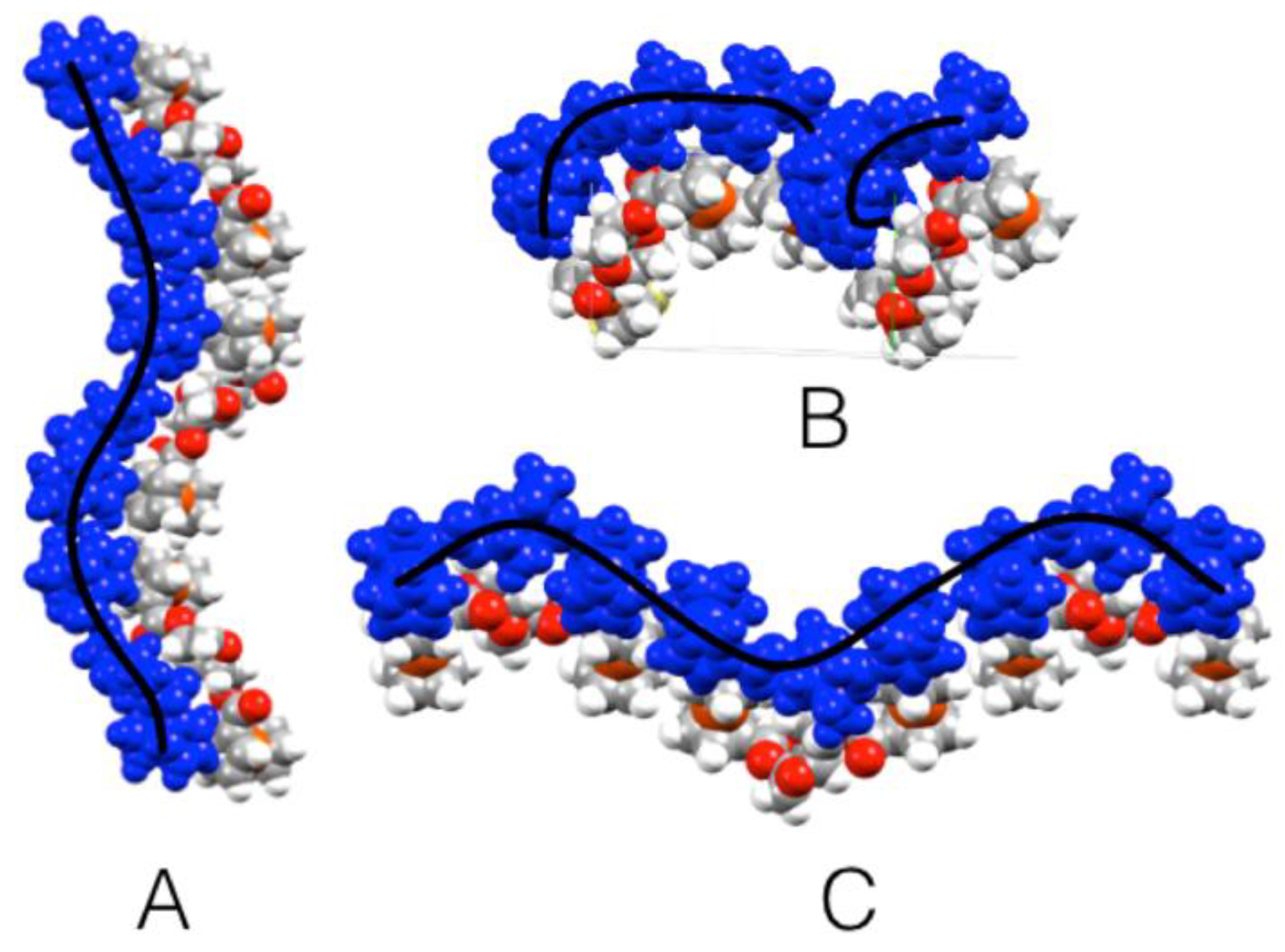
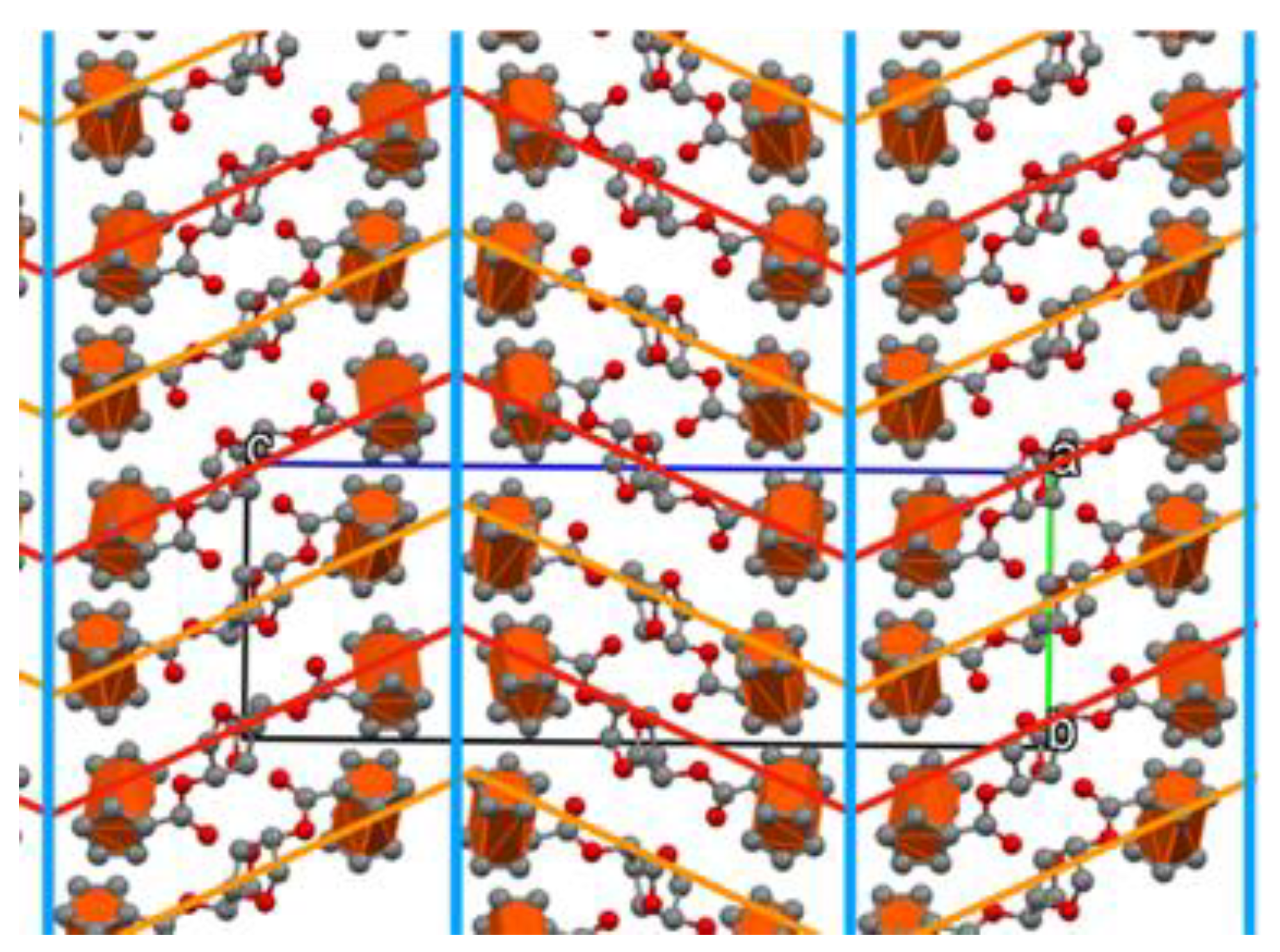
3.2. Crystal Structure of Ferrocenyl Derivatives 3 and 4
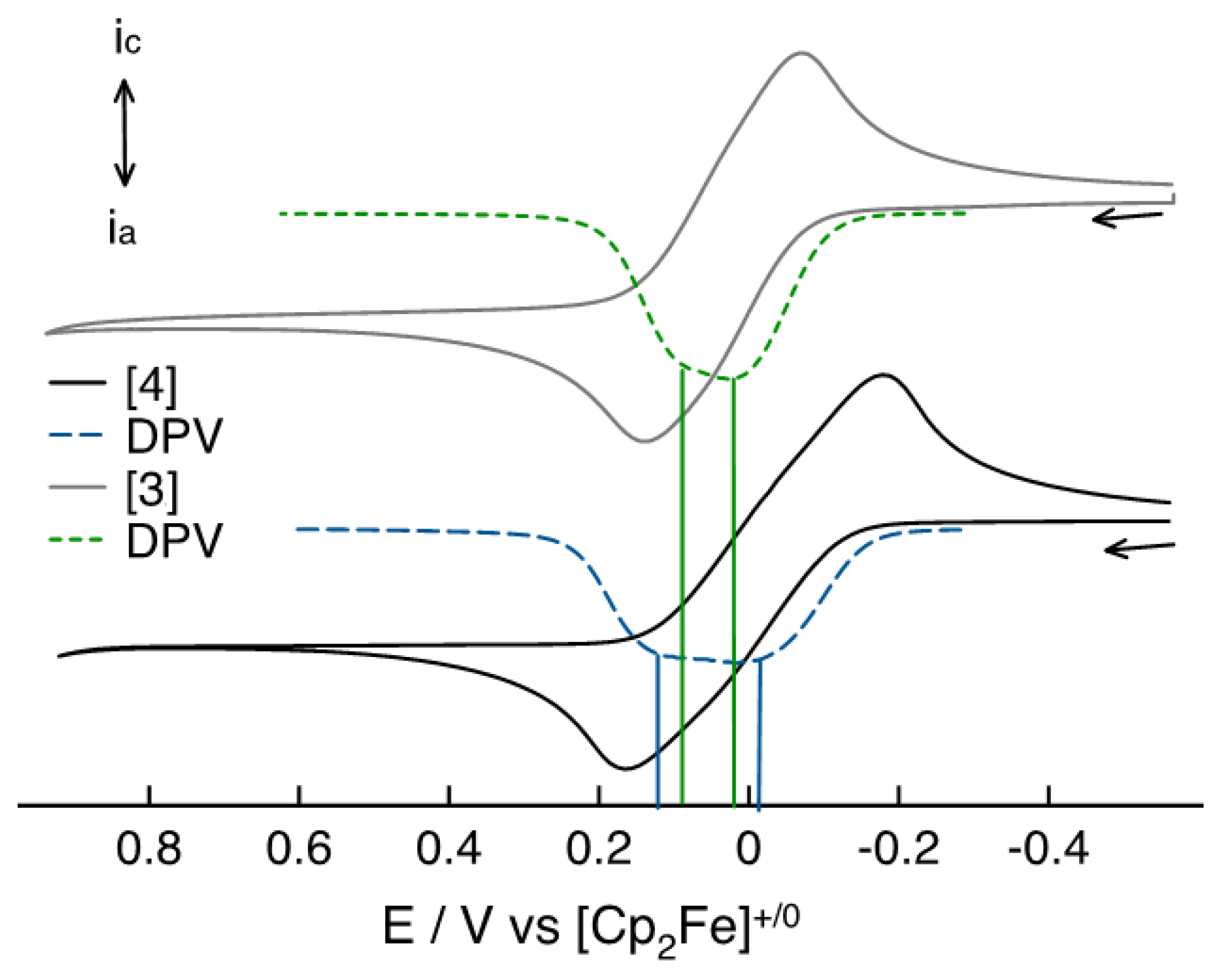
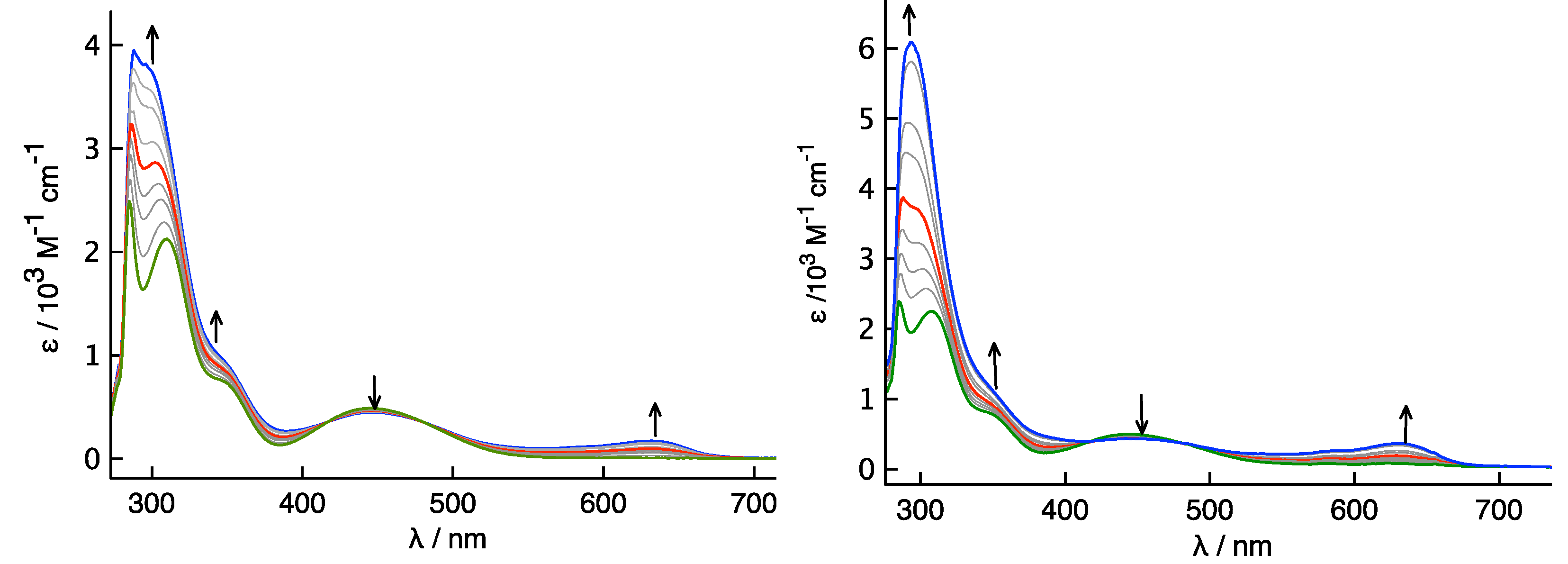
3.3. Electrochemical and Spectroelectrochemical Studies of Compounds 3 and 4
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Richard Keene, F. Isolation and characterisation of stereoisomers in di- And tri-nuclear complexes. Chem. Soc. Rev. 1998, 27, 185–193. [Google Scholar] [CrossRef]
- Otón, F.; Ratera, I.; Espinosa, A.; Tárraga, A.; Veciana, J.; Molina, P. Conformationally modulated intramolecular electron transfer process in a diaza[2,2]ferrocenophane. Inorg. Chem. 2010, 49, 3183–3191. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Mandal, A.; Paretzki, A.; Beyer, K.; Kaim, W.; Lahiri, G.K. Isomeric Diruthenium Complexes of a Heterocyclic and Quinonoid Bridging Ligand: Valence and Spin Alternatives for the Metal/Ligand/Metal Arrangement. Inorg. Chem. 2016, 55, 12357–12365. [Google Scholar] [CrossRef]
- Ma, X.; Lin, C.S.; Hu, S.M.; Tan, C.H.; Wen, Y.H.; Sheng, T.L.; Wu, X.T. Influence of central metalloligand geometry on electronic communication between metals: Syntheses, crystal structures, mmct properties of isomeric cyanido-bridged Fe2Ru complexes, and TDDFT calculations. Chem.—A Eur. J. 2014, 20, 7025–7036. [Google Scholar] [CrossRef]
- Hildebrandt, A.; Lang, H. (Multi)ferrocenyl Five-Membered heterocycles: Excellent connecting units for electron transfer studies. Organometallics 2013, 32, 5640–5653. [Google Scholar] [CrossRef]
