
Crystal Engineering of Hydrogen Bonding for Direct Air Capture of CO2: A Quantum Crystallography Perspective


Abstract
:
1. Introduction

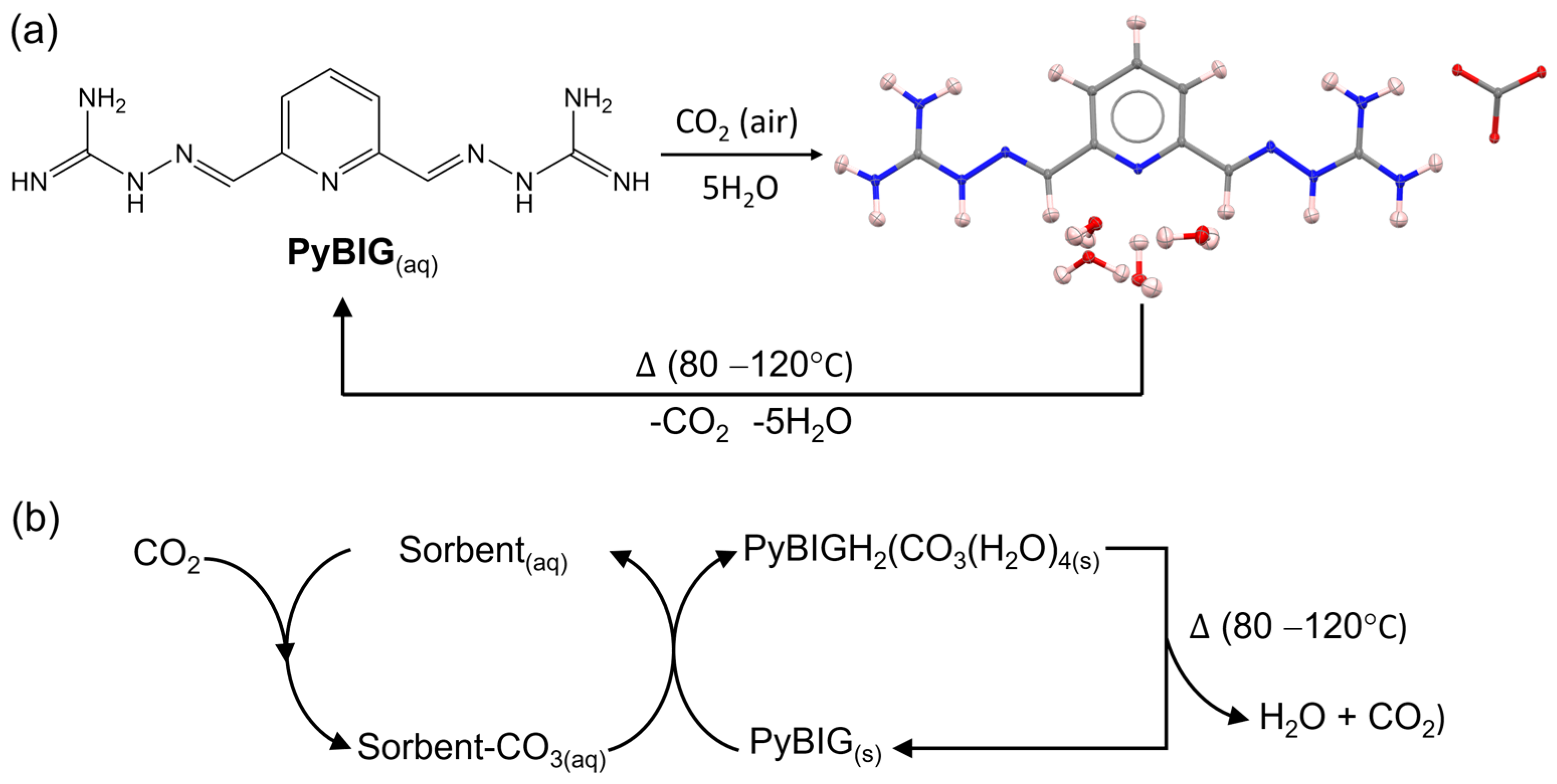
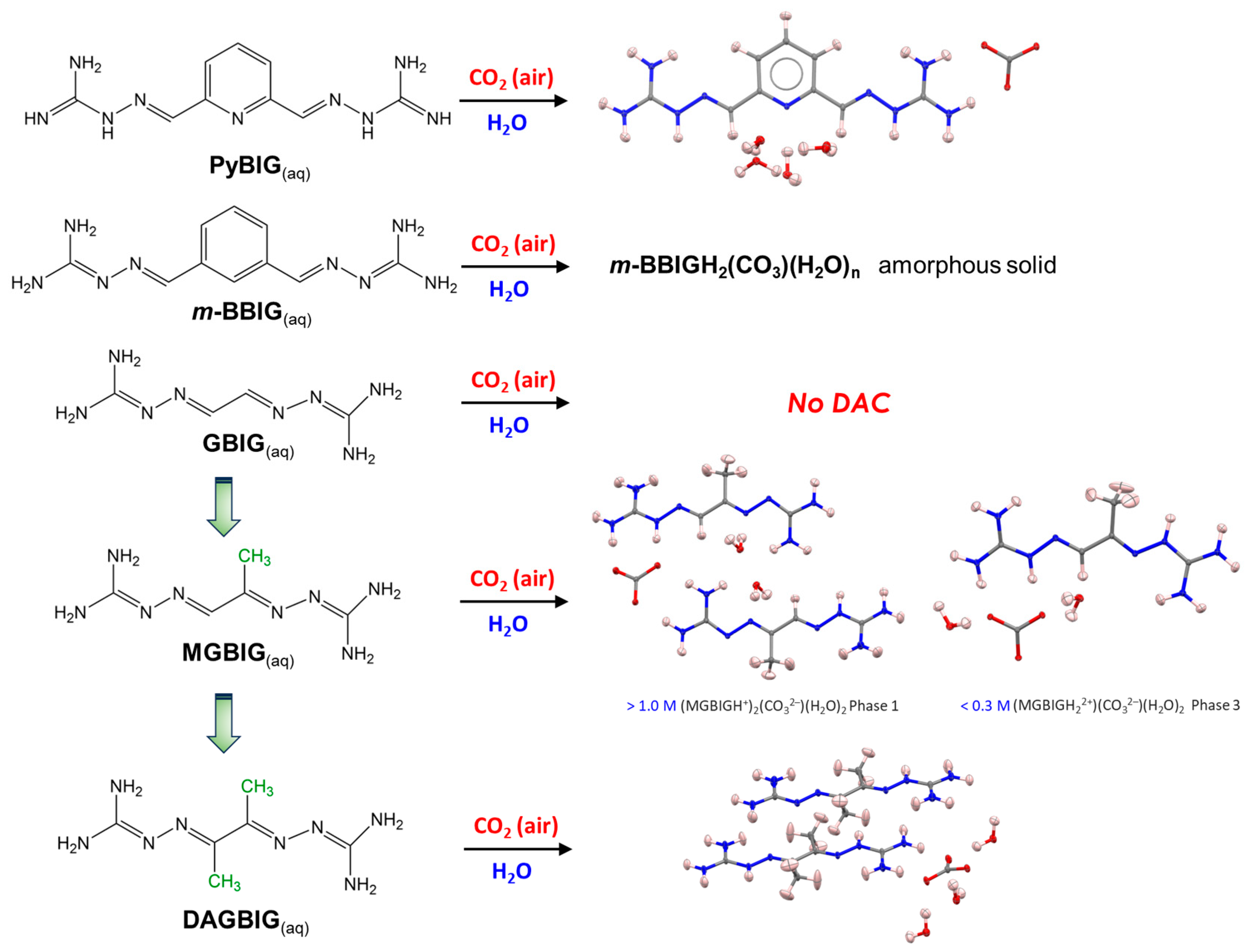
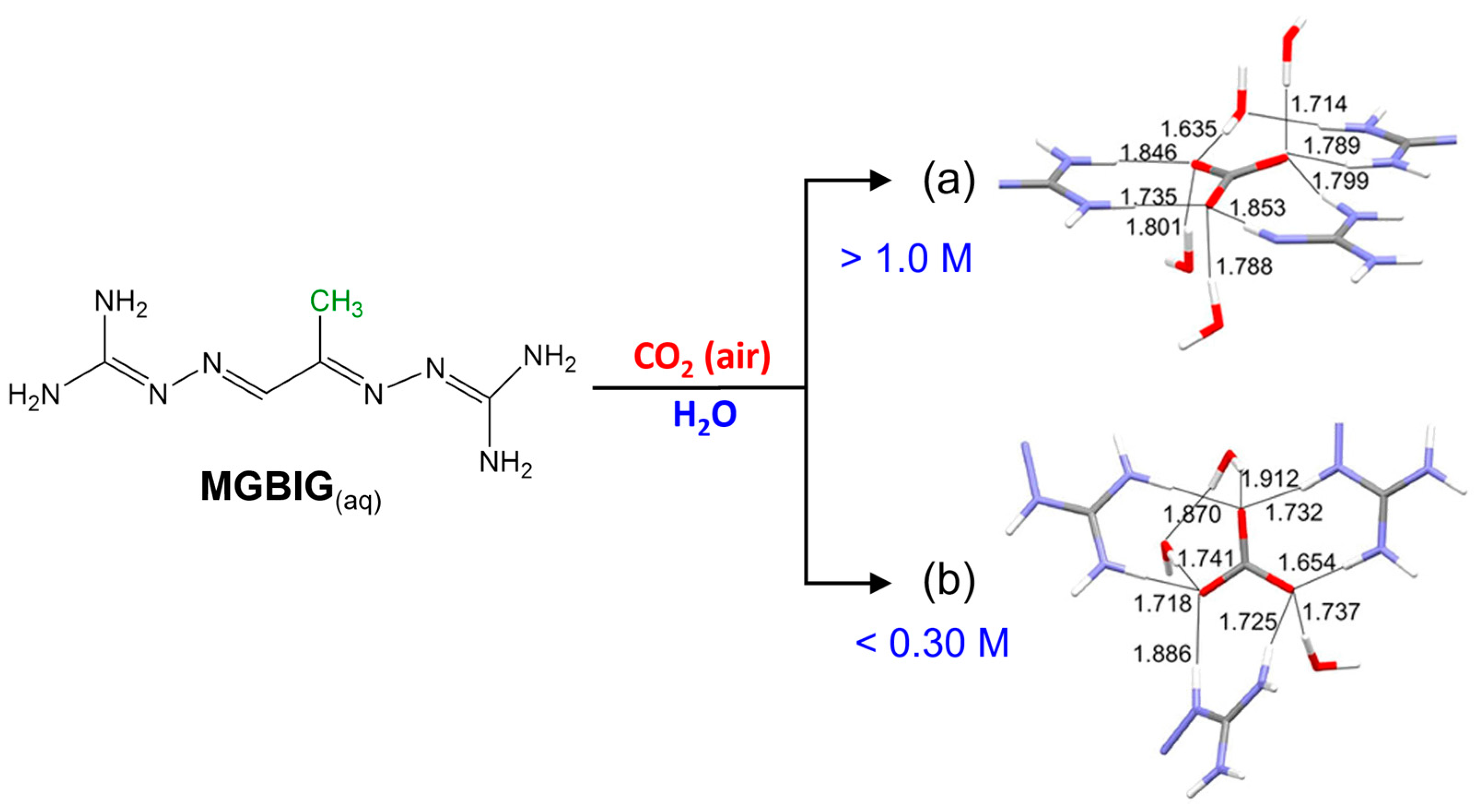
2. Family of Bis-Iminoguanidine Carbonate Salts Used for DAC Materials
