




QM/MM Study of a Nucleophilic Substitution Reaction Catalyzed by Uridine Phosphorylase from Vibrio cholerae
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation and Parameterization
2.2. Levels of Theory Used
2.3. Calculations of the Reaction Path and the Transition State
2.4. Calculations of Thermochemical Parameters
2.5. Calculations for the Nucleophilic Attack Reaction without an Enzyme
3. Results and Discussion
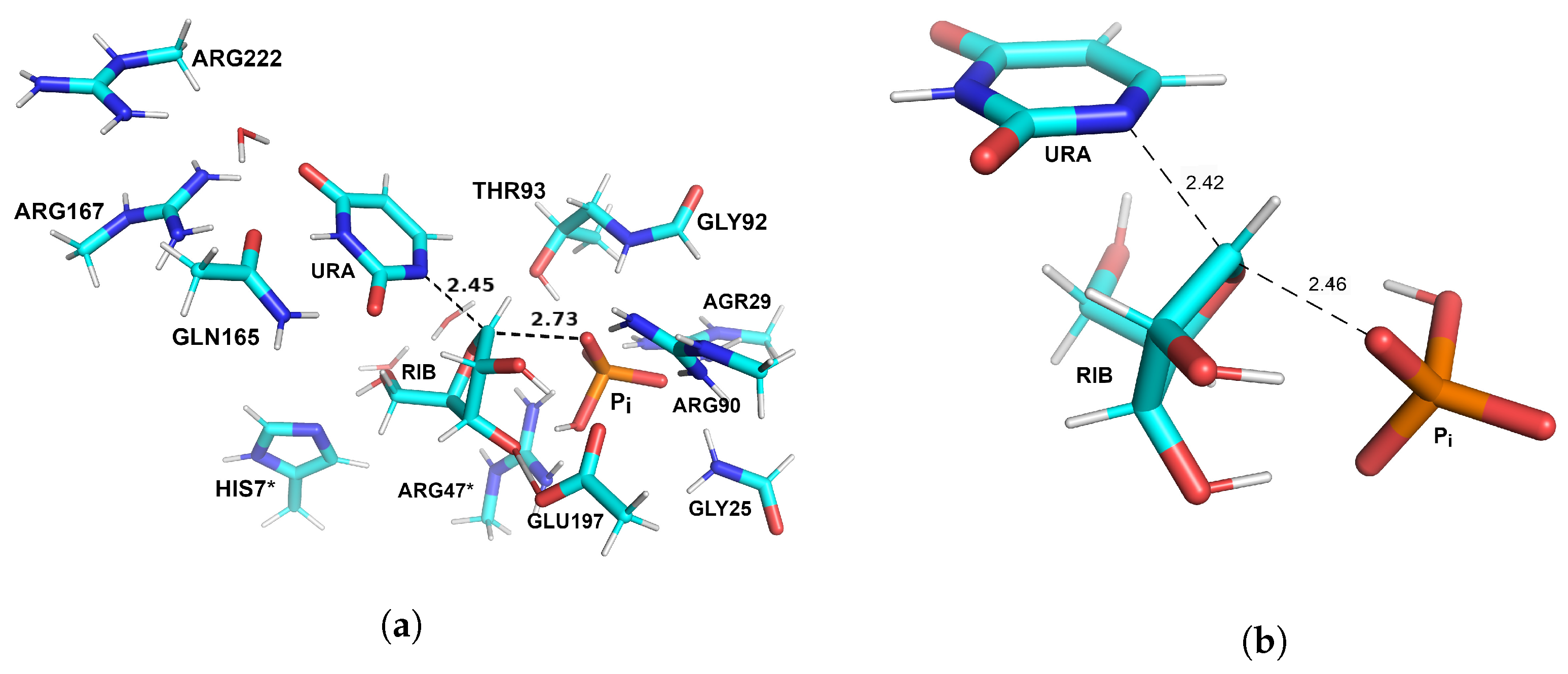
3.1. Optimized Structures of Substrate Complexes and Products with UP
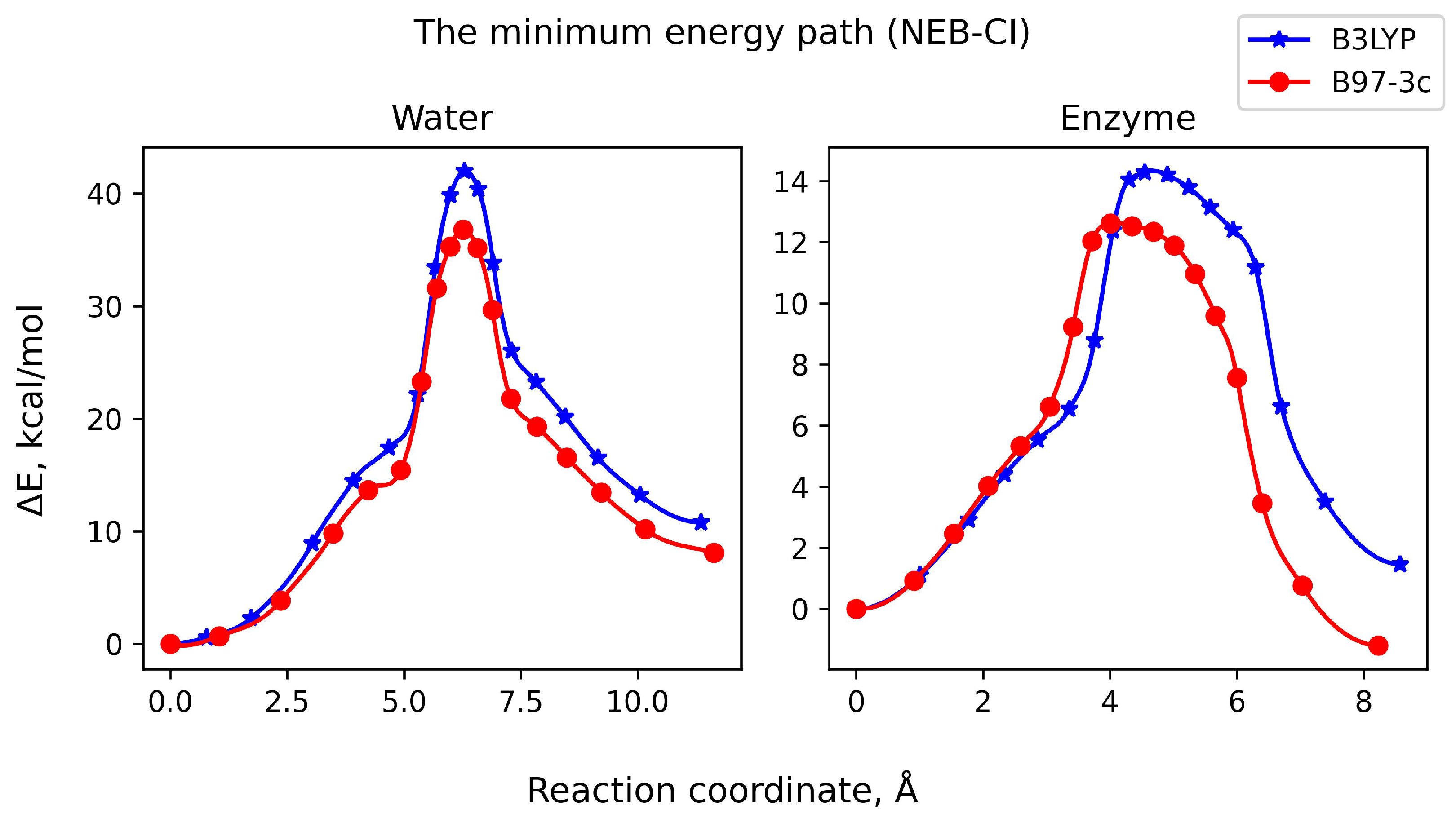
3.2. Reaction Pathway and Transition State for the Nucleophilic Attack of Hydrogen Phosphate Ion on Uridine
3.3. Nucleophilic Substitution Reaction Involving Hydrogen Orthovanadat Anion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| QM/MM | Quantum Mechanics/Molecular Modelling |
| UP | Uridine Phosphorylase |
| aa | amino acid residues |
| xTB | extended Tight-binding method |
| DFT | Density Functional Theory |
| MEP | Minimum Energy Path |
| NEB-CI | Climbing Image Nudged Elastic Band method |
| EF | Eigenvector Following method |
| IDPP | Image-Dependent-Pair-Potential method |
| PHVA | Partial Hessian |
| qRRHO | quasi-Rigid Rotor Harmonic Oscillator |
| URA | Uracil moiety |
| RIB | Ribosyl moiety |
| PME | Particle-Mesh Evald method |
| DPLNO | Domain-based Local Pair Natural Orbital computational scheme |
| CCSD(T) | Coupled Cluster Single-Double-Triple method |
| SCF | Self-Consistent Field |
References
- Lashkov, A.A.; Zhukhlistova, N.E.; Seregina, T.A.; Gabdulkhakov, A.G.; Mikhailov, A.M. Uridine phosphorylase in biomedical, structural, and functional aspects: A review. Crystallogr. Rep. 2011, 56, 560–589. [Google Scholar] [CrossRef]
- Paul, D.; Oleary, S.E.; Rajashankar, K.; Bu, W.; Toms, A.; Settembre, E.C.; Sanders, J.M.; Begley, T.P.; Ealick, S.E. Glycal formation in crystals of uridine phosphorylase. Biochemistry 2010, 49, 3499–3509. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.G.; Vetticatt, M.J.; Merino, E.F.; Cassera, M.B.; Schramm, V.L. Transition-state analysis of Trypanosoma cruzi uridine phosphorylase- catalyzed arsenolysis of uridine. J. Am. Chem. Soc. 2011, 133, 9923–9931. [Google Scholar] [CrossRef]