- Ma, X.; Lin, C.S.; Zhu, X.Q.; Hu, S.M.; Sheng, T.L.; Wu, X.T. An Unusually Delocalized Mixed-Valence State of a Cyanidometal-Bridged Compound Induced by Thermal Electron Transfer. Angew. Chem.—Int. Ed. 2017, 56, 1605–1609. [Google Scholar] [CrossRef]
- Packheiser, R.; Jakob, A.; Ecorchard, P.; Walfort, B.; Lang, H. Diphenylphosphinoferrocene gold(I) acetylides: Synthesis of heterotri- And heterotetrametallic transition metal complexes. Organometallics 2008, 27, 1214–1226. [Google Scholar] [CrossRef]
- Rößler, K.; Rüffer, T.; Walfort, B.; Packheiser, R.; Holze, R.; Zharnikov, M.; Lang, H. Synthesis, characterization and electrochemical behavior of unsymmetric transition metal-terminated biphenyl ethynyl thiols. J. Organomet. Chem. 2007, 692, 1530–1545. [Google Scholar] [CrossRef]
- Klenk, S.; Rupf, S.; Suntrup, L.; Van Der Meer, M.; Sarkar, B. The Power of Ferrocene, Mesoionic Carbenes, and Gold: Redox-Switchable Catalysis. Organometallics 2017, 36, 2026–2035. [Google Scholar] [CrossRef]
- D’Alessandro, D.M.; Kelso, L.S.; Keene, F.R. Stereochemical influences on intervalence charge transfer in homodinuclear complexes of ruthenium. Inorg. Chem. 2001, 40, 6841–6844. [Google Scholar] [CrossRef]
- D’Alessandro, D.M.; Keene, F.R. Intervalence charge transfer (IVCT) in trinuclear and tetranuclear complexes of iron, ruthenium, and osmium. Chem. Rev. 2006, 106, 2270–2298. [Google Scholar] [CrossRef]
