NMR Crystallography of the Polymorphs of Metergoline

Abstract
:1. Introduction
2. Materials and Methods
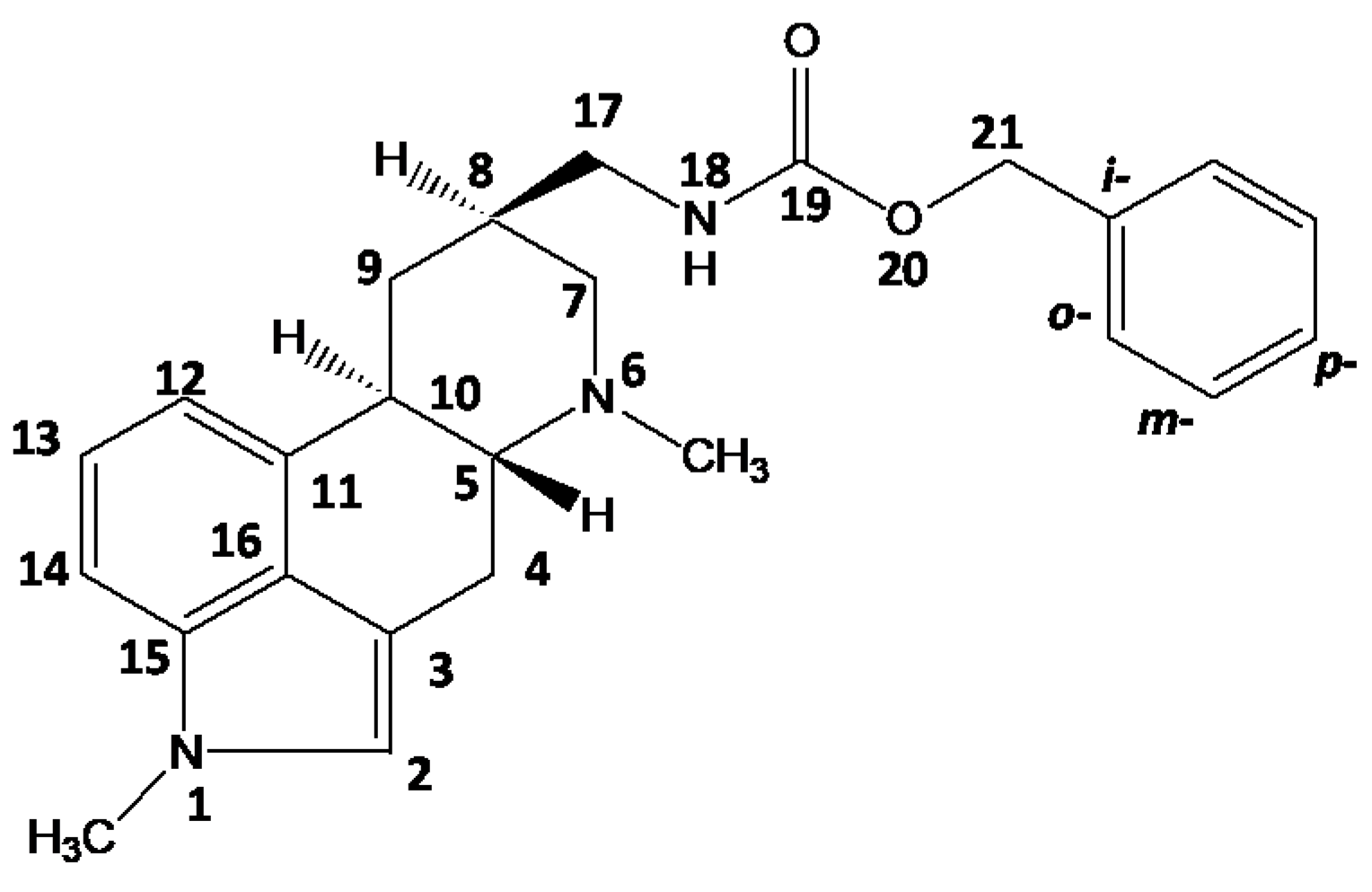
2.1. Metergoline Structures
2.2. Solid-State NMR Experiments
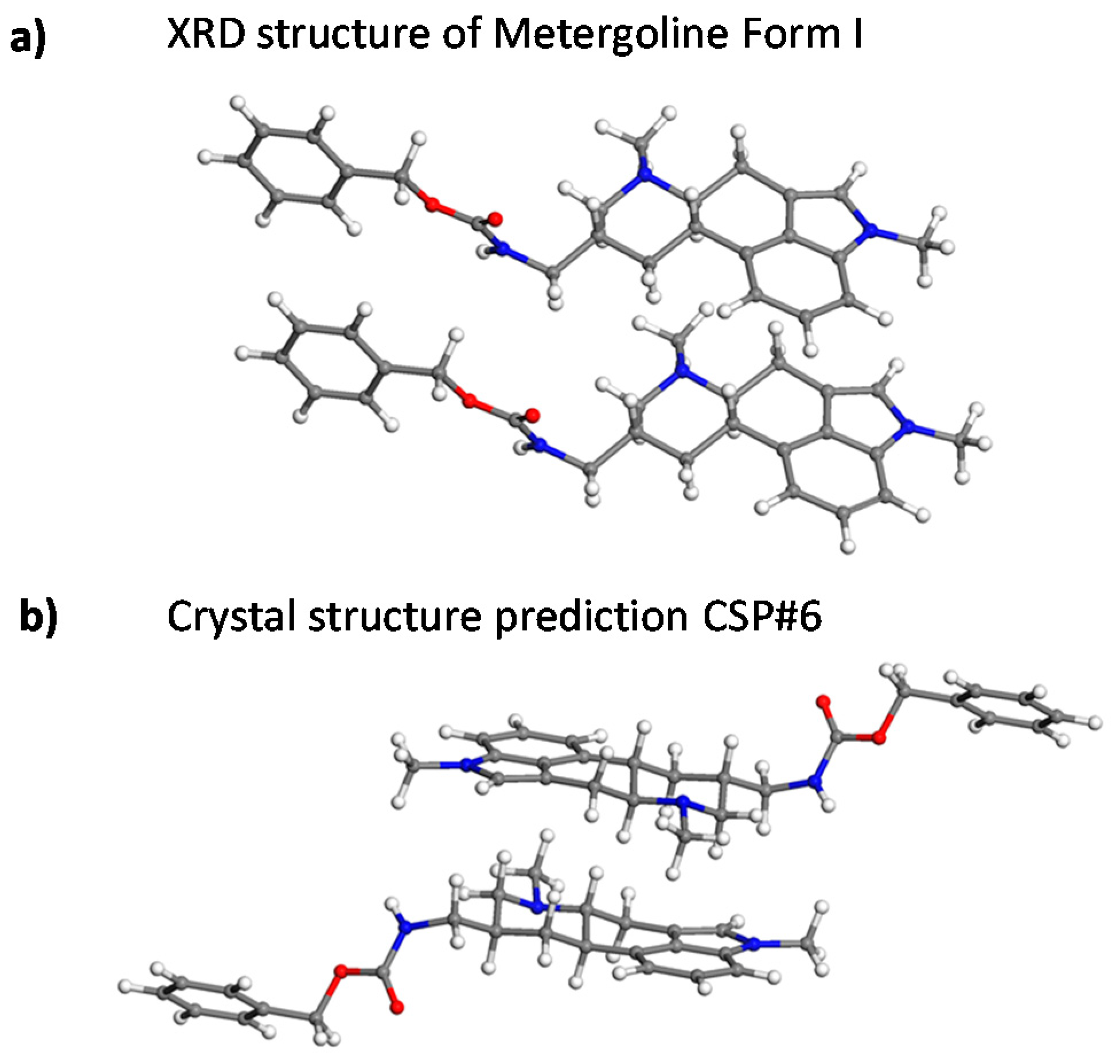
2.3. Crystal Structure Predictions
2.4. DFT Calculations
3. Results
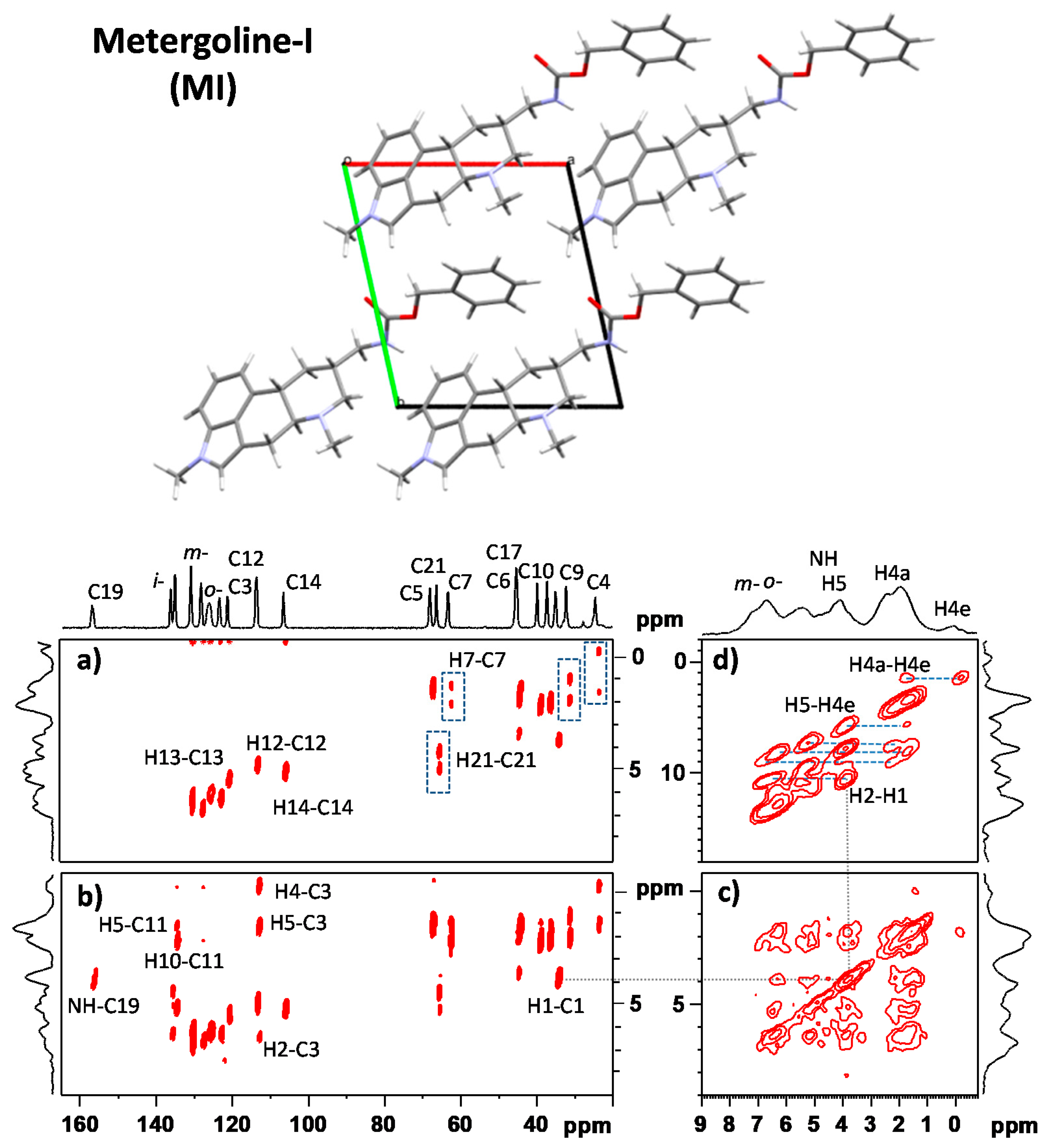
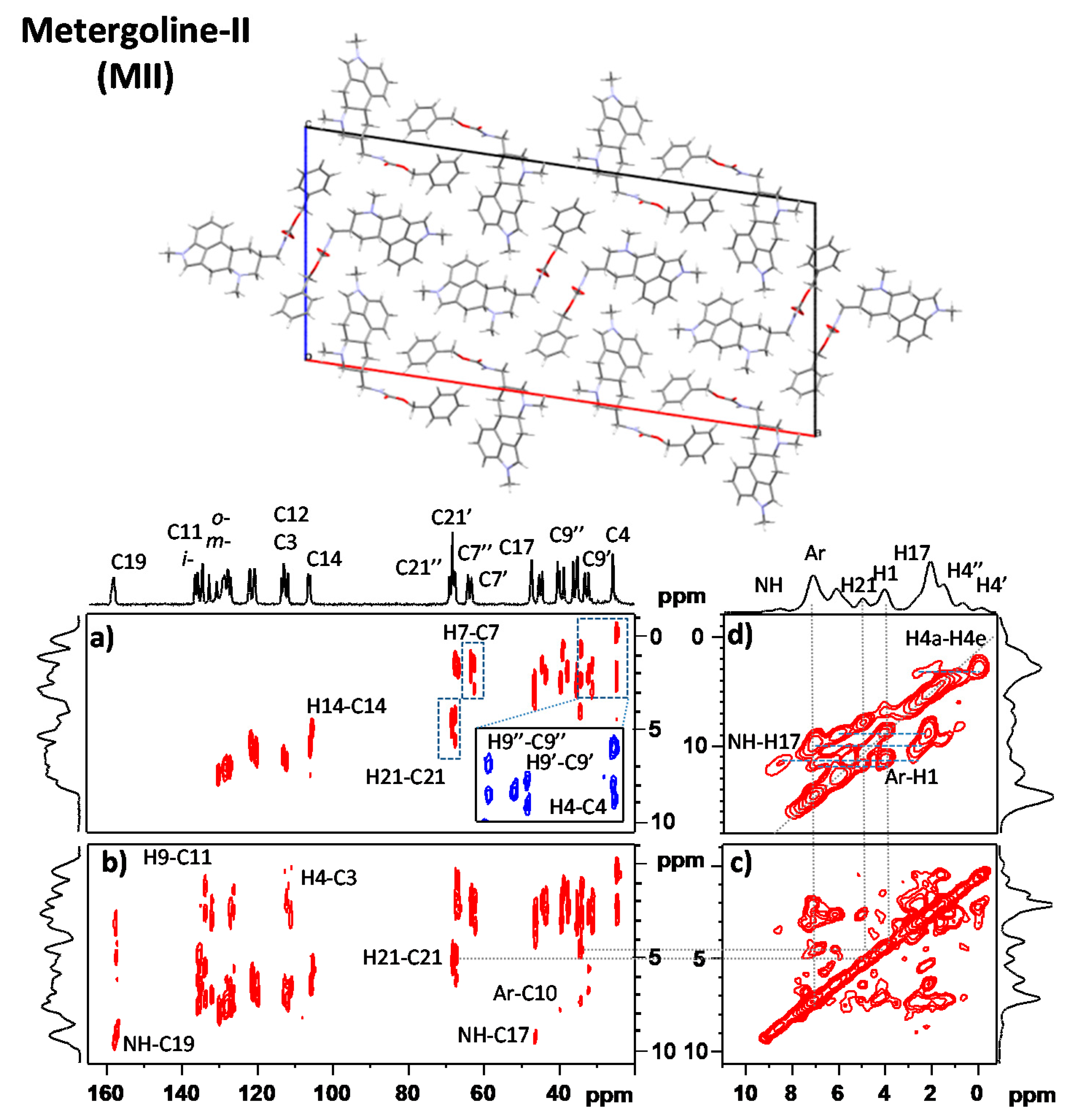
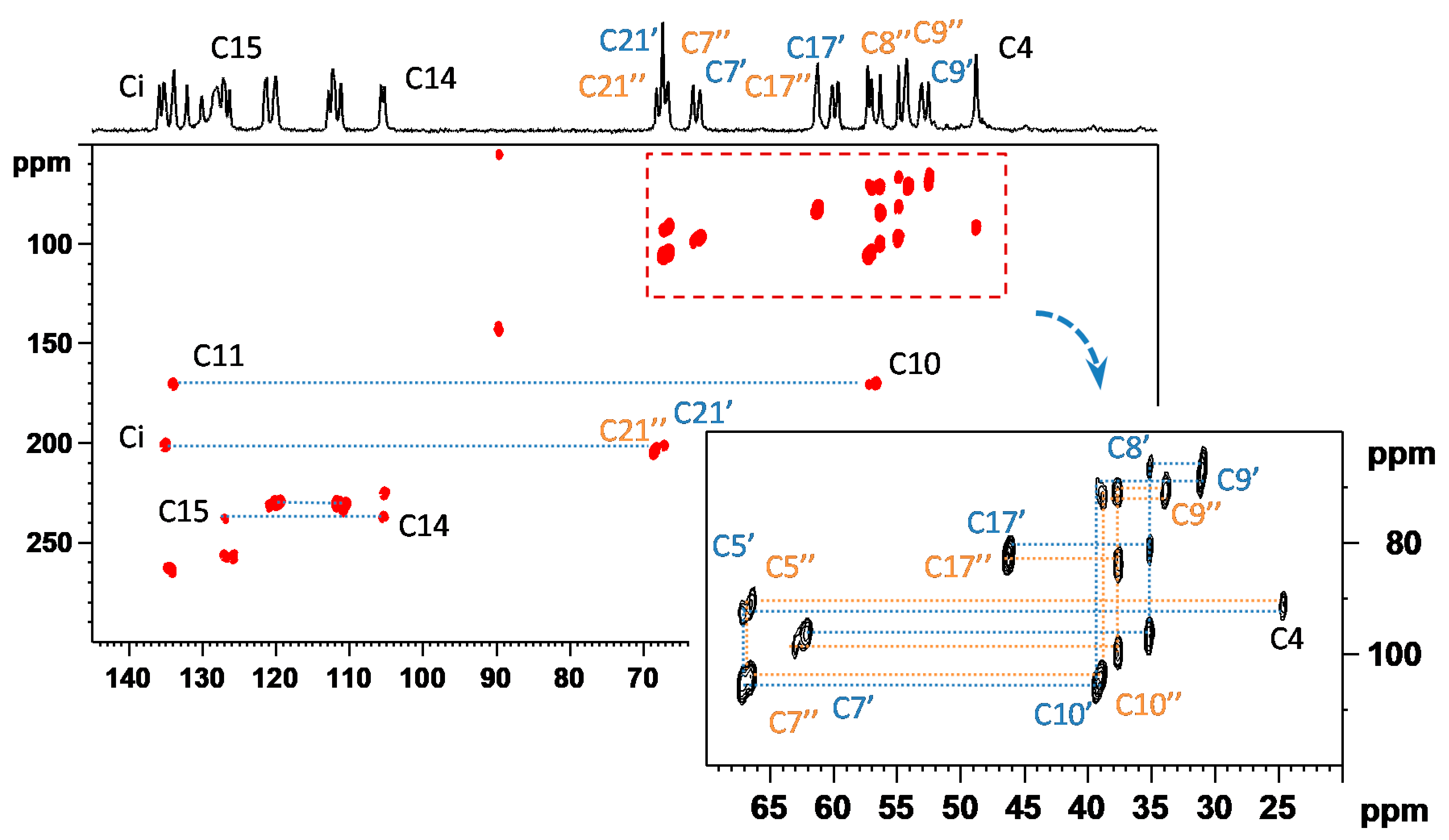
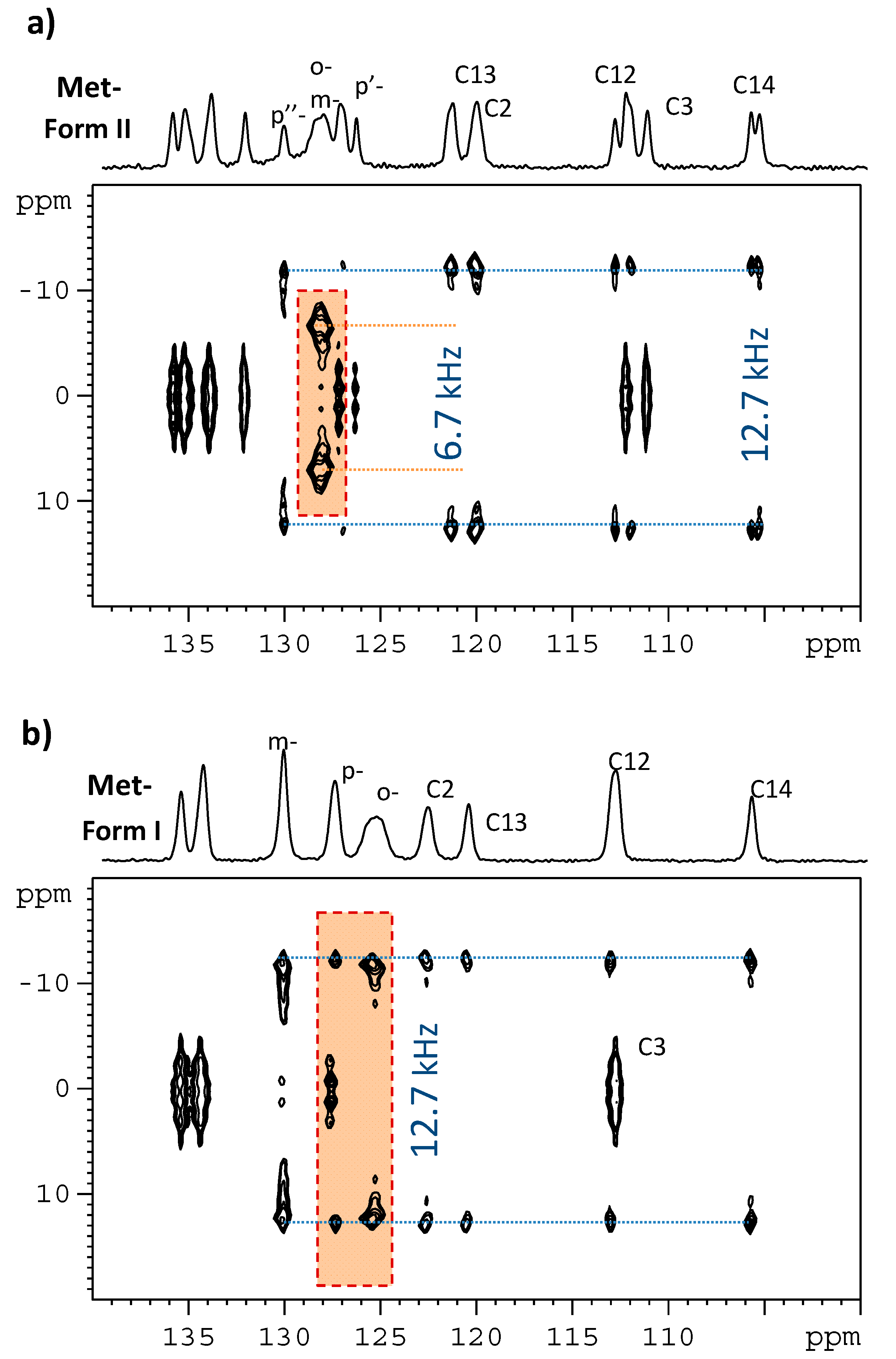
3.1. Signal Assignment of the Polymorphs
3.2. Segmental Dynamics
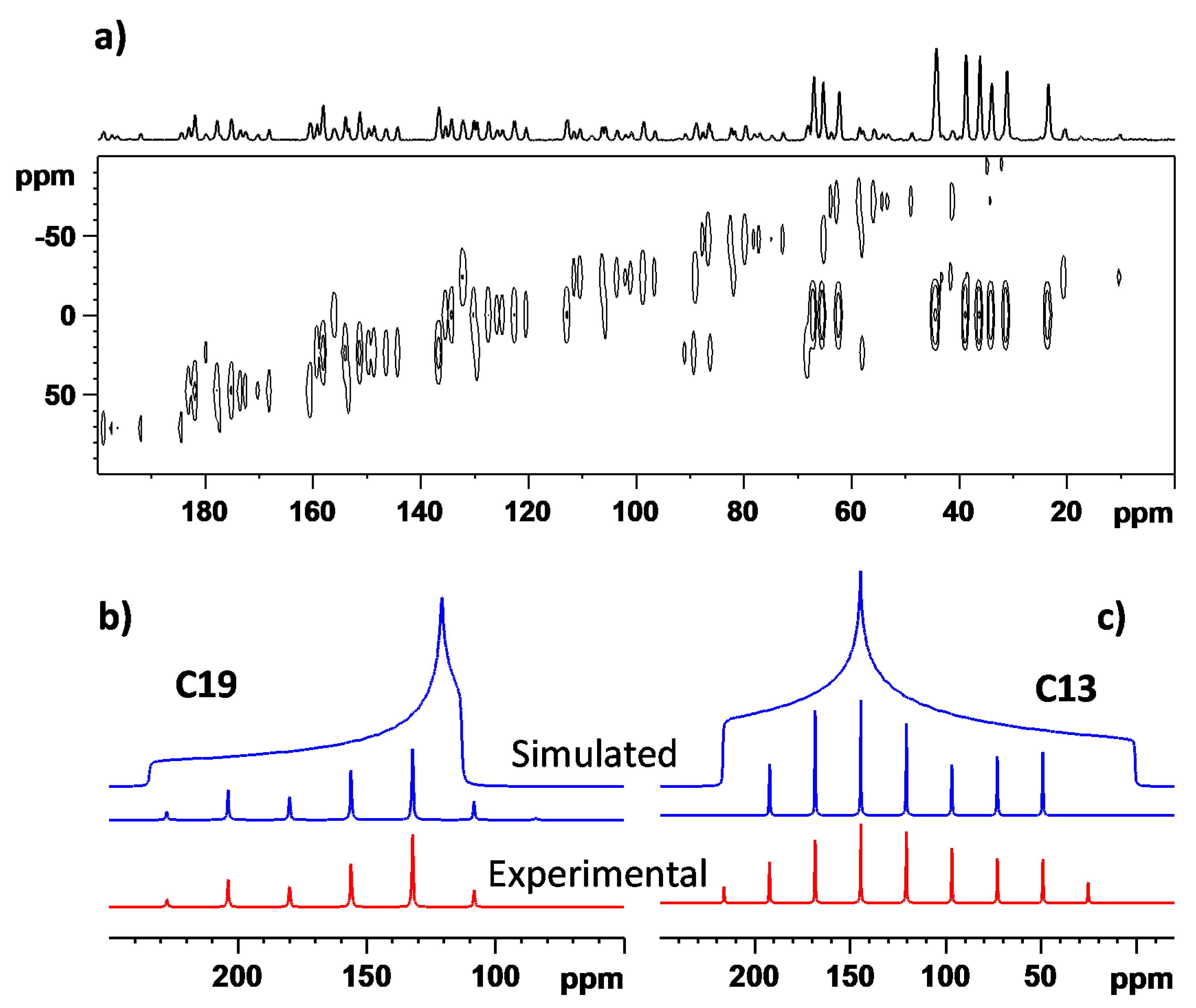
3.3. The 13C NMR Chemical Shift Tensors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Harris, R.K.; Wasylishen, R.E.; Duer, M.J. NMR Crystallography; Wile: Hoboken, NJ, USA, 2010. [Google Scholar]
- Salager, E.; Day, G.M.; Stein, R.S.; Pickard, C.J.; Elena, B.; Emsley, L. Powder crystallography by combined crystal structure prediction and high-resolution 1H solid-state NMR spectroscopy. J. Am. Chem. Soc. 2010, 132, 2564–2566. [Google Scholar] [CrossRef] [PubMed]
- Reilly, A.M.; Cooper, R.I.; Adjiman, C.S.; Bhattacharya, S.; Boese, A.D.; Brandenburg, J.G.; Bygrave, P.J.; Bylsma, R.; Campbell, J.E.; Car, R.; et al. Report on the sixth blind test of organic crystal structure prediction methods. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 439–459. [Google Scholar] [CrossRef] [PubMed]