- Silva, R.G.; Kipp, D.R.; Schramm, V.L. Constrained Bonding Environment in the Michaelis Complex of Trypanosoma cruzi Uridine Phosphorylase. Biochemistry 2012, 51, 6715–6717. [Google Scholar] [CrossRef] [PubMed]
- Prokofev, I.I.; Lashkov, A.A.; Gabdulkhakov, A.G.; Balaev, V.V.; Seregina, T.A.; Mironov, A.S.; Betzel, C.; Mikhailov, A.M. X-ray structures of uridine phosphorylase from Vibrio cholerae in complexes with uridine, thymidine, uracil, thymine, and phosphate anion: Substrate specificity of bacterial uridine phosphorylases. Crystallogr. Rep. 2016, 61, 954–973. [Google Scholar] [CrossRef]
- Eistrikh-Heller, P.A.; Rubinsky, S.V.; Samygina, V.R.; Gabdulkhakov, A.G.; Kovalchuk, M.V.; Mironov, A.S.; Lashkov, A.A. Crystallization in Microgravity and the Atomic-Resolution Structure of Uridine Phosphorylase from Vibrio cholerae. Crystallogr. Rep. 2021, 66, 777–785. [Google Scholar] [CrossRef]
- Caradoc-Davies, T.T.; Cutfield, S.M.; Lamont, I.L.; Cutfield, J.F. Crystal structures of Escherichia coli uridine phosphorylase in two native and three complexed forms reveal basis of substrate specificity, induced conformational changes and influence of potassium. J. Mol. Biol. 2004, 337, 337–354. [Google Scholar] [CrossRef]
- Antipov Alexey, N.; Okorokova Natalya, A.; Safonova Tatyana, N.; Veiko Vladimir, P. Vanadate as a new substrate for nucleoside phosphorylases. JBIC J. Biol. Inorg. Chem. 2022, 27, 221–227. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef]
- Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended tight-binding quantum chemistry methods. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, 11, e1493. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Brandenburg, J.G.; Bannwarth, C.; Hansen, A.; Grimme, S. B97-3c: A revised low-cost variant of the B97-D density functional method. J. Chem. Phys. 2018, 148, 064104. [Google Scholar] [CrossRef]