- Carter, C.; Kratish, Y.; Jurca, T.; Gao, Y.; Marks, T.J. Bis-Ferrocenyl-Pyridinediimine Trinuclear Mixed-Valent Complexes with Metal-Binding Dependent Electronic Coupling: Synthesis, Structures, and Redox-Spectroscopic Characterization. J. Am. Chem. Soc. 2020, 142, 18715–18729. [Google Scholar] [CrossRef] [PubMed]
- Roeser, S.; Maji, S.; Benet-Buchholz, J.; Pons, J.; Llobet, A. Synthesis, characterization, reactivity, and linkage isomerization of Ru(Cl)2(L)(DMSO)2 complexes. Eur. J. Inorg. Chem. 2013, 2, 232–240. [Google Scholar] [CrossRef]
- Ringenberg, M.R.; Holz, J.; Peters, R. Intervalence of two planar chiral 2-methylferrocenyl groups over a diaurum bridge. Dalt. Trans. 2018, 47, 12873–12878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayerbe Garcia, M.; Frey, W.; Ringenberg, M.R.; Schwilk, M.; Peters, R. Dinuclear planar chiral ferrocenyl gold(i) & gold(ii) complexes. Chem. Commun. 2015, 51, 16806–16809. [Google Scholar] [CrossRef] [Green Version]
- Fenouillot, F.; Rousseau, A.; Colomines, G.; Saint-Loup, R.; Pascault, J.P. Polymers from renewable 1,4:3,6-dianhydrohexitols (isosorbide, isomannide and isoidide): A review. Prog. Polym. Sci. 2010, 35, 578–622. [Google Scholar] [CrossRef]
- Shou, Q.; Yuan, R.; Ma, G.; Liang, X.; Wan, J.; Xian, M.; Wang, Q. Chiral nanostructures of isosorbide- and isomannide-based polyurethanes. Polymer 2019, 164, 118–125. [Google Scholar] [CrossRef]
- Wu, J.; Eduard, P.; Jasinska-Walc, L.; Rozanski, A.; Noordover, B.A.J.; Van Es, D.S.; Koning, C.E. Fully isohexide-based polyesters: Synthesis, characterization, and structure-properties relations. Macromolecules 2013, 46, 384–394. [Google Scholar] [CrossRef]
