3.1. Crystal Structures of the Cu4 and Cu6 Complexes
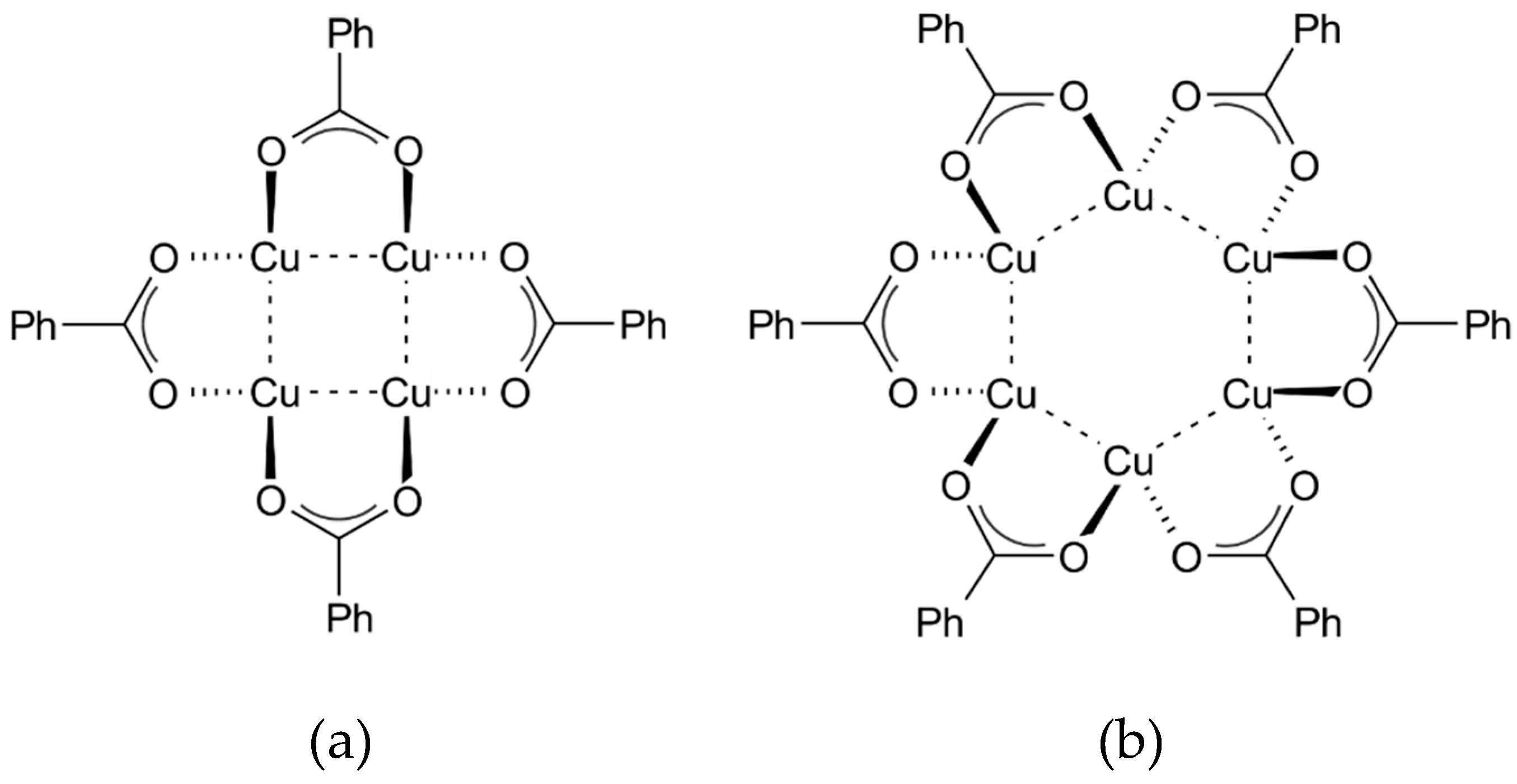
The studied tetranuclear
Cu4 complex (i.e., [(PhCO
2)
4Cu
4]) crystallises in the triclinic
space group with two molecules in the asymmetric unit (
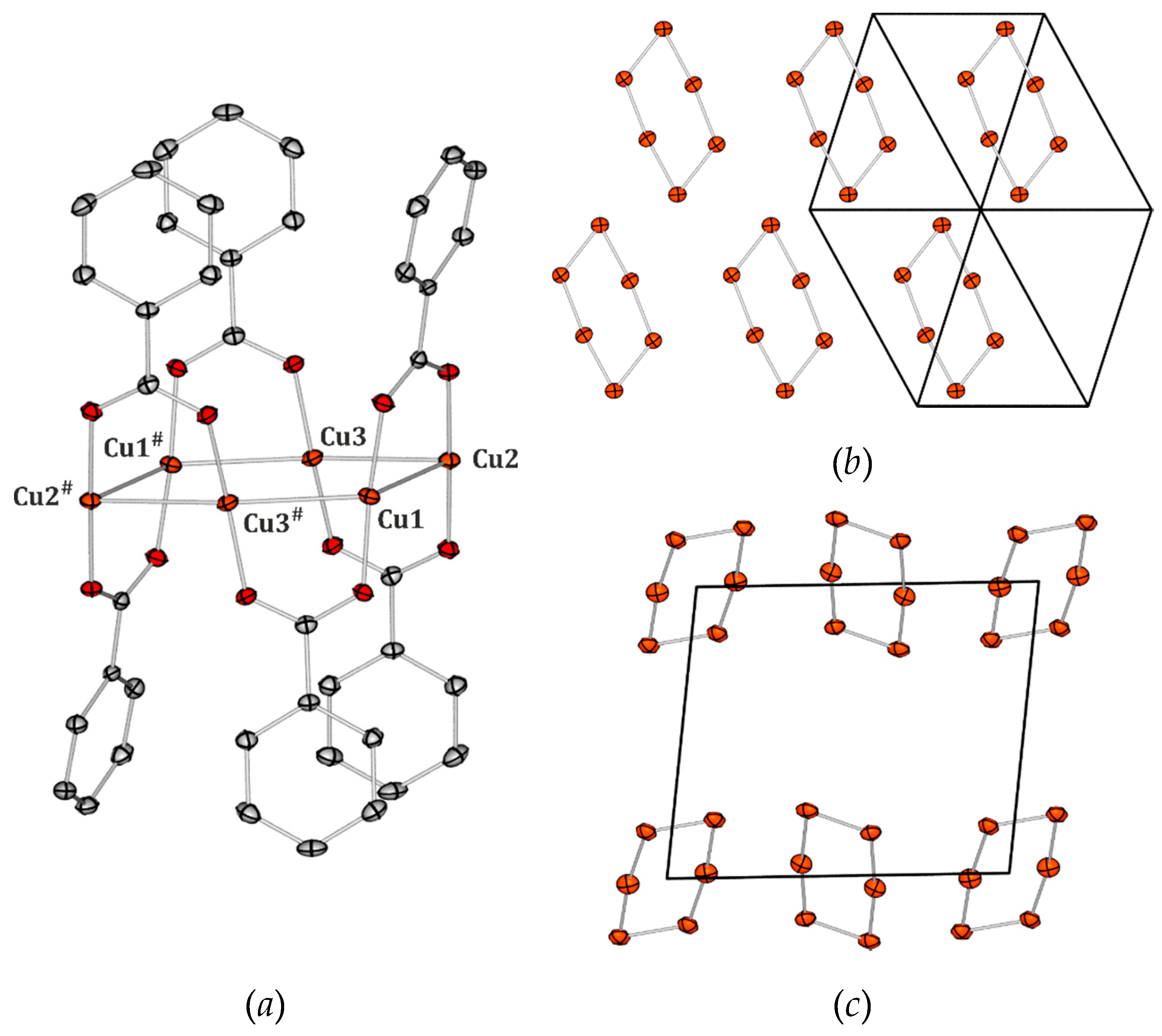
Figure 1). These molecules differ somewhat in the geometry of the metal core (
Table 1), but they share the general structural features. The metal core of the molecule A is characterised by, on the average, slightly longer Cu−Cu bonds, however, the shorter diagonal of the formed rhomboid can still be classified as a metal-metal bond at 90 K (based on the van der Waals radii criteria), which is not the case as far as the second symmetry-independent molecule B is concerned. The metal core of the molecule A is also more regular and resembles a parallelogram with two longer (ca. 2.73 Å at 90 K) and two shorter edges (ca. 2.71 Å at 90 K). For the purpose of this study, the