- Bonhomme, C.; Gervais, C.; Babonneau, F.; Coelho, C.; Pourpoint, F.; Azaïs, T.; Ashbrook, S.E.; Griffin, J.M.; Yates, J.R.; Mauri, F.; et al. First-principles calculation of NMR parameters using the gauge including projector augmented wave method: A chemist’s point of view. Chem. Rev. 2012, 112, 5733–5779. [Google Scholar] [CrossRef] [PubMed]
- Ashbrook, S.E.; McKay, D. Combining solid-state NMR spectroscopy with first-principles calculations—A guide to NMR crystallography. Chem. Commun. (Camb.) 2016, 52, 7186–7204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryce, D.L. NMR crystallography: Structure and properties of materials from solid-state nuclear magnetic resonance observables. IUCrj 2017, 4, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Tatton, A.S.; Blade, H.; Brown, S.P.; Hodgkinson, P.; Hughes, L.P.; Lill, S.O.N.; Yates, J.R. Improving confidence in crystal structure solutions using NMR crystallography: The case of β-piroxicam. Cryst. Growth Des. 2018, 18, 3339–3351. [Google Scholar] [CrossRef]
- Nilsson Lill, S.O.; Widdifield, C.M.; Pettersen, A.; Svensk Ankarberg, A.; Lindkvist, M.; Aldred, P.; Gracin, S.; Shankland, N.; Shankland, K.; Schantz, S.; et al. Elucidating an amorphous form stabilization mechanism for Tenapanor hydrochloride: Crystal structure analysis using X-ray diffraction, NMR crystallography, and molecular modeling. Mol. Pharm. 2018, 15, 1476–1487. [Google Scholar] [CrossRef] [PubMed]