- Osterkamp, F.; Wehlan, H.; Koert, U.; Wiesner, M.; Raddatz, P.; Goodman, S.L. Synthesis and biological evaluation of dianhydrohexitol integrin antagonists. Tetrahedron 1999, 55, 10713–10734. [Google Scholar] [CrossRef]
- Freitas, R.F.; Teixeira, T.S.P.; Barros, T.G.; Santos, J.A.N.; Kondo, M.Y.; Juliano, M.A.; Juliano, L.; Blaber, M.; Antunes, O.A.C.; Abrahão, O.; et al. Isomannide derivatives as new class of inhibitors for human kallikrein 7. Bioorg. Med. Chem. Lett. 2012, 22, 6072–6075. [Google Scholar] [CrossRef]
- Dhasaiyan, P.; Parekh, N.; Vijai Kumar Reddy, T.; Sandhya Rani, G.; Prabhavathi Devi, B.L.A.; Prasad, B.L.V. Self-assembly of isomannide-based monoesters of C18-fatty acids and their cellular uptake studies. RSC Adv. 2016, 6, 72074–72079. [Google Scholar] [CrossRef]
- Koert, U. Isomannide and Isosorbide. In Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons, Ltd.: Chichester, UK, 2012; pp. 1–5. ISBN 047084289X. [Google Scholar]
- Kadraoui, M.; Maunoury, T.; Derriche, Z.; Guillarme, S.; Saluzzo, C. Isohexides as versatile scaffolds for asymmetric catalysis. Eur. J. Org. Chem. 2015, 2015, 441–457. [Google Scholar] [CrossRef]
- Rose, M.; Palkovits, R. Isosorbide as a renewable platform chemical for versatile applications-quo vadis? ChemSusChem 2012, 5, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Eduard, P.; Thiyagarajan, S.; Van Haveren, J.; Van Es, D.S.; Koning, C.E.; Lutz, M.; Fonseca