Cu4 crystal structure was determined also at 225 K, 250 K, and RT in order to catch any structural changes which could be related to the temperature spectroscopic behaviour of the system. It appeared that, the analysed crystal structure does not undergo any phase transition in the temperature range between 90 K and room temperature (RT). In general, temperature influences the lattice and geometrical parameters in a typical way. Regarding the core shapes, the copper–copper distances get elongated except for the longer diagonal in the B molecule, which shortens with temperature rise. Furthermore, both metal cores become slightly more ‘square-like’, i.e., the obtuse angles diminish, whereas the acute ones increase (the average changes though do not exceed about 2°).
As already mentioned, the crystal structure of the
Cu6 complex (
Figure 2) behaves unlike that of
Cu4 with changes of temperature. Although it also forms the triclinic
crystal lattice, it is differently packed depending on temperature (
Figure 2). Below 253 K the asymmetric unit contains a half of the
Cu6 molecule, which is located at the centre of symmetry. Between 253 K and 263 K a phase transition is observed, as described in the literature [
11], leading to two independent halves of the
Cu6 complex comprising the asymmetric unit at higher temperatures (
Figure 2). The crystal packing similarities and differences between the 293 K and at 173 K crystal structures of
Cu6 have already been discussed by Filatov et al. [
11]. Our X-ray diffraction measurement conducted at 90 K confirms that there are no other phase transitions down to this temperature point. Geometrical parameters characterising the
Cu6 copper cores at 90 K and at RT are collected in
Table 2. Again, the copper–copper distances tend to elongate with increasing temperature. The
Cu6 molecules do not exhibit the inter-core Cu⋯Cu short diagonal contacts, as indicated by the respective distances. The Cu−Cu bonds in
Cu6, however, are on average shorter than in the case of
Cu4. Additionally, in the case of the high-temperature phase, the symmetry-independent molecules are very much alike, in contrast to two molecules comprising ASU in the
Cu4 crystal structure. Furthermore, the geometries of all the
Cu6 species at both high- and low-temperature crystal forms resemble each other more than the
Cu4 symmetry-independent molecules.