- Kobera, L.; Rohlicek, J.; Czernek, J.; Abbrent, S.; Streckova, M.; Sopcak, T.; Brus, J. Unexpected crystallization patterns of zinc boron imidazolate framework ZBIF-1: NMR crystallography of integrated metal-organic frameworks. ChemPhysChem 2017, 18, 3576–3582. [Google Scholar] [CrossRef] [PubMed]
- Czernek, J.; Pawlak, T.; Potrzebowski, M.J.; Brus, J. The comparison of approaches to the solid-state NMR-based structural refinement of vitamin B1 hydrochloride and of its monohydrate. Chem. Phys. Lett. 2013, 555, 135–140. [Google Scholar] [CrossRef]
- Hušák, M.; Jegorov, A.; Rohlíček, J.; Fitch, A.; Czernek, J.; Kobera, L.; Brus, J. Determining the crystal structures of peptide analogs of boronic acid in the absence of single crystals: Intricate motifs of ixazomib citrate revealed by XRPD guided by ss-NMR. Cryst. Growth Des. 2018, 18, 3616–3625. [Google Scholar] [CrossRef]
- Brus, J.; Czernek, J.; Kobera, L.; Urbanova, M.; Abbrent, S.; Husak, M. Predicting the crystal structure of decitabine by powder NMR crystallography: Influence of long-range molecular packing symmetry on NMR parameters. Cryst. Growth Des. 2016, 16, 7102–7111. [Google Scholar] [CrossRef]
- Brus, J.; Czernek, J.; Hruby, M.; Svec, P.; Kobera, L.; Abbrent, S.; Urbanova, M. Efficient strategy for determining the atomic-resolution structure of micro- and nanocrystalline solids within polymeric microbeads: Domain-edited NMR crystallography. Macromolecules 2018, 51, 5364–5374. [Google Scholar] [CrossRef]