- Riplinger, C.; Pinski, P.; Becker, U.; Valeev, E.F.; Neese, F. Sparse maps—A systematic infrastructure for reduced-scaling electronic structure methods. II. Linear scaling domain based pair natural orbital coupled cluster theory. J. Chem. Phys. 2016, 144, 024109. [Google Scholar] [CrossRef]
- Zheng, J.; Xu, X.; Truhlar, D.G. Minimally augmented Karlsruhe basis sets. Theor. Chem. Acc. 2011, 128, 295–305. [Google Scholar] [CrossRef]
- Ásgeirsson, V.; Birgisson, B.O.; Bjornsson, R.; Becker, U.; Neese, F.; Riplinger, C.; Jónsson, H. Nudged Elastic Band Method for Molecular Reactions Using Energy-Weighted Springs Combined with Eigenvector following. J. Chem. Theory Comput. 2021, 17, 4929–4945. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. Climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Smidstrup, S.; Pedersen, A.; Stokbro, K.; Jónsson, H. Improved initial guess for minimum energy path calculations. J. Chem. Phys. 2014, 140, 214106. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Jensen, J.H. Partial Hessian vibrational analysis: The localization of the molecular vibrational energy and entropy. Theor. Chem. Acc. 2002, 107, 211–219. [Google Scholar] [CrossRef]
- Grimme, S. Supramolecular binding thermodynamics by dispersion-corrected density functional theory. Chem.—Eur. J. 2012, 18, 9955–9964. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Yildirim, I.; Stern, H.A.; Kennedy, S.D.; Tubbs, J.D.; Turner, D.H. Reparameterization of RNA ξ Torsion Parameters for the AMBER Force Field and Comparison to NMR Spectra for Cytidine and Uridine. J. Chem. Theory Comput. 2010, 6, 1520–1531. [Google Scholar] [CrossRef]
- Ryu, H.; Park, J.; Kim, H.K.; Park, J.Y.; Kim, S.T.; Baik, M.H. Pitfalls in Computational Modeling of Chemical Reactions and How To Avoid Them. Organometallics 2018, 37, 3228–3239. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Calculation Methods | ER (s/e) | EA (s/e) | ER (DFT) | EA (DFT) | ER (CC) | EA (CC) | GR (CC) | GA (CC) |