Guerra, C. Isohexide derivatives from renewable resources as chiral building blocks. ChemSusChem 2011, 4, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Ramachandran, U. The synthesis and applications of asymmetric phase-transfer catalysts derived from isomannide and isosorbide. Tetrahedron 2005, 61, 4141–4148. [Google Scholar] [CrossRef]
- M’Sahel, M.; Obadia, M.M.; Medimagh, R.; Serghei, A.; Said Zina, M.; Drockenmuller, E.; Malek, M.; Obadia, M.M.; Medimagh, R.; Serghei, A.; et al. Biosourced 1,2,3-triazolium ionic liquids derived from isosorbide. New J. Chem. 2016, 40, 740–747. [Google Scholar] [CrossRef]
- Ibrahim, H.; Bournaud, C.; Guillot, R.; Toffano, M.; Vo-Thanh, G. Synthesis of novel chiral monophosphine ligands derived from isomannide and isosorbide. Application to enantioselective hydrogenation of olefins. Tetrahedron Lett. 2012, 53, 4900–4902. [Google Scholar] [CrossRef]
- Loupy, A.; Monteux, D.A. Isomannide and isosorbide as new chiral auxiliaries for the stereoselective synthesis of tertiary α-hydroxy acids. Tetrahedron 2002, 58, 1541–1549. [Google Scholar] [CrossRef]
- Zullo, V.; Górecki, M.; Guazzelli, L.; Mezzetta, A.; Pescitelli, G.; Iuliano, A. Exploiting isohexide scaffolds for the preparation of chiral ionic liquids tweezers. J. Mol. Liq. 2021, 322, 114528. [Google Scholar] [CrossRef]
- Zullo, V.; Grecchi, S.; Araneo, S.; Galli, M.; Arnaboldi, S.; Micheli, L.; Mezzetta, A.; Guazzelli, L.; Iuliano, A.; Mussini, P.R. Electroactive bio-based chiral tweezers: Attractive selectors for enantioselective voltammetry. Electrochim. Acta 2022, 436, 141191. [Google Scholar] [CrossRef]