- Turner, E.H.; Schwartz, P.J.; Lowe, C.H.; Nawab, S.S.; Feldman-Naim, S.; Drake, C.L.; Myers, F.S.; Barnett, R.L.; Rosenthal, N.E. Double-blind, placebo-controlled study of single-dose Metergoline in depressed patients with seasonal affective disorder. J. Clin. Psychopharmacol. 2002, 22, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Serantoni, E.F.; Sabatino, P.; Riva di Sanseverino, L.; Sheldrick, G.M. 6-Dimethyl-8β-[(benzyloxycarbonyl) aminomethyl]-10α-ergoline, ‘Lyserdol’. Acta Crystallogr. B 1977, 33, 2899–2902. [Google Scholar] [CrossRef]
- Czernek, J.; Brus, J. Theoretical predictions of the two-dimensional solid-state NMR spectra: A case study of the 13C–1H correlations in metergoline. Chem. Phys. Lett. 2013, 586, 56–60. [Google Scholar] [CrossRef]
- Beran, G.J.O. Modeling polymorphic molecular crystals with electronic structure theory. Chem. Rev. 2016, 116, 5567–5613. [Google Scholar] [CrossRef] [PubMed]
- Czernek, J.; Brus, J. The covariance of the differences between experimental and theoretical chemical shifts as an aid for assigning two-dimensional heteronuclear correlation solid-state NMR spectra. Chem. Phys. Lett. 2014, 608, 334–339. [Google Scholar] [CrossRef]
- Czernek, J. On the solid-state NMR spectra of naproxen. Chem. Phys. Lett. 2015, 619, 230–235. [Google Scholar] [CrossRef]