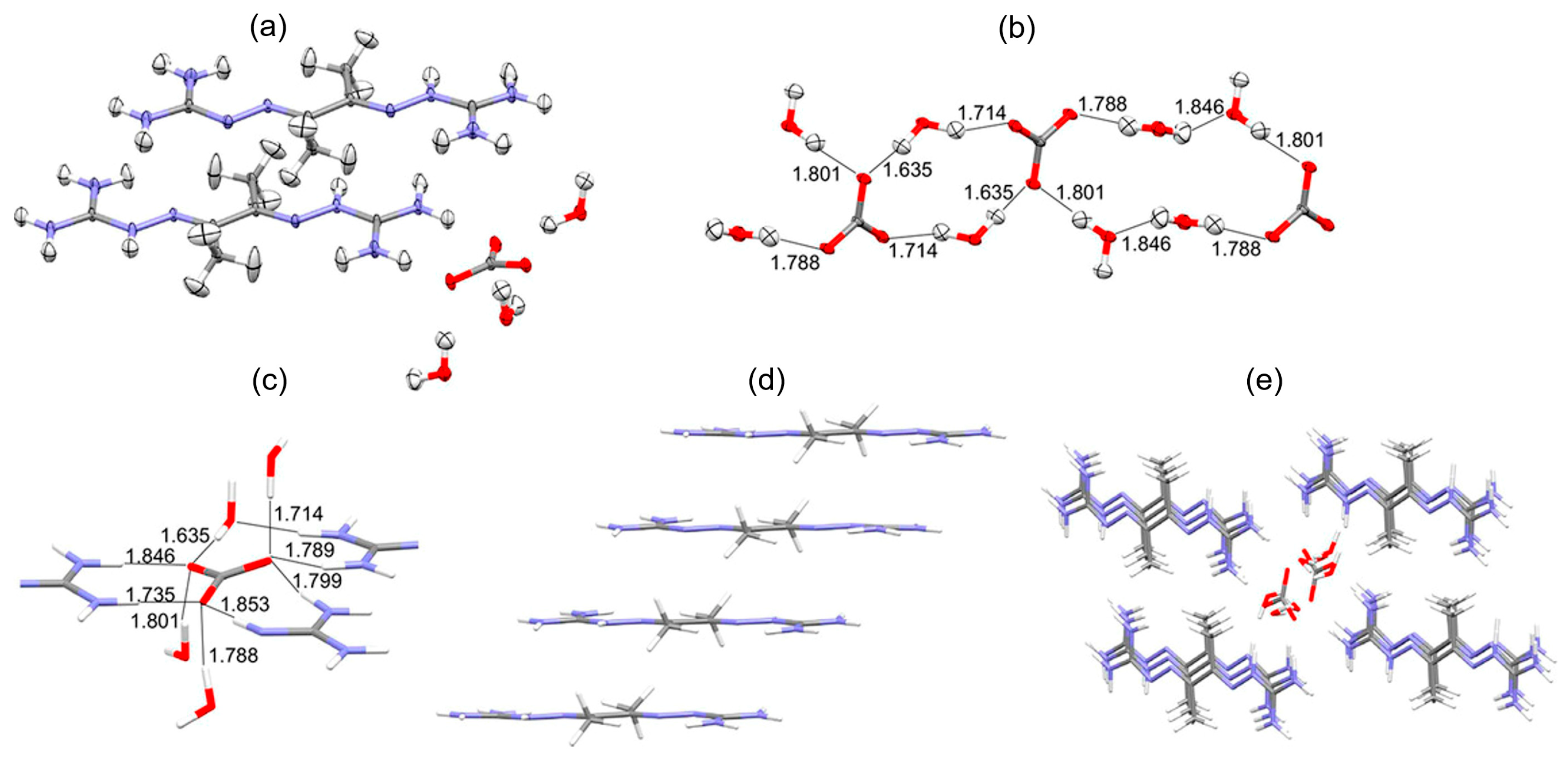
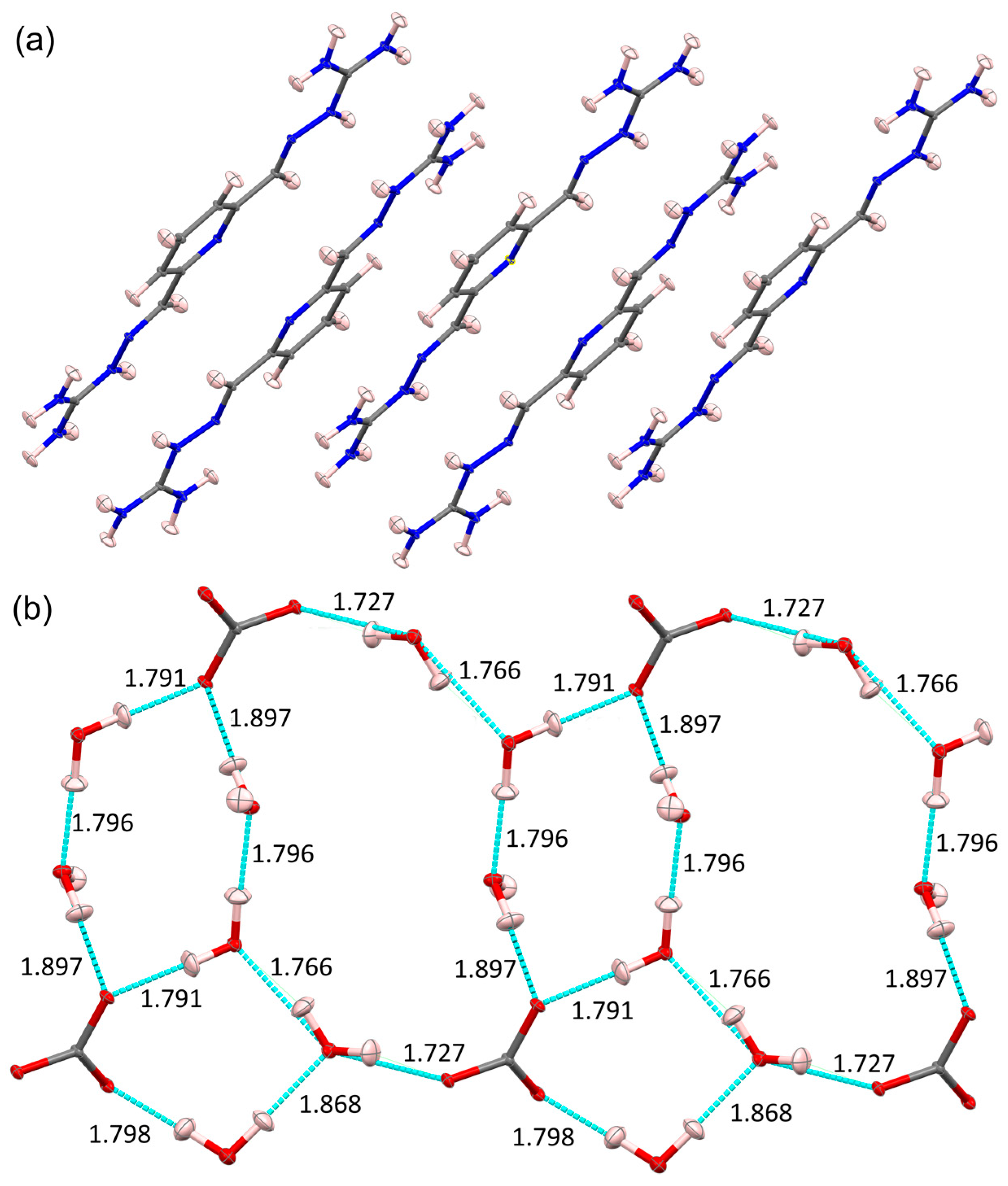
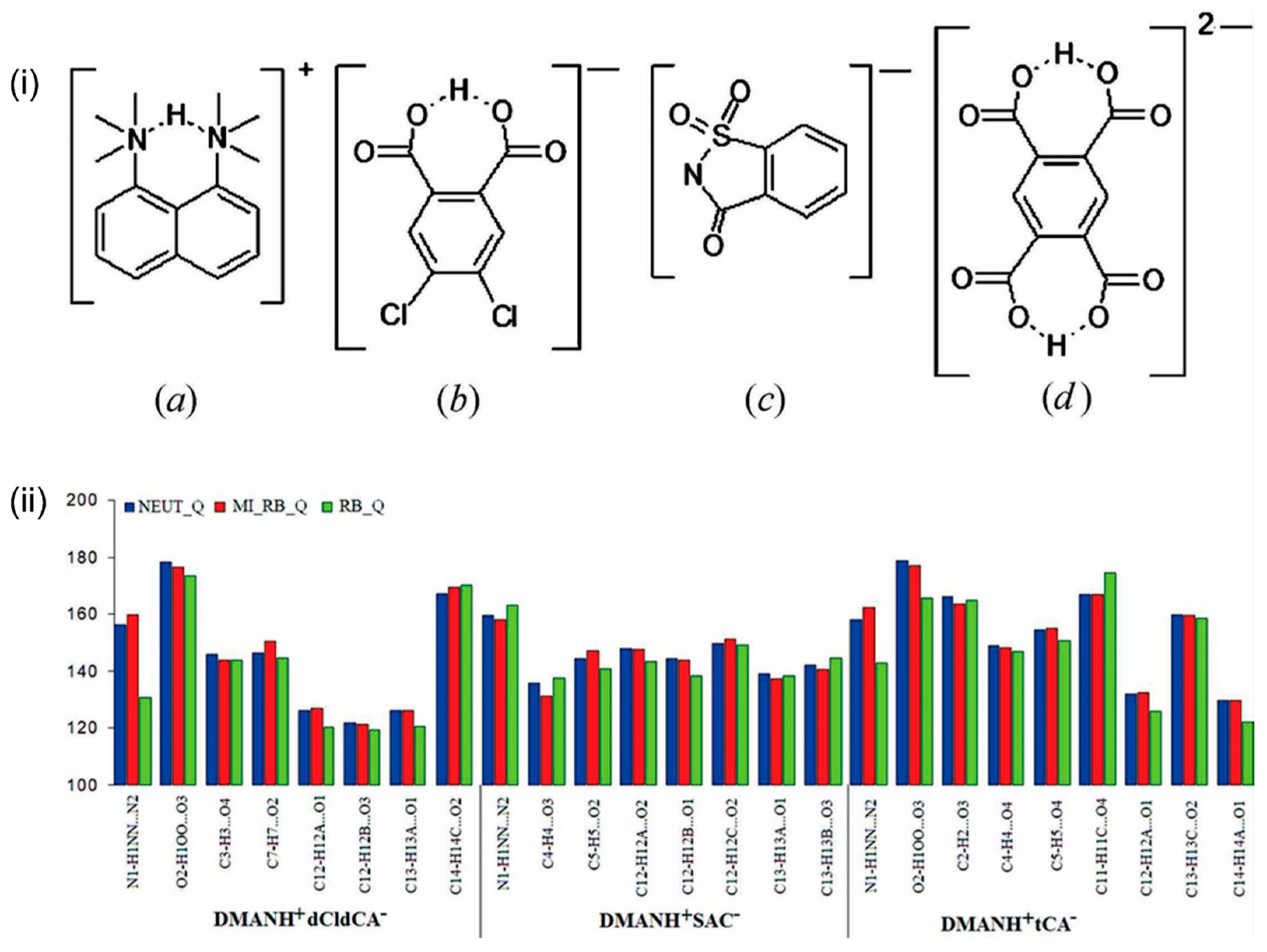
3. DAC Studied by Single-Crystal Neutron Diffraction




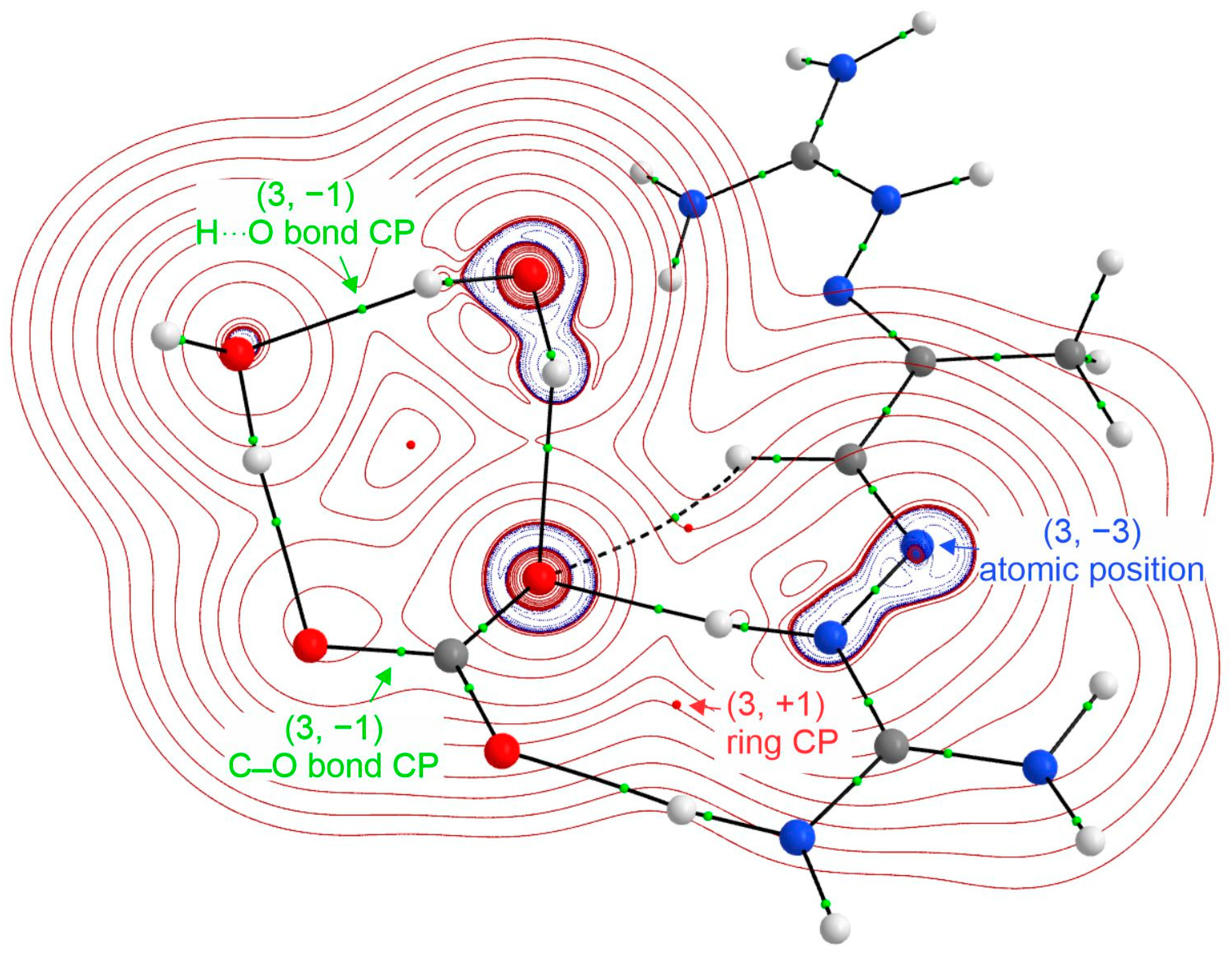
4. DAC Studied by High-Resolution Single-Crystal X-ray Diffraction Quantum Crystallography Studies
5. Accurate Hydrogen Atom Positions from Quantum Crystallography Studies
- N is the number of atoms in the unit cell;
- N is the number of cells in the crystal;
- e(k|jq) represents the kth component of a normalized complex eigenvector e(jq);
- ωj is the frequency of mode j;
- mk is the mass of atom k;
- Ej(q) is the energy of the mode.