3.2. Single-Crystal Luminescence Characterisation
In order to quantitatively relate spectroscopic features with the X-ray-derived crystal structures, multi-temperature time-resolved single-crystal spectroscopic experiments were performed for the studied systems. Such proceedings also eliminated problems encountered for powder samples which may contain amorphous phase impurities that affect their properties (e.g., excited states’ lifetimes). Our experiments revealed that crystals of the
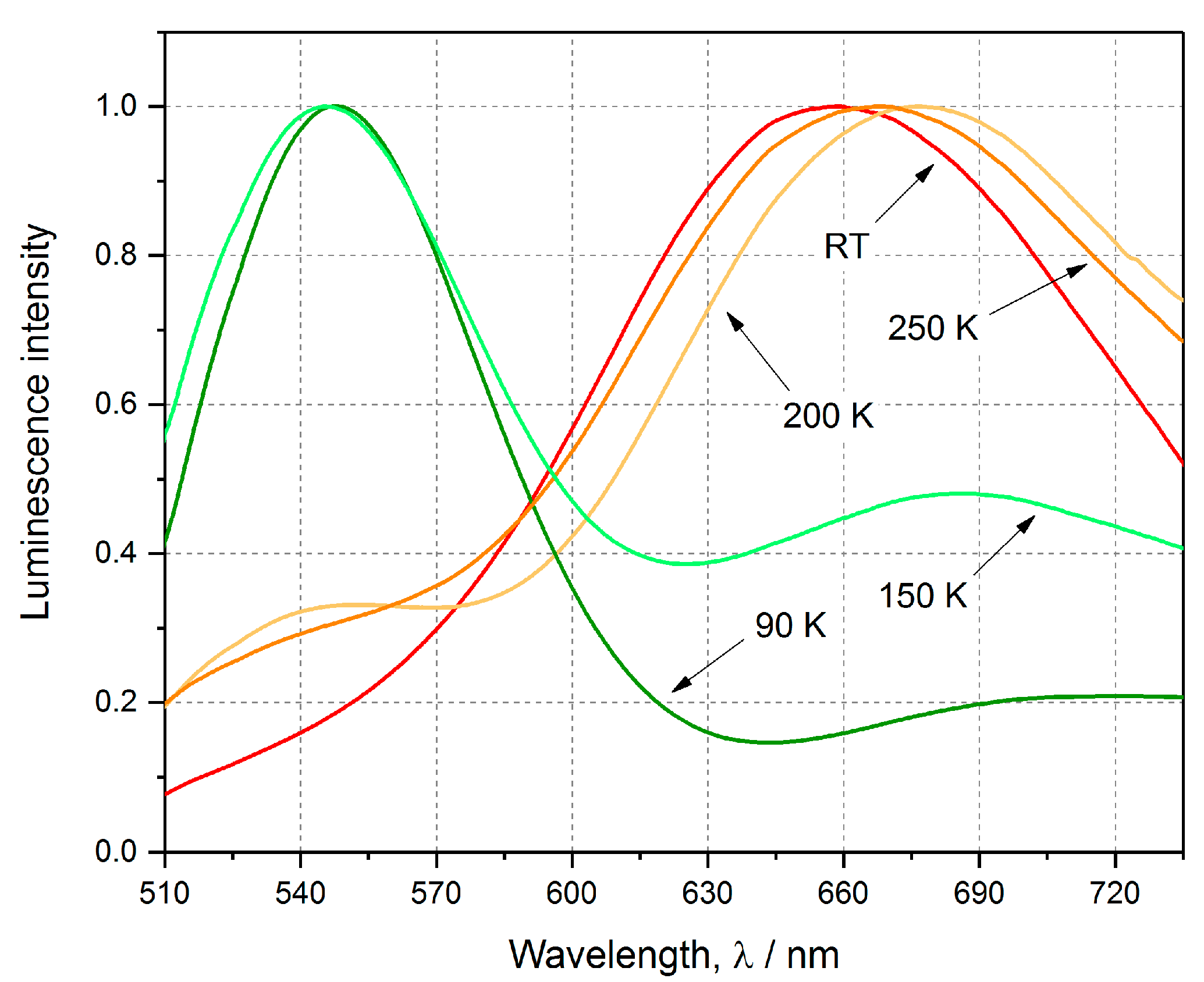
Cu4 complex exhibit two emission bands in the visible-light spectral region, namely the red band, ~680 nm, and the green band, 546 nm (
Figure 3,
Table 3). At high-temperature, the emission is clearly orange (250 K) to red (RT), but at lower temperatures it becomes yellow, yellow-green to intensively green at 90 K (
Figure S1, Supplementary Materials [
48]). Indeed, when taking a closer look at the emission spectra at different temperatures, at RT almost exclusively the low-energy red band is visible, whereas the very weak green-band signal can be spotted only in the short time scale. In turn, while the temperature is being decreased, the high-energy green band gradually becomes stronger. At 200 K, the green band is still much less intensive than the other one, however, below 150 K it is clearly dominant. At 90 K, the high-energy band is very strong, while the low-energy band is almost fully suppressed, which is reflected in a brightly green emission of the sample (
Supplementary Materials). The numerical parameters describing the emission maxima and the estimated emission decay lifetimes at different temperatures are presented in
Table 3.