- Asakura, T.; Ohata, T.; Kametani, S.; Okushita, K.; Yazawa, K.; Nishiyama, Y.; Nishimura, K.; Aoki, A.; Suzuki, F.; Kaji, H.; et al. Intermolecular packing in B. mori silk fibroin: Multinuclear NMR study of the model peptide (Ala-Gly) 15 defines a heterogeneous antiparallel antipolar mode of assembly in the silk II form. Macromolecules 2015, 48, 28–36. [Google Scholar] [CrossRef]
- Hušák, M.; Jegorov, A.; Brus, J.; van Beek, W.; Pattison, P.; Christensen, M.; Favre-Nicolin, V.; Maixner, J. Metergoline II: Structure solution from powder diffraction data with preferred orientation and from microcrystal. Struct. Chem. 2008, 19, 517–525. [Google Scholar] [CrossRef]
- Sakellariou, D.; Lesage, A.; Hodgkinson, P.; Emsley, L. Homonuclear dipolar decoupling in solid-state NMR using continuous phase modulation. Chem. Phys. Lett. 2000, 319, 253–260. [Google Scholar] [CrossRef]
- Wu, X.L.; Burns, S.T.; Zilm, K.W. Spectral editing in CPMAS NMR. Generating subspectra based on proton multiplicities. J. Magn. Reson. Ser. A 1994, 111, 29–36. [Google Scholar] [CrossRef]
- Van Rossum, B.-J.; Förster, H.; de Groot, H.J.M. High-field and high-speed CP-MAS13C NMR heteronuclear dipolar-correlation spectroscopy of solids with frequency-switched Lee–Goldburg homonuclear decoupling. J. Magn. Reson. 1997, 124, 516–519. [Google Scholar] [CrossRef]