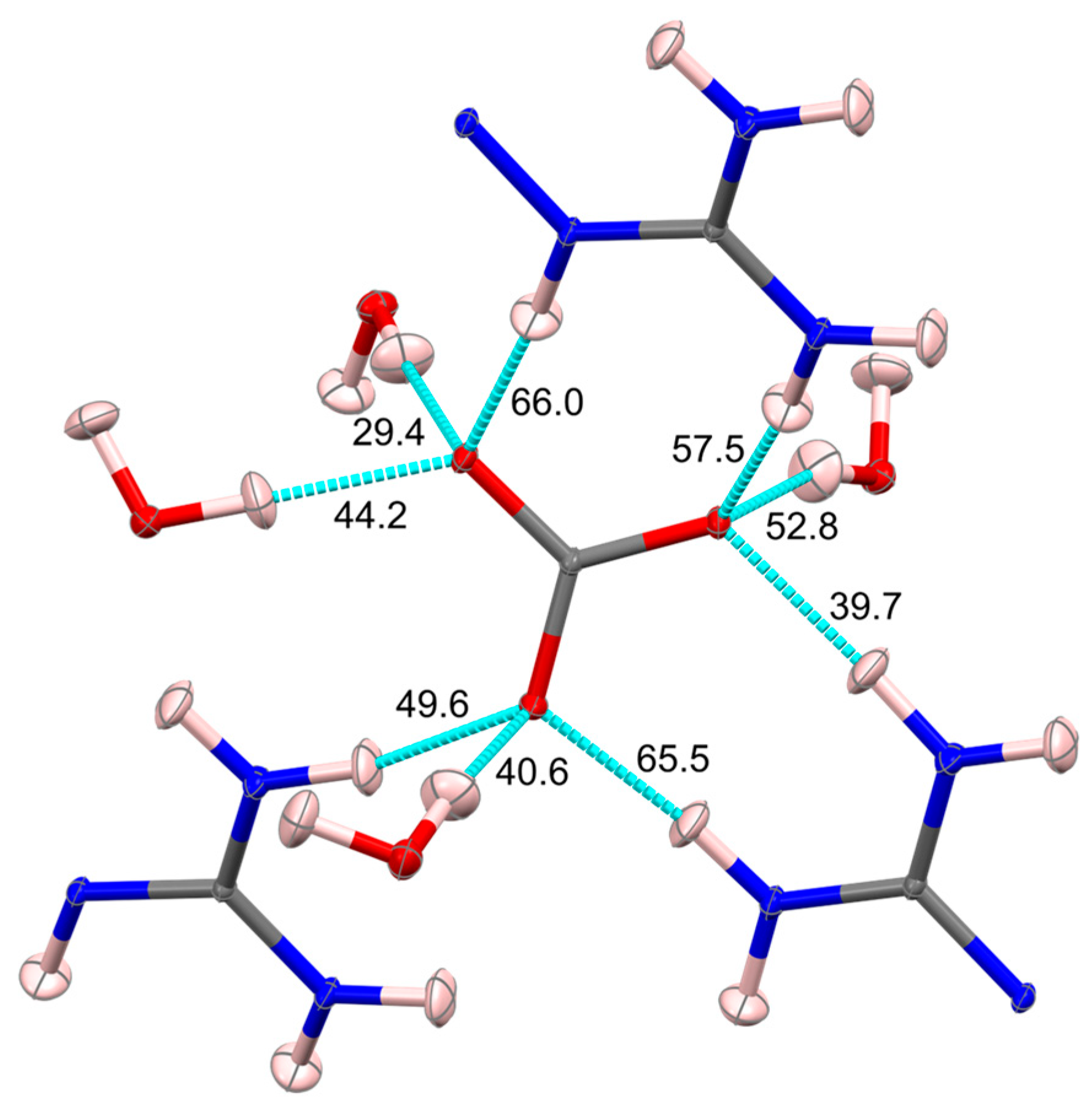
6. Interaction Energies from X-ray Electron Density Study
7. Challenges
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviation | Description |
| ADPs | Anisotropic Displacement Parameters |
| BCPs | Bond Critical Points |
| BIGs | Bis-iminoguanidines |
| CSD | Cambridge Structural Database |
| DABIG | Diacetyl bis(iminoguanidine) |
| DAC | Direct Air Capture |
| DFT | Density Functional Theory |
| DSC | Differential Scanning Calorimetry |
| ELMOs | Extremely Localized Molecular Orbitals |
| EML | Espinosa–Molins–Lecomte approach |
| EPMM | Exact Potential and Multipole Moments |
| FTIR | Fourier Transform Infrared Spectroscopy |
| FuBIG | 2,5-Furan-bis(iminoguanidine) |
| GBIG | Glyoxal-bis(iminoguanidine) Compound |
| HAR | Hirshfeld Atom Refinement |
| HBA | Hydrogen Bond Acceptor |
| HBD | Hydrogen Bond Donor |
| HBFs | Hydrogen-Bonded Frameworks |
| IAM | Independent Atom Model |
| m-BBIG | Meta-benzene-bis(iminoguanidine) |
| MEA | Monoethanolamine |
| MGBIG | Methyl-glyoxal-bis(iminoguanidine) |
| MSDs | Mean Square Displacements |
| NoMoRe | Normal-Mode Refinement |
| P1 | Phase 1 of the MGBIG |
| P2 | Phase 2 of the MGBIG |
| P3 | Phase 3 of the MGBIG |
| PyBIG | 2,6-Pyridine-bis(iminoguanidine) |
| TAAM | Transferable Atom Refinement |
| QC | Quantum Crystallography |
| QTAIM | Quantum Theory of Atoms in Molecules |
| TGA | Thermogravimetric Analysis |
| XAS | X-ray Absorption Spectroscopy |
| XRD | X-ray Diffraction |
| XWR | X-ray Wavefunction Refinement |
References
- Mansfield, L.A.; Nowack, P.J.; Kasoar, M.; Everitt, R.G.; Collins, W.J.; Voulgarakis, A. Predicting Global Patterns of Long-Term Climate Change from Short-Term Simulations Using Machine Learning. Npj Clim. Atmos. Sci. 2020, 3, 44. [Google Scholar] [CrossRef]
- Abbass, K.; Qasim, M.Z.; Song, H.; Murshed, M.; Mahmood, H.; Younis, I. A Review of the Global Climate Change Impacts, Adaptation, and Sustainable Mitigation Measures. Environ. Sci. Pollut. Res. 2022, 29, 42539–42559. [Google Scholar] [CrossRef] [PubMed]
- Erans, M.; Sanz-Pérez, E.S.; Hanak, D.P.; Clulow, Z.; Reiner, D.M.; Mutch, G.A. Direct Air Capture: Process Technology, Techno-Economic and Socio-Political Challenges. Energy Environ. Sci. 2022, 15, 1360–1405. [Google Scholar] [CrossRef]
- Sanz-Pérez, E.S.; Murdock, C.R.; Didas, S.A.; Jones, C.W. Direct Capture of CO2 from Ambient Air. Chem. Rev. 2016, 116, 11840–11876. [Google Scholar] [CrossRef] [PubMed]
- Keith, D.W.; Holmes, G.; St. Angelo, D.; Heidel, K. A Process for Capturing CO2 from the Atmosphere. Joule 2018, 2, 1573–1594. [Google Scholar] [CrossRef]
- Yusov, A.; Dillon, A.M.; Ward, M.D. Hydrogen Bonded Frameworks: Smart Materials Used Smartly. Mol. Syst. Des. Eng. 2021, 6, 756–778. [Google Scholar] [CrossRef]
- Custelcean, R. Direct Air Capture of CO2 via Crystal Engineering. Chem. Sci. 2021, 12, 12518–12528. [Google Scholar] [CrossRef]
- Custelcean, R.; Williams, N.J.; Wang, X.; Garrabrant, K.A.; Martin, H.J.; Kidder, M.K.; Ivanov, A.S.; Bryantsev, V.S. Dialing in Direct Air Capture of CO2 by Crystal Engineering of Bisiminoguanidines. ChemSusChem 2020, 13, 6381–6390. [Google Scholar] [CrossRef]
- Liu, S.; Chen, Y.; Yue, B.; Wang, C.; Qin, B.; Chai, Y.; Wu, G.; Li, J.; Han, X.; da-Silva, I.; et al. Regulating Extra-Framework Cations in Faujasite Zeolites for Capture of Trace Carbon Dioxide. Chem. Eur. J. 2022, 28, e202201659. [Google Scholar] [CrossRef]
- Grabowsky, S.; Genoni, A.; Bürgi, H.-B. Quantum Crystallography. Chem. Sci. 2017, 8, 4159–4176. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; International Series of Monographs on Chemistry; Oxford University Press: Oxford, NY, USA, 1994; ISBN 978-0-19-855865-1. [Google Scholar]
- Axe, J.D. Neutron Scattering: Progress and Prospects. Science 1991, 252, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.L.; White, N.G. Hydrogen Atoms in Supramolecular Chemistry: A Structural Perspective. Where Are They, and Why Does It Matter? Chem. Soc. Rev. 2023, 52, 6254–6269. [Google Scholar] [CrossRef]
- Bhadra, M.; Lee, J.Y.C.; Cowley, R.E.; Kim, S.; Siegler, M.A.; Solomon, E.I.; Karlin, K.D. Intramolecular Hydrogen Bonding Enhances Stability and Reactivity of Mononuclear Cupric Superoxide Complexes. J. Am. Chem. Soc. 2018, 140, 9042–9045. [Google Scholar] [CrossRef]
- Nichols, D.A.; Hargis, J.C.; Sanishvili, R.; Jaishankar, P.; Defrees, K.; Smith, E.W.; Wang, K.K.; Prati, F.; Renslo, A.R.; Woodcock, H.L.; et al. Ligand-Induced Proton Transfer and Low-Barrier Hydrogen Bond Revealed by X-ray Crystallography. J. Am. Chem. Soc. 2015, 137, 8086–8095. [Google Scholar] [CrossRef]
- Oksanen, E.; Chen, J.C.-H.; Fisher, S.Z. Neutron Crystallography for the Study of Hydrogen Bonds in Macromolecules. Mol. J. Synth. Chem. Nat. Prod. Chem. 2017, 22, 596. [Google Scholar] [CrossRef] [PubMed]
- Pflug, C.; Rudolph, D.; Schleid, T.; Kohlmann, H. Hydrogenation Reaction Pathways and Crystal Structures of La2H2Se, La2H3Se and La2H4Se. Eur. J. Inorg. Chem. 2022, 2022, e202101095. [Google Scholar] [CrossRef]
- Anderson, A.C. The Process of Structure-Based Drug Design. Chem. Biol. 2003, 10, 787–797. [Google Scholar] [CrossRef]
- Manzoni, F.; Wallerstein, J.; Schrader, T.E.; Ostermann, A.; Coates, L.; Akke, M.; Blakeley, M.P.; Oksanen, E.; Logan, D.T. Elucidation of Hydrogen Bonding Patterns in Ligand-Free, Lactose- and Glycerol-Bound Galectin-3C by Neutron Crystallography to Guide Drug Design. J. Med. Chem. 2018, 61, 4412–4420. [Google Scholar] [CrossRef]
- Zhao, C.; Chen, L.; Che, Y.; Pang, Z.; Wu, X.; Lu, Y.; Liu, H.; Day, G.M.; Cooper, A.I. Digital Navigation of Energy–Structure–Function Maps for Hydrogen-Bonded Porous Molecular Crystals. Nat. Commun. 2021, 12, 817. [Google Scholar] [CrossRef]
- Arnold, J.E.; Day, G.M. Crystal Structure Prediction of Energetic Materials. Cryst. Growth Des. 2023, 23, 6149–6160. [Google Scholar] [CrossRef]
- Sato, T.; Mochizuki, T.; Ikeda, K.; Honda, T.; Otomo, T.; Sagayama, H.; Yang, H.; Luo, W.; Lombardo, L.; Züttel, A.; et al. Crystal Structural Investigations for Understanding the Hydrogen Storage Properties of YMgNi4-Based Alloys. ACS Omega 2020, 5, 31192–31198. [Google Scholar] [CrossRef] [PubMed]
- Hathwar, V.R.; Sist, M.; Jørgensen, M.R.V.; Mamakhel, A.H.; Wang, X.; Hoffmann, C.M.; Sugimoto, K.; Overgaard, J.; Iversen, B.B. Quantitative Analysis of Intermolecular Interactions in Orthorhombic Rubrene. IUCrJ 2015, 2, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, M.R.V.; Hathwar, V.R.; Sist, M.; Wang, X.; Hoffmann, C.M.; Briseno, A.L.; Overgaard, J.; Iversen, B.B. Accurate Atomic Displacement Parameters from Time-of-Flight Neutron-Diffraction Data at TOPAZ. Acta Crystallogr. Sect. Found. Adv. 2014, 70, 679–681. [Google Scholar] [CrossRef]
- Grosjean, A.; Spackman, P.R.; Edwards, A.J.; Tolborg, K.; Vosegaard, E.S.; Koutsantonis, G.A.; Iversen, B.B.; Spackman, M.A. Insights into Host–Guest Binding in Hydroquinone Clathrates: Single-Crystal X-ray and Neutron Diffraction, and Complementary Computational Studies on the Hydroquinone-CO2 Clathrate. Cryst. Growth Des. 2021, 21, 3477–3486. [Google Scholar] [CrossRef]
- Carrington, E.J.; Vitórica-Yrezábal, I.J.; Brammer, L. Crystallographic Studies of Gas Sorption in Metal–Organic Frameworks. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2014, 70, 404–422. [Google Scholar] [CrossRef] [PubMed]