|---|---|---|---|---|---|---|---|---|
| XTB-GFN2 (MEP 1, OPT 2, FREQ 3) | −2.6 | 31.2 | 3.3 | 13.4 | 1.9 | 20.2 | 3.6 | 17.7 |
| XTB-GFN2 (MEP, FREQ) + B3LYP 4 (OPT) | −1.0 | 32.0 | 1.5 | 10.5 | 0.2 | 16.6 | 0.6 | 14.0 |
| B3LYP (MEP, OPT) + XTB-GFN2(FREQ) | −1.0 | 32.5 | 1.5 | 10.6 | 0.2 | 16.7 | 0.6 | 14.0 |
| B97-3c | - | - | −1.2 | 9.1 | 0.2 | 16.4 | 0.1 | 14.7 |
| Solvent (water, C-PCM) w/o enzime | ||||||||
| B3LYP | - | - | 10.7 | 33.6 | 9.0 | 38.4 | 6.8 | 35.3 |
| B3LYP, each reactant/product separately | - | - | −3.4 | 13.8 | −3.4 | 21.5 | −2.6 | 35.6 |
| B97-3c | - | - | 7.9 | 24.8 | 8.6 | 34.4 | 8.0 | 31.6 |
| B97-3c, each reactant/product separately | - | - | - | - | −8.3 | 13.0 | −7.5 | 26.5 |
| Nucleophilic substitution reaction involving hydroorthovanadat-ion | ||||||||
| B97-3c | - | - | −6.1 | 7.3 | −3.8 | 14.4 | −3.7 | 12.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lashkov, A.A.; Eistrich-Geller, P.A.; Samygina, V.R.; Rubinsky, S.V. QM/MM Study of a Nucleophilic Substitution Reaction Catalyzed by Uridine Phosphorylase from Vibrio cholerae. Crystals 2023, 13, 803. https://doi.org/10.3390/cryst13050803
Lashkov AA, Eistrich-Geller PA, Samygina VR, Rubinsky SV. QM/MM Study of a Nucleophilic Substitution Reaction Catalyzed by Uridine Phosphorylase from Vibrio cholerae. Crystals. 2023; 13(5):803. https://doi.org/10.3390/cryst13050803
Chicago/Turabian StyleLashkov, Alexander A., Polina A. Eistrich-Geller, Valeriya R. Samygina, and Sergey V. Rubinsky. 2023. "QM/MM Study of a Nucleophilic Substitution Reaction Catalyzed by Uridine Phosphorylase from Vibrio cholerae" Crystals 13, no. 5: 803. https://doi.org/10.3390/cryst13050803
APA StyleLashkov, A. A., Eistrich-Geller, P. A., Samygina, V. R., & Rubinsky, S. V. (2023). QM/MM Study of a Nucleophilic Substitution Reaction Catalyzed by Uridine Phosphorylase from Vibrio cholerae. Crystals, 13(5), 803. https://doi.org/10.3390/cryst13050803