- Balzano, F.; Iuliano, A.; Uccello-Barretta, G.; Zullo, V. Renewable Resources for Enantiodiscrimination: Chiral Solvating Agents for NMR Spectroscopy from Isomannide and Isosorbide. J. Org. Chem. 2022, 87, 12698–12709. [Google Scholar] [CrossRef]
- Zullo, V.; Petri, A.; Iuliano, A. An Efficient and Practical Chemoenzymatic Route to (3 R,3a R,6 R,6a R)-Hexahydrofuro[3,2- b]furan-6-amino-3-ol (6-Aminoisomannide) from Renewable Sources. SynOpen 2021, 5, 161–166. [Google Scholar] [CrossRef]
- Zullo, V.; Iuliano, A.; Pescitelli, G.; Zinna, F. Tunable excimer circularly polarized luminescence in isohexide derivatives from renewable resources. Chem.—A Eur. J. 2022, 28, e202104226. [Google Scholar] [CrossRef]
- Zullo, V.; Iuliano, A.; Guazzelli, L. Sugar-Based Ionic Liquids: Multifaceted Challenges and Intriguing Potential. Molecules 2021, 26, 2052. [Google Scholar] [CrossRef] [PubMed]
- Zullo, V.; Iuliano, A. Rh-Catalyzed Asymmetric Conjugate Addition of Arylboronic Acids to 3-Arylpropenoates: Enantioselective Synthesis of (R)-Tolterodine. Eur. J. Org. Chem. 2019, 2019, 1377–1384. [Google Scholar] [CrossRef]
- Krejčik, M.; Daněk, M.; Hartl, F. Simple construction of an infrared optically transparent thin-layer electrochemical cell. J. Electroanal. Chem. Interfacial Electrochem. 1991, 317, 179–187. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R.; Gmbh, F.K. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Takano, Y.; Houk, K.N. Benchmarking the conductor-like polarizable continuum model (CPCM) for aqueous solvation free energies of neutral and ionic organic molecules. J. Chem. Theory Comput. 2005, 1, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Janiak, C. A critical account on n-n stacking in metal complexes with aromatic nitrogen-containing ligands. J. Chem. Soc. Dalt. Trans. 2000, 2000, 3885–3896. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zullo, V.; Guo, T.; Iuliano, A.; Ringenberg, M.R. Control of Molecular Packing in Crystal and Electron Communication of Two Ferrocenyl Moieties across Chiral Isomannide or Isosorbide Bridge. Crystals 2023, 13, 520. https://doi.org/10.3390/cryst13030520
Zullo V, Guo T, Iuliano A, Ringenberg MR. Control of Molecular Packing in Crystal and Electron Communication of Two Ferrocenyl Moieties across Chiral Isomannide or Isosorbide Bridge. Crystals. 2023; 13(3):520. https://doi.org/10.3390/cryst13030520
Chicago/Turabian StyleZullo, Valerio, Tianao Guo, Anna Iuliano, and Mark R. Ringenberg. 2023. "Control of Molecular Packing in Crystal and Electron Communication of Two Ferrocenyl Moieties across Chiral Isomannide or Isosorbide Bridge" Crystals 13, no. 3: 520. https://doi.org/10.3390/cryst13030520
APA StyleZullo, V., Guo, T., Iuliano, A., & Ringenberg, M. R. (2023). Control of Molecular Packing in Crystal and Electron Communication of Two Ferrocenyl Moieties across Chiral Isomannide or Isosorbide Bridge. Crystals, 13(3), 520. https://doi.org/10.3390/cryst13030520