- Takamizawa, S.; Nakata, E.; Yokoyama, H.; Mochizuki, K.; Mori, W. Carbon Dioxide Inclusion Phases of a Transformable 1D Coordination Polymer Host [Rh2(O2CPh)4(Pyz)]n. Angew. Chem. Int. Ed. 2003, 42, 4331–4334. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.A.; Mullangi, D.; Deng, Z.; Wang, Y.; Peh, S.B.; Wei, F.; Wang, J.; Brown, C.M.; Zhao, D.; Canepa, P.; et al. Aluminum Formate, Al(HCOO)3: An Earth-Abundant, Scalable, and Highly Selective Material for CO2 Capture. Sci. Adv. 2022, 8, eade1473. [Google Scholar] [CrossRef]
- Capelli, S.C.; Bürgi, H.-B.; Dittrich, B.; Grabowsky, S.; Jayatilaka, D. Hirshfeld Atom Refinement. IUCrJ 2014, 1, 361–379. [Google Scholar] [CrossRef]
- Jayatilaka, D. Wave Function for Beryllium from X-ray Diffraction Data. Phys. Rev. Lett. 1998, 80, 798–801. [Google Scholar] [CrossRef]
- Meyer, B.; Genoni, A. Libraries of Extremely Localized Molecular Orbitals. 3. Construction and Preliminary Assessment of the New Databanks. J. Phys. Chem. A 2018, 122, 8965–8981. [Google Scholar] [CrossRef]
- Gianopoulos, C.G.; Chua, Z.; Zhurov, V.V.; Seipp, C.A.; Wang, X.; Custelcean, R.; Pinkerton, A.A. Direct Air Capture of CO 2—Topological Analysis of the Experimental Electron Density (QTAIM) of the Highly Insoluble Carbonate Salt of a 2,6-Pyridine-Bis(Iminoguanidine), (PyBIGH2)(CO3)(H2O)4. IUCrJ 2019, 6, 56–65. [Google Scholar] [CrossRef]
- Custelcean, R. Direct Air Capture with Bis-Iminoguanidines: From Discovery to Commercialization. Chem 2021, 7, 2848–2852. [Google Scholar] [CrossRef]
- Seipp, C.A.; Williams, N.J.; Kidder, M.K.; Custelcean, R. CO2 Capture from Ambient Air by Crystallization with a Guanidine Sorbent. Angew. Chem. Int. Ed. 2017, 56, 1042–1045. [Google Scholar] [CrossRef] [PubMed]
- Brethomé, F.M.; Williams, N.J.; Seipp, C.A.; Kidder, M.K.; Custelcean, R. Direct Air Capture of CO2 via Aqueous-Phase Absorption and Crystalline-Phase Release Using Concentrated Solar Power. Nat. Energy 2018, 3, 553–559. [Google Scholar] [CrossRef]
- Williams, N.J.; Seipp, C.A.; Brethomé, F.M.; Ma, Y.-Z.; Ivanov, A.S.; Bryantsev, V.S.; Kidder, M.K.; Martin, H.J.; Holguin, E.; Garrabrant, K.A.; et al. CO2 Capture via Crystalline Hydrogen-Bonded Bicarbonate Dimers. Chem 2019, 5, 719–730. [Google Scholar] [CrossRef]
- Ma, C.; Pietrucci, F.; Andreoni, W. Capture and Release of CO2 in Monoethanolamine Aqueous Solutions: New Insights from First-Principles Reaction Dynamics. J. Chem. Theory Comput. 2015, 11, 3189–3198. [Google Scholar] [CrossRef] [PubMed]
- Custelcean, R.; Garrabrant, K.A.; Agullo, P.; Williams, N.J. Direct Air Capture of CO2 with Aqueous Peptides and Crystalline Guanidines. Cell Rep. Phys. Sci. 2021, 2, 100385. [Google Scholar] [CrossRef]
- Kasturi, A.; Gabitto, J.; Tsouris, C.; Custelcean, R. Carbon Dioxide Capture with Aqueous Amino Acids: Mechanistic Study of Amino Acid Regeneration by Guanidine Crystallization and Process Intensification. Sep. Purif. Technol. 2021, 271, 118839. [Google Scholar] [CrossRef]
- Zhang, Q.; Jiang, Y.; Li, Y.; Song, X.; Luo, X.; Ke, Z.; Zou, Y. Design, Synthesis, and Physicochemical Study of a Biomass-Derived CO2 Sorbent 2,5-Furan-Bis(Iminoguanidine). iScience 2021, 24, 102263. [Google Scholar] [CrossRef]
- Stamberga, D.; Thiele, N.A.; Custelcean, R. Synergistic Direct Air Capture of CO2 with Aqueous Guanidine/Amino Acid Solvents. MRS Adv. 2022, 7, 399–403. [Google Scholar] [CrossRef]
- Jang, G.G.; Kasturi, A.; Stamberga, D.; Custelcean, R.; Keum, J.K.; Yiacoumi, S.; Tsouris, C. Ultra-Fast Microwave Regeneration of CO2 Solid Sorbents for Energy-Efficient Direct Air Capture. Sep. Purif. Technol. 2023, 309, 123053. [Google Scholar] [CrossRef]
- Kasturi, A.; Gug Jang, G.; Stamberga, D.; Custelcean, R.; Yiacoumi, S.; Tsouris, C. Determination of the Regeneration Energy of Direct Air Capture Solvents/Sorbents Using Calorimetric Methods. Sep. Purif. Technol. 2023, 310, 123154. [Google Scholar] [CrossRef]
- Espinosa, E.; Souhassou, M.; Lachekar, H.; Lecomte, C. Topological Analysis of the Electron Density in Hydrogen Bonds. Acta Crystallogr. B 1999, 55, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen Bond Strengths Revealed by Topological Analyses of Experimentally Observed Electron Densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Allen, F.H.; Bruno, I.J. Bond Lengths in Organic and Metal-Organic Compounds Revisited: X—H Bond Lengths from Neutron Diffraction Data. Acta Crystallogr. B 2010, 66, 380–386. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Madsen, A.Ø.; Sørensen, H.O.; Flensburg, C.; Stewart, R.F.; Larsen, S. Modeling of the Nuclear Parameters for H Atoms in X-ray Charge-Density Studies. Acta Crystallogr. A 2004, 60, 550–561. [Google Scholar] [CrossRef]
- Madsen, A.Ø.; Hoser, A.A. SHADE3 Server: A Streamlined Approach to Estimate H-Atom Anisotropic Displacement Parameters Using Periodic Ab Initio Calculations or Experimental Information. J. Appl. Crystallogr. 2014, 47, 2100–2104. [Google Scholar] [CrossRef]
- Hoser, A.A.; Dominiak, P.M.; Woźniak, K. Towards the Best Model for H Atoms in Experimental Charge-Density Refinement. Acta Crystallogr. A 2009, 65, 300–311. [Google Scholar] [CrossRef]
- Coppens, P. X-ray Charge Densities and Chemical Bonding; International Union of Crystallography: Chester, UK, 1997; ISBN 978-0-19-535694-6. [Google Scholar]
- Pawlędzio, S.; Makal, A.; Plażuk, D.; Woźniak, K. Experimental Charge Density of Ferrocenyl Derivative of β-Lactam. J. Mol. Struct. 2020, 1217, 128274. [Google Scholar] [CrossRef]
- Korlyukov, A.A.; Malinska, M.; Vologzhanina, A.V.; Goizman, M.S.; Trzybinski, D.; Wozniak, K. Charge Density View on Bicalutamide Molecular Interactions in the Monoclinic Polymorph and Androgen Receptor Binding Pocket. IUCrJ 2020, 7, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Tchoń, D.; Makal, A.; Gutmann, M.; Woźniak, K. Doxycycline Hydrate and Doxycycline Hydrochloride Dihydrate—Crystal Structure and Charge Density Analysis. Z. Krist. Cryst. Mater. 2018, 233, 649–661. [Google Scholar] [CrossRef]
- Cole, J.M.; Goeta, A.E.; Howard, J.a.K.; McIntyre, G.J. X-ray and Neutron Diffraction Studies of the Non-Linear Optical Compounds MBANP and MBADNP at 20 K: Charge-Density and Hydrogen-Bonding Analyses. Acta Crystallogr. B 2002, 58, 690–700. [Google Scholar] [CrossRef]
- Mallinson, P.R.; Smith, G.T.; Wilson, C.C.; Grech, E.; Wozniak, K. From Weak Interactions to Covalent Bonds: A Continuum in the Complexes of 1,8-Bis(Dimethylamino)Naphthalene. J. Am. Chem. Soc. 2003, 125, 4259–4270. [Google Scholar] [CrossRef]
- Dominiak, P.M.; Makal, A.; Mallinson, P.R.; Trzcinska, K.; Eilmes, J.; Grech, E.; Chruszcz, M.; Minor, W.; Woźniak, K. Continua of Interactions between Pairs of Atoms in Molecular Crystals. Chem. Weinh. Bergstr. Ger. 2006, 12, 1941–1949. [Google Scholar] [CrossRef] [PubMed]
- Munshi, P.; Madsen, A.Ø.; Spackman, M.A.; Larsen, S.; Destro, R. Estimated H-Atom Anisotropic Displacement Parameters: A Comparison between Different Methods and with Neutron Diffraction Results. Acta Crystallogr. A 2008, 64, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, K.; Mallinson, P.R.; Smith, G.T.; Wilson, C.C.; Grech, E. Role of C-H···O Hydrogen Bonds in the Ionic Complexes of 1,8-Bis(Dimethylamino)Naphthalene. J. Phys. Org. Chem. 2003, 16, 764–771. [Google Scholar] [CrossRef]