- Lesage, A.; Bardet, M.; Emsley, L. Through-Bond Carbon−Carbon Connectivities in Disordered Solids by NMR. J. Am. Chem. Soc. 1999, 121, 10987–10993. [Google Scholar] [CrossRef]
- Brus, J.; Jegorov, A. Through-bonds and through-space solid-state NMR correlations at natural isotopic abundance: Signal assignment and structural study of simvastatin. J. Phys. Chem. A 2004, 108, 3955–3964. [Google Scholar] [CrossRef]
- Sakellariou, D.; Lesage, A.; Emsley, L. Proton-proton constraints in powdered solids from 1H–1H–1H and 1H–1H–13C three-dimensional NMR chemical shift correlation spectroscopy. J. Am. Chem. Soc. 2001, 123, 5604–5605. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.P.; Lesage, A.; Elena, B.; Emsley, L. Probing proton-proton proximities in the solid state: High-resolution two-dimensional 1H–1H double-quantum CRAMPS NMR spectroscopy. J. Am. Chem. Soc. 2004, 126, 13230–13231. [Google Scholar] [CrossRef] [PubMed]
- Hohwy, M.; Rienstra, C.M.; Jaroniec, C.P.; Griffin, R.G. Fivefold symmetric homonuclear dipolar recoupling in rotating solids: Application to double quantum spectroscopy. J. Chem. Phys. 1999, 110, 7983–7992. [Google Scholar] [CrossRef]
- Antzutkin, O.N.; Shekar, S.C.; Levitt, M.H. Two-dimensional sideband separation in magic-angle-spinning NMR. J. Magn. Reson. Ser. A 1995, 115, 7–19. [Google Scholar] [CrossRef]