- Hoser, A.A.; Madsen, A.Ø. Dynamic Quantum Crystallography: Lattice-Dynamical Models Refined against Diffraction Data. I. Theory. Acta Crystallogr. Sect. Found. Adv. 2016, 72, 206–214. [Google Scholar] [CrossRef]
- Hoser, A.A.; Rekis, T.; Madsen, A.Ø. Dynamics and Disorder: On the Stability of Pyrazinamide Polymorphs. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2022, 78, 416–424. [Google Scholar] [CrossRef]
- Woińska, M.; Hoser, A.A.; Chodkiewicz, M.L.; Woźniak, K. Enhancing Hydrogen Positions in X-ray Structures of Transition Metal Hydride Complexes with Dynamic Quantum Crystallography. IUCrJ 2024, 11, 45–56. [Google Scholar] [CrossRef]
- Pichon-Pesme, V.; Jelsch, C.; Guillot, B.; Lecomte, C. A Comparison between Experimental and Theoretical Aspherical-Atom Scattering Factors for Charge-Density Refinement of Large Molecules. Acta Crystallogr. A 2004, 60, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Zarychta, B.; Pichon-Pesme, V.; Guillot, B.; Lecomte, C.; Jelsch, C. On the Application of an Experimental Multipolar Pseudo-Atom Library for Accurate Refinement of Small-Molecule and Protein Crystal Structures. Acta Crystallogr. A 2007, 63, 108–125. [Google Scholar] [CrossRef] [PubMed]
- Domagała, S.; Jelsch, C. Optimal Local Axes and Symmetry Assignment for Charge-Density Refinement. J. Appl. Crystallogr. 2008, 41, 1140–1149. [Google Scholar] [CrossRef]
- Rybicka, P.M.; Kulik, M.; Chodkiewicz, M.L.; Dominiak, P.M. Multipolar Atom Types from Theory and Statistical Clustering (MATTS) Data Bank: Impact of Surrounding Atoms on Electron Density from Cluster Analysis. J. Chem. Inf. Model. 2022, 62, 3766–3783. [Google Scholar] [CrossRef] [PubMed]
- Dominiak, P.M.; Volkov, A.; Li, X.; Messerschmidt, M.; Coppens, P. A Theoretical Databank of Transferable Aspherical Atoms and Its Application to Electrostatic Interaction Energy Calculations of Macromolecules. J. Chem. Theory Comput. 2007, 3, 232–247. [Google Scholar] [CrossRef] [PubMed]
- Koritsanszky, T.; Volkov, A.; Coppens, P. Aspherical-Atom Scattering Factors from Molecular Wave Functions. 1. Transferability and Conformation Dependence of Atomic Electron Densities of Peptides within the Multipole Formalism. Acta Crystallogr. A 2002, 58, 464–472. [Google Scholar] [CrossRef]
- Dittrich, B.; Koritsánszky, T.; Luger, P. A Simple Approach to Nonspherical Electron Densities by Using Invarioms. Angew. Chem. Int. Ed. 2004, 43, 2718–2721. [Google Scholar] [CrossRef]
- Dittrich, B.; Hübschle, C.B.; Luger, P.; Spackman, M.A. Introduction and Validation of an Invariom Database for Amino-Acid, Peptide and Protein Molecules. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 1325–1335. [Google Scholar] [CrossRef]
- Bąk, J.M.; Domagala, S.; Dittrich, B.; Jelsch, C.; Hübschle, C.; Dominiak, P.M. Verification of Structural and Electrostatic Properties Obtained by the Use of Different Pseudoatom Databases. Acta Crystallogr. Sect. Found. Crystallogr. 2011, B67, 141–153. [Google Scholar] [CrossRef]
- Jayatilaka, D.; Dittrich, B. X-ray Structure Refinement Using Aspherical Atomic Density Functions Obtained from Quantum-Mechanical Calculations. Acta Crystallogr. Sect. A 2008, 64, 383–393. [Google Scholar] [CrossRef]
- Jayatilaka, D.; Grimwood, D.J. Wavefunctions Derived from Experiment. I. Motivation and Theory. Acta Crystallogr. Sect. A 2001, 57, 76–86. [Google Scholar] [CrossRef]
- Malaspina, L.A.; Wieduwilt, E.K.; Bergmann, J.; Kleemiss, F.; Meyer, B.; Ruiz-López, M.F.; Pal, R.; Hupf, E.; Beckmann, J.; Piltz, R.O.; et al. Fast and Accurate Quantum Crystallography: From Small to Large, from Light to Heavy. J. Phys. Chem. Lett. 2019, 10, 6973–6982. [Google Scholar] [CrossRef] [PubMed]
- Chodkiewicz, M.L.; Woińska, M.; Woźniak, K. Hirshfeld Atom like Refinement with Alternative Electron Density Partitions. IUCrJ 2020, 7, 1199–1215. [Google Scholar] [CrossRef] [PubMed]
- Malaspina, L.A.; Genoni, A.; Jayatilaka, D.; Turner, M.J.; Sugimoto, K.; Nishibori, E.; Grabowsky, S. The Advanced Treatment of Hydrogen Bonding in Quantum Crystallography. J. Appl. Crystallogr. 2021, 54, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Malaspina, L.A.; Hoser, A.A.; Edwards, A.J.; Woińska, M.; Turner, M.J.; Price, J.R.; Sugimoto, K.; Nishibori, E.; Bürgi, H.-B.; Jayatilaka, D.; et al. Hydrogen Atoms in Bridging Positions from Quantum Crystallographic Refinements: Influence of Hydrogen Atom Displacement Parameters on Geometry and Electron Density. CrystEngComm 2020, 22, 4778–4789. [Google Scholar] [CrossRef]
- Woinska, M.; Jayatilaka, D.; Dittrich, B.; Flaig, R.; Luger, P.; Woźniak, K.; Dominiak, P.M.; Grabowsky, S. Validation of X-Ray Wavefunction Refinement. Chemphyschem Eur. J. Chem. Phys. Phys. Chem. 2017, 18, 3334–3351. [Google Scholar] [CrossRef] [PubMed]
- Woińska, M.; Jayatilaka, D.; Spackman, M.A.; Edwards, A.J.; Dominiak, P.M.; Woźniak, K.; Nishibori, E.; Sugimoto, K.; Grabowsky, S. Hirshfeld Atom Refinement for Modelling Strong Hydrogen Bonds. Acta Crystallogr. Sect. A 2014, 70, 483–498. [Google Scholar] [CrossRef]
- Woińska, M.; Grabowsky, S.; Dominiak, P.M.; Woźniak, K.; Jayatilaka, D. Hydrogen Atoms Can Be Located Accurately and Precisely by X-ray Crystallography. Sci. Adv. 2016, 2, e1600192. [Google Scholar] [CrossRef]
- Woinska, M.; Wanat, M.; Taciak, P.; Pawinski, T.; Minor, W.; Wozniak, K. Energetics of Interactions in the Solid State of 2-Hydroxy-8-X-Quinoline Derivatives (X = Cl, Br, I, S-Ph): Comparison of Hirshfeld Atom, X-ray Wavefunction and Multipole Refinements. IUCrJ 2019, 6, 868–883. [Google Scholar] [CrossRef]
- Wanat, M.; Malinska, M.; Hoser, A.A.; Woźniak, K. Further Validation of Quantum Crystallography Approaches. Molecules 2021, 26, 3730. [Google Scholar] [CrossRef]
- Genoni, A. X-Ray Constrained Extremely Localized Molecular Orbitals: Theory and Critical Assessment of the New Technique. J. Chem. Theory Comput. 2013, 9, 3004–3019. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.; Genoni, A.; Macchi, P. Analysis of Crystal Field Effects and Interactions Using X-ray Restrained ELMOs. J. Mol. Struct. 2020, 1209, 127975. [Google Scholar] [CrossRef]
- Wieduwilt, E.K.; Macetti, G.; Scatena, R.; Macchi, P.; Genoni, A. Extending Libraries of Extremely Localized Molecular Orbitals to Metal Organic Frameworks: A Preliminary Investigation. Crystals 2021, 11, 207. [Google Scholar] [CrossRef]
- Wieduwilt, E.K.; Macetti, G.; Malaspina, L.A.; Jayatilaka, D.; Grabowsky, S.; Genoni, A. Post-Hartree-Fock Methods for Hirshfeld Atom Refinement: Are They Necessary? Investigation of a Strongly Hydrogen-Bonded Molecular Crystal. J. Mol. Struct. 2020, 1209, 127934. [Google Scholar] [CrossRef]
- Dovesi, R.; Orlando, R.; Civalleri, B.; Roetti, C.; Saunders, V.R.; Zicovich-Wilson, C.M. CRYSTAL: A Computational Tool for the Ab Initio Study of the Electronic Properties of Crystals. Z. Krist. Cryst. Mater. 2009, 220, 571–573. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A Program for Hirshfeld Surface Analysis, Visualization and Quantitative Analysis of Molecular Crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Gavezzotti, A. Calculation of Lattice Energies of Organic Crystals: The PIXEL Integration Method in Comparison with More Traditional Methods. Z. Krist. Cryst. Mater. 2005, 220, 499–510. [Google Scholar] [CrossRef]
- Gavezzotti, A. Efficient Computer Modeling of Organic Materials. The Atom–Atom, Coulomb–London–Pauli (AA-CLP) Model for Intermolecular Electrostatic-Polarization, Dispersion and Repulsion Energies. New J. Chem. 2011, 35, 1360–1368. [Google Scholar] [CrossRef]
- Volkov, A.; King, H.F.; Coppens, P.; Farrugia, L.J. On the Calculation of the Electrostatic Potential, Electric Field and Electric Field Gradient from the Aspherical Pseudoatom Model. Acta Crystallogr. Sect. A 2006, 62, 400–408. [Google Scholar] [CrossRef]
- Volkov, A.; Macchi, C.; Farrugia, L.J.; Gatti, P.; Mallinson, P.R.; Richter, T.; Koritsanszky, T. XD2016—A Computer Program Package for Multipole Refinement, Topological Analysis of Charge Densities and Evaluation of Intermolecular Energies from Experimental or Theoretical Structure Factors; University at Buffalo: Amherst, NY, USA, 2016. [Google Scholar]
- Jelsch, C.; Guillot, B.; Lagoutte, A.; Lecomte, C. Advances in Protein and Small-Molecule Charge-Density Refinement Methods Using MoPro. J. Appl. Crystallogr. 2005, 38, 38–54. [Google Scholar] [CrossRef]
- Guillot, B.; Viry, L.; Guillot, R.; Lecomte, C.; Jelsch, C. Refinement of Proteins at Subatomic Resolution with MOPRO. J. Appl. Crystallogr. 2001, 34, 214–223. [Google Scholar] [CrossRef]
- Volkov, A.; Li, X.; Koritsanszky, T.; Coppens, P. Ab Initio Quality Electrostatic Atomic and Molecular Properties Including Intermolecular Energies from a Transferable Theoretical Pseudoatom Databank. J. Phys. Chem. A 2004, 108, 4283–4300. [Google Scholar] [CrossRef]
- Volkov, A.; Koritsanszky, T.; Coppens, P. Combination of the Exact Potential and Multipole Methods (EP/MM) for Evaluation of Intermolecular Electrostatic Interaction Energies with Pseudoatom Representation of Molecular Electron Densities. Chem. Phys. Lett. 2004, 391, 170–175. [Google Scholar] [CrossRef]
- Li, X.; Volkov, A.V.; Szalewicz, K.; Coppens, P. Interaction Energies between Glycopeptide Antibiotics and Substrates in Complexes Determined by X-ray Crystallography: Application of a Theoretical Databank of Aspherical Atoms and a Symmetry-Adapted Perturbation Theory-Based Set of Interatomic Potentials. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Kulik, M.; Goral, A.M.; Jasiński, M.; Dominiak, P.M.; Trylska, J. Electrostatic Interactions in Aminoglycoside-RNA Complexes. Biophys. J. 2015, 108, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Budniak, U.A.; Karolak, N.K.; Kulik, M.; Młynarczyk, K.; Górna, M.W.; Dominiak, P.M. The Role of Electrostatic Interactions in IFIT5-RNA Complexes Predicted by the UBDB+EPMM Method. J. Phys. Chem. B 2022, 126, 9152–9167. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Mills, M.J.L.; Popelier, P.L.A. Multipolar Electrostatics for Proteins: Atom–Atom Electrostatic Energies in Crambin. J. Comput. Chem. 2014, 35, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Malińska, M.; Jarzembska, K.N.; Goral, A.M.; Kutner, A.; Woźniak, K.; Dominiak, P.M. Sunitinib: From Charge-Density Studies to Interaction with Proteins. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 1257–1270. [Google Scholar] [CrossRef]
- Falcicchio, A.; Lill, S.O.N.; Perna, F.M.; Salomone, A.; Coppi, D.I.; Cuocci, C.; Stalke, D.; Capriati, V. Organotrifluoroborates as Attractive Self-Assembling Systems: The Case of Bifunctional Dipotassium Phenylene-1,4-Bis(Trifluoroborate). Dalton Trans. 2015, 44, 19447–19450. [Google Scholar] [CrossRef]
- Özbek, F.E.; Sertçelik, M.; Yüksek, M. Hirshfeld Surface Analysis and Interaction Energy Calculations of Bis(4-Chlorophenylacetate)Bis(Pyridine-4-Carboxamide)Zinc(II). Cauc. J. Sci. 2020, 7, 83–91. [Google Scholar] [CrossRef]
- Kumar, P.; Bojarowski, S.A.; Jarzembska, K.N.; Domagała, S.; Vanommeslaeghe, K.; MacKerell, A.D.; Dominiak, P.M. A Comparative Study of Transferable Aspherical Pseudoatom Databank and Classical Force Fields for Predicting Electrostatic Interactions in Molecular Dimers. J. Chem. Theory Comput. 2014, 10, 1652–1664. [Google Scholar] [CrossRef] [PubMed]
- Munshi, P.; Guru Row, T.N. Topological Analysis of Charge Density Distribution in Concomitant Polymorphs of 3-Acetylcoumarin, A Case of Packing Polymorphism. Cryst. Growth Des. 2006, 6, 708–718. [Google Scholar] [CrossRef]
- Munshi, P.; Guru Row, T.N. Intra- and Intermolecular Interactions in Small Bioactive Molecules: Cooperative Features from Experimental and Theoretical Charge-Density Analysis. Acta Crystallogr. B 2006, 62, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Bilal, A.; Mehmood, A.; Noureen, S.; Lecomte, C.; Ahmed, M. Crystal Engineering of a Co-Crystal of Antipyrine and 2-Chlorobenzoic Acid: Relative Energetic Contributions Based on Multipolar Refinement. CrystEngComm 2022, 24, 7758–7770. [Google Scholar] [CrossRef]
- Pawlędzio, S.; Makal, A.; Trzybiński, D.; Woźniak, K. Crystal Structure, Interaction Energies and Experimental Electron Density of the Popular Drug Ketoprophen. IUCrJ 2018, 5, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Dominiak, P.M.; Grech, E.; Barr, G.; Teat, S.; Mallinson, P.; Woźniak, K. Neutral and Ionic Hydrogen Bonding in Schiff Bases. Chem. Eur. J. 2003, 9, 963–970. [Google Scholar] [CrossRef]
- Koch, U.; Popelier, P.L.A. Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Jabłoński, M.; Monaco, G. Different Zeroes of Interaction Energies as the Cause of Opposite Results on the Stabilizing Nature of C–H···O Intramolecular Interactions. J. Chem. Inf. Model. 2013, 53, 1661–1675. [Google Scholar] [CrossRef]
- Gatti, C.; May, E.; Destro, R.; Cargnoni, F. Fundamental Properties and Nature of CH··O Interactions in Crystals on the Basis of Experimental and Theoretical Charge Densities. The Case of 3,4-Bis(Dimethylamino)-3-Cyclobutene-1,2-Dione (DMACB) Crystal. J. Phys. Chem. A 2002, 106, 2707–2720. [Google Scholar] [CrossRef]
- Nikolaienko, T.Y.; Bulavin, L.A.; Hovorun, D.M. Bridging QTAIM with Vibrational Spectroscopy: The Energy of Intramolecular Hydrogen Bonds in DNA-Related Biomolecules. Phys. Chem. Chem. Phys. 2012, 14, 7441–7447. [Google Scholar] [CrossRef]
- Kuznetsov, M.L. Relationships between Interaction Energy and Electron Density Properties for Homo Halogen Bonds of the [(A)nY–X···X–Z(B)m] Type (X = Cl, Br, I). Molecules 2019, 24, 2733. [Google Scholar] [CrossRef] [PubMed]
- Mata, I.; Alkorta, I.; Espinosa, E.; Molins, E. Relationships between Interaction Energy, Intermolecular Distance and Electron Density Properties in Hydrogen Bonded Complexes under External Electric Fields. Chem. Phys. Lett. 2011, 507, 185–189. [Google Scholar] [CrossRef]
- Afonin, A.V.; Vashchenko, A.V.; Sigalov, M.V. Estimating the Energy of Intramolecular Hydrogen Bonds from 1H NMR and QTAIM Calculations. Org. Biomol. Chem. 2016, 14, 11199–11211. [Google Scholar] [CrossRef] [PubMed]
- Gavezzotti, A. Calculation of Intermolecular Interaction Energies by Direct Numerical Integration over Electron Densities. I. Electrostatic and Polarization Energies in Molecular Crystals. J. Phys. Chem. B 2002, 106, 4145–4154. [Google Scholar] [CrossRef]
- Gavezzotti, A. Towards a Realistic Model for the Quantitative Evaluation of Intermolecular Potentials and for the Rationalization of Organic Crystal Structures. Part, I. Philosophy. CrystEngComm 2003, 5, 429–438. [Google Scholar] [CrossRef]