- Antzutkin, O.N.; Lee, Y.K.; Levitt, M.H. 13C and 15N-chemical shift anisotropy of ampicillin and penicillin-V studied by 2D-PASS and CP/MAS NMR. J. Magn. Reson. 1998, 135, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Ladizhansky, V.; Vega, S. Polarization transfer dynamics in Lee-Goldburg Cross polarization nuclear magnetic resonance experiments on rotating solids. J. Chem. Phys. 2000, 112, 7158–7168. [Google Scholar] [CrossRef]
- Hong, M.; Yao, X.L.; Jakes, K.; Huster, D. Investigation of molecular motions by Lee-Goldburg cross-polarization NMR Spectroscopy. J. Phys. Chem. B 2002, 106, 7355–7364. [Google Scholar] [CrossRef]
- Ramamoorthy, A.; Wei, Y.F.; Lee, D.K. PISEMA solid-state NMR spectroscopy. Ann. Rep. NMR Spectrosc. 2004, 52, 1–52. [Google Scholar] [CrossRef]
- Brus, J.; Urbanova, M. Selective measurement of heteronuclear 1H–13C dipolar couplings in motionally heterogeneous semicrystalline polymer systems. J. Phys. Chem. A 2005, 109, 5050–5054. [Google Scholar] [CrossRef] [PubMed]
- Langer, B.; Schnell, I.; Spiess, H.W.; Grimmer, A.R. Temperature calibration under ultrafast MAS conditions. J. Magn. Reson. 1999, 138, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Brus, J. Heating of samples induced by fast magic-angle spinning. Solid State Nucl. Magn. Reson. 2000, 16, 151–160. [Google Scholar] [CrossRef]
- Materials Studio 5.0. Available online: http://www.3dsbiovia.com/products/collaborative-science/biovia-materials-studio (accessed on 24 September 2018).