- Gavezzotti, A. Quantitative Ranking of Crystal Packing Modes by Systematic Calculations on Potential Energies and Vibrational Amplitudes of Molecular Dimers. J. Chem. Theory Comput. 2005, 1, 834–840. [Google Scholar] [CrossRef]
- Gavezzotti, A. The Lines-of-Force Landscape of Interactions between Molecules in Crystals; Cohesive versus Tolerant and ‘collateral Damage’ Contact. Acta Crystallogr. B 2010, 66, 396–406. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16, Revision, C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. A Direct MP2 Gradient Method. Chem. Phys. Lett. 1990, 166, 275–280. [Google Scholar] [CrossRef]
- Funnell, N.P.; Bull, C.L.; Ridley, C.J.; Capelli, S. Structural Behaviour of OP-ROY at Extreme Conditions. CrystEngComm 2019, 21, 4473–4483. [Google Scholar] [CrossRef]
- Maschio, L.; Civalleri, B.; Ugliengo, P.; Gavezzotti, A. Intermolecular Interaction Energies in Molecular Crystals: Comparison and Agreement of Localized Møller-Plesset 2, Dispersion-Corrected Density Functional, and Classical Empirical Two-Body Calculations. J. Phys. Chem. A 2011, 115, 11179–11186. [Google Scholar] [CrossRef]
- Carlucci, L.; Gavezzotti, A. A Quantitative Measure of Halogen Bond Activation in Cocrystallization. Phys. Chem. Chem. Phys. 2017, 19, 18383–18388. [Google Scholar] [CrossRef] [PubMed]
- Dunitz, J.D.; Gavezzotti, A. Proteogenic Amino Acids: Chiral and Racemic Crystal Packings and Stabilities. J. Phys. Chem. B 2012, 116, 6740–6750. [Google Scholar] [CrossRef]
- Hutchison, I.B.; Bull, C.L.; Marshall, W.G.; Parsons, S.; Urquhart, A.J.; Oswald, I.D.H. Compression of Glycolide-H4 to 6 GPa. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2017, 73, 1151–1157. [Google Scholar] [CrossRef]
- Reeves, M.G.; Wood, P.A.; Parsons, S. MrPIXEL: Automated Execution of Pixel Calculations via the Mercury Interface. J. Appl. Crystallogr. 2020, 53, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld Surface Analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Jayatilaka, D.; Spackman, M.A. Towards Quantitative Analysis of Intermolecular Interactions with Hirshfeld Surfaces. Chem. Commun. 2007, 0, 3814–3816. [Google Scholar] [CrossRef]
- Spackman, M.A.; McKinnon, J.J. Fingerprinting Intermolecular Interactions in Molecular Crystals. CrystEngComm 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Turner, M.J.; Thomas, S.P.; Shi, M.W.; Jayatilaka, D.; Spackman, M.A. Energy Frameworks: Insights into Interaction Anisotropy and the Mechanical Properties of Molecular Crystals. Chem. Commun. 2015, 51, 3735–3738. [Google Scholar] [CrossRef]
- Ziemniak, M.; Pawlędzio, S.; Zawadzka-Kaźmierczuk, A.; Dominiak, P.M.; Trzybiński, D.; Koźmiński, W.; Zieliński, R.; Fokt, I.; Priebe, W.; Woźniak, K.; et al. X-Ray Wavefunction Refinement and Comprehensive Structural Studies on Bromo-Substituted Analogues of 2-Deoxy-D-Glucose in Solid State and Solution. RSC Adv. 2022, 12, 8345–8360. [Google Scholar] [CrossRef]
- Pawlędzio, S.; Malinska, M.; Kleemiss, F.; Grabowsky, S.; Woźniak, K. Influence of Modelling Disorder on Hirshfeld Atom Refinement Results of an Organo-Gold(I) Compound. IUCrJ 2022, 9, 497–507. [Google Scholar] [CrossRef]
- Fugel, M.; Jayatilaka, D.; Hupf, E.; Overgaard, J.; Hathwar, V.R.; Macchi, P.; Turner, M.J.; Howard, J.A.K.; Dolomanov, O.V.; Puschmann, H.; et al. Probing the Accuracy and Precision of Hirshfeld Atom Refinement with HARt Interfaced with Olex2. IUCrJ 2018, 5, 32–44. [Google Scholar] [CrossRef]
- Köhler, C.; Lübben, J.; Krause, L.; Hoffmann, C.; Herbst-Irmer, R.; Stalke, D. Comparison of Different Strategies for Modelling Hydrogen Atoms in Charge Density Analyses. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2019, 75, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A. How Reliable Are Intermolecular Interaction Energies Estimated from Topological Analysis of Experimental Electron Densities? Cryst. Growth Des. 2015, 15, 5624–5628. [Google Scholar] [CrossRef]
- Khalilov, L.M.; Mescheryakova, E.S.; Bikmukhametov, K.S.; Makhmudiyarova, N.N.; Shangaraev, K.R.; Tulyabaev, A.R. Twist-Chair Conformation of the Tetraoxepane Ring Remains Unchanged in Tetraoxaspirododecane Diamines. Acta Crystallogr. Sect. C Struct. Chem. 2020, 76, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Kowsalya, P.S.; Bhuvanesh, N.S.P.; Neelakantan, M.A. Chemical Reactivity and Quantifying the Intra- and Intermolecular Interactions in Zwitterionic Compounds. ChemistrySelect 2018, 3, 2045–2052. [Google Scholar] [CrossRef]
- Matta, C.F.; Castillo, N.; Boyd, R.J. Extended Weak Bonding Interactions in DNA: π-Stacking (Base−Base), Base−Backbone, and Backbone−Backbone Interactions. J. Phys. Chem. B 2006, 110, 563–578. [Google Scholar] [CrossRef]
- Kumar, P.; Cabaj, M.K.; Dominiak, P.M. Intermolecular Interactions in Ionic Crystals of Nucleobase Chlorides—Combining Topological Analysis of Electron Densities with Energies of Electrostatic Interactions. Crystals 2019, 9, 668. [Google Scholar] [CrossRef]
- Alkorta, I.; Mata, I.; Molins, E.; Espinosa, E. Charged versus Neutral Hydrogen-Bonded Complexes: Is There a Difference in the Nature of the Hydrogen Bonds? Chem. Eur. J. 2016, 22, 9226–9234. [Google Scholar] [CrossRef]
- Bauer, C.A.; Schneider, G.; Göller, A.H. Machine Learning Models for Hydrogen Bond Donor and Acceptor Strengths Using Large and Diverse Training Data Generated by First-Principles Interaction Free Energies. J. Cheminform. 2019, 11, 59. [Google Scholar] [CrossRef]
- Xu, X.; Song, C.; Andresen, J.M.; Miller, B.G.; Scaroni, A.W. Novel Polyethylenimine-Modified Mesoporous Molecular Sieve of MCM-41 Type as High-Capacity Adsorbent for CO2 Capture. Energy Fuels 2002, 16, 1463–1469. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | HBs from Cation | HBs from Water | HBs from Anion |
|---|---|---|---|
| PyBIG | 5 | 4 | 0 |
| GBIG | 3 | 2 | 2 |
| MGBIG P1 | 8 | 0 | 0 |
| MGBIG P3 | 6 | 3 | 0 |
| DABIG | 5 | 4 | 0 |
| Neutron [a] | XRD_(BIGs) [b] | Neutron_(BIGs) [b] | |||
|---|---|---|---|---|---|
| Substructure | d_mean | d_mean | d_median | d_mean | d_median |
| C=Csp2—H | 1.082 | 0.981 | 0.951 | 1.096 | 1.096 |
| C(ar)—H | 1.083 | 0.993 | 0.950 | 1.086 | 1.086 |
| C—Csp3—H3 | 1.077 | 0.975 | 0.980 | - | - |
| Csp2—N—H2 | 1.013 | 0.893 | 0.880 | 1.021 | 1.021 |
| Z2—N—H | 1.027 | 0.927 | 0.920 | 1.049 | 1.050 |
| Z—O—H | 0.983 | 0.905 | 0.900 | 0.976 | 0.974 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawlędzio, S.; Wang, X. Crystal Engineering of Hydrogen Bonding for Direct Air Capture of CO2: A Quantum Crystallography Perspective. Crystals 2024, 14, 77. https://doi.org/10.3390/cryst14010077
Pawlędzio S, Wang X. Crystal Engineering of Hydrogen Bonding for Direct Air Capture of CO2: A Quantum Crystallography Perspective. Crystals. 2024; 14(1):77. https://doi.org/10.3390/cryst14010077
Chicago/Turabian StylePawlędzio, Sylwia, and Xiaoping Wang. 2024. "Crystal Engineering of Hydrogen Bonding for Direct Air Capture of CO2: A Quantum Crystallography Perspective" Crystals 14, no. 1: 77. https://doi.org/10.3390/cryst14010077
APA StylePawlędzio, S., & Wang, X. (2024). Crystal Engineering of Hydrogen Bonding for Direct Air Capture of CO2: A Quantum Crystallography Perspective. Crystals, 14(1), 77. https://doi.org/10.3390/cryst14010077