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef] [Green Version]
- Shankland, N.; Wilson, C.C.; Florence, A.J.; Cox, P.J. Refinement of ibuprofen at 100 K by single-crystal pulsed neutron diffraction. Acta Crystallogr. C 1997, 53, 951–954. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Pickard, C.J.; Mauri, F. All-electron magnetic response with pseudopotentials: NMR chemical shifts. Phys. Rev. B 2001, 63, 245101. [Google Scholar] [CrossRef]
- Yates, J.R.; Pickard, C.J.; Mauri, F. Calculation of NMR chemical shifts for extended systems using ultrasoft pseudopotentials. Phys. Rev. B 2007, 76, 024401. [Google Scholar] [CrossRef]
- Carignani, E.; Borsacchi, S.; Marini, A.; Mennucci, B.; Geppi, M. 13C chemical shielding tensors: A combined solid-state NMR and DFT study of the role of small-amplitude motions. J. Phys. Chem. C 2011, 115, 25023–25029. [Google Scholar] [CrossRef]
- Dračínský, M.; Hodgkinson, P. A molecular dynamics study of the effects of fast molecular motions on solid-state NMR parameters. CrystEngComm 2013, 15, 8705–8712. [Google Scholar] [CrossRef] [Green Version]
- Pawlak, T.; Trzeciak-Karlikowska, K.; Czernek, J.; Ciesielski, W.; Potrzebowski, M.J. Computed and experimental chemical shift parameters for rigid and flexible YAF peptides in the solid state. J. Phys. Chem. B 2012, 116, 1974–1983. [Google Scholar] [CrossRef] [PubMed]
- Brus, J.; Urbanová, M.; Kelnar, I.; Kotek, J. A solid-state NMR study of structure and segmental dynamics of semicrystalline elastomer-toughened nanocomposites. Macromolecules 2006, 39, 5400–5409. [Google Scholar] [CrossRef]
- Brus, J.; Urbanová, M.; Strachota, A. Epoxy networks reinforced with polyhedral oligomeric silsesquioxanes: Structure and segmental dynamics as studied by solid-state NMR. Macromolecules 2008, 41, 372–386. [Google Scholar] [CrossRef]
- Brus, J.; Jakeš, J. Geometry of multiple-spin systems as reflected in 13C–{1H} dipolar spectra measured at Lee-Goldburg cross-polarization. Solid State Nucl. Magn. Reson. 2005, 27, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.G.; Williams, J.; McDermott, A. Nuclear magnetic resonance studies of biopolymer dynamics. J. Phys. Chem. 1996, 100, 13293–13310. [Google Scholar] [CrossRef]
- Lipari, G.; Szabo, A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 1982, 104, 4546–4559. [Google Scholar] [CrossRef]
- Carignani, E.; Borsacchi, S.; Geppi, M. Detailed characterization of the dynamics of ibuprofen in the solid state by a multi-technique NMR approach. ChemPhysChem 2011, 12, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Carignani, E.; Borsacchi, S.; Concistrè, M.; Johannessen, O.G.; Geppi, M. Direct observation of the effects of small-amplitude motions on 13C nuclear shielding tensors by means of low-temperature 2D MAS NMR spectroscopy. Chem. Phys. Lett. 2018, 706, 107–112. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Polymorph/Simulated Spectrum | |||
|---|---|---|---|---|
| MI/13C 1D | MI/1H 1D | MII/13C 1D | MII/1H 1D | |
| slope | −1.0191 | −1.0464 | −1.0198 | −0.9237 |
| standard error of slope | 0.0052 | 0.0192 | 0.0049 | 0.0218 |
| intercept/(ppm) | 173.40 | 32.20 | 173.00 | 30.84 |
| standard error of intercept/(ppm) | 0.53 | 0.10 | 0.49 | 0.11 |
| standard deviation/(ppm) | 1.05 | 0.23 | 1.38 | 0.39 |
| average abs. deviation/(ppm) | 0.77 | 0.19 | 0.99 | 0.28 |
| maximum abs. deviation/(ppm) | 2.34 | 0.44 | 4.18 | 1.29 |
| adjusted R2 | 0.99941 | 0.99231 | 0.99898 | 0.97353 |
| number of data points | 24 | 24 | 46 | 50 |
| MI/{1H, 13C} 2D | MII/{1H, 13C} 2D | |||
| covariance/((ppm)2) | 0.0468 | 0.125 | ||
| number of pairs | 18 | 40 | ||
| Parameter | Metergoline Structure | Ibuprofen | ||
|---|---|---|---|---|
| XRD | CSP#6 | CSP#15 | ||
| slope | −1.0496 | −1.0576 | −1.0439 | −1.0744 |
| standard error of slope | 0.0081 | 0.0103 | 0.0112 | 0.0113 |
| intercept/(ppm) | 176.08 | 176.18 | 174.86 | 177.93 |
| standard error of intercept/(ppm) | 0.94 | 1.19 | 1.29 | 1.36 |
| standard deviation/(ppm) | 4.53 | 5.72 | 6.21 | 5.66 |
| average abs. deviation/(ppm) | 1.15 | 1.50 | 1.56 | 1.55 |
| maximum abs. deviation/(ppm) | 13.5 | 16.0 | 15.7 | 12.5 |
| adjusted R2 | 0.99664 | 0.99472 | 0.99364 | 0.99585 |
| number of data points | 57 | 57 | 57 | 39 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czernek, J.; Urbanova, M.; Brus, J. NMR Crystallography of the Polymorphs of Metergoline. Crystals 2018, 8, 378. https://doi.org/10.3390/cryst8100378
Czernek J, Urbanova M, Brus J. NMR Crystallography of the Polymorphs of Metergoline. Crystals. 2018; 8(10):378. https://doi.org/10.3390/cryst8100378
Chicago/Turabian StyleCzernek, Jiri, Martina Urbanova, and Jiri Brus. 2018. "NMR Crystallography of the Polymorphs of Metergoline" Crystals 8, no. 10: 378. https://doi.org/10.3390/cryst8100378
APA StyleCzernek, J., Urbanova, M., & Brus, J. (2018). NMR Crystallography of the Polymorphs of Metergoline. Crystals, 8(10), 378. https://doi.org/10.3390/cryst8100378