It appears that the emission decay can be efficiently modelled by a bi-exponential function in the case of both band types. The temperature influences strongly the emission decay lifetimes, which are significantly shorter at high temperatures. The green band maximum does not shift with temperature, while the red band maximum shifts notably from about 715 nm at 90 K to 660 K determined at RT.
Interestingly, in the case of the related
Cu6 compound, crystals of which undergo the phase transition between 253 and 263 K, the emission maximum shifts only from 586 nm at 90 K to 577 nm at RT (
Figure 4). Furthermore, the temperature has also weaker effect on the emission decay lifetime when compared to
Cu4 (
Table 4).
Finally, it should be noted that significant Stokes shift and relatively long emission decay times (μs regime) observed for the studied complexes’ crystals, i.e., Cu4 and Cu6, suggest that in both cases, the emissive state is of triplet nature.
3.3. Theoretical Modelling
For the purpose of the described emission bands’ nature investigation, theoretical modelling and computations were performed. At first, it was checked how the ground state optimised molecular geometries compare to those found in the crystal lattice. The ground-state geometry optimizations were conducted using crystal structures as a starting point. Two approaches were compared, i.e., optimization of a single
Cu4 moiety in the gas phase and optimization of both
Cu4 molecules forming the asymmetric unit in the QM/MM approach using the ONIOM electronic embedding to emulate the effect of the crystal environment. The results of theoretical calculations are shown in
Table 5, and in the
Supplementary Materials.
In general, the agreement between the experiment and theory appears to be satisfactory. Both tested range-separated functionals, ωB97XD and CAM-B3LYP, yielded similar optimised gas-phase geometry of the
1A ground state exhibiting the
symmetry (
in crystallographic notation), which closely resembles the experimental geometries. In turn, QM/MM correctly imitates distortions from the
symmetry of both independent molecules constituting the asymmetric unit (the A and B molecules), including different shapes of the Cu4-cores. The most significant differences between the gas phase and QM/MM results, however, concern the organic ligands, as illustrated in
Figure 5.
Having received the above overview of the approach and functional performance for the ground-state of
Cu4, lowest excited states of the studied complex were derived using the TDDFT method.
Table 6 presents the first ten triplet excited states calculated for the
Cu4 species, both in the gas phase and in the molecular environment.
The excitation energies obtained for the isolated complex in the
symmetry are comparable to these derived for the embedded complexes with distorted-core geometries. The first excited
3A state involves excitations from HOMO, HOMO-1, and HOMO-2 to LUMO and several higher-lying orbitals. The frontier molecular orbitals of
Cu4 are shown in
Figure 6, i.e., HOMO of the antibonding character and the bonding LUMO (see also
Figure S5). As indicated by the orbital population analysis, both the initial and final molecular orbitals are composed mainly of copper d orbitals, which suggests that
3A has a distinct cluster-centred character. Directly above 1
3A, there is a set of close-lying excited states, three of the
3B symmetry and one of the
3A symmetry. The corresponding excitation amplitudes are distributed among configurations of a ligand-ligand and core-ligand character.
To gain insight into the nature of the computed excited states, the one-particle transition density matrix (1-TDM) obtained at the TDDFT level of theory was analysed. In particular, the concept of charge transfer numbers introduced by Plasser & Lishka [
31] was employed, as well as its extension to a transition-metal complex case [
32]. When two molecular fragments A and B are considered, the charge transfer number is defined as:
where the sum runs over all atomic basis functions
and
on fragments A and B, respectively.
stands for 1-TDM, while
is the overlap matrix. The total charge transfer (CT) character is given as the sum of the off-diagonal elements of the CT matrix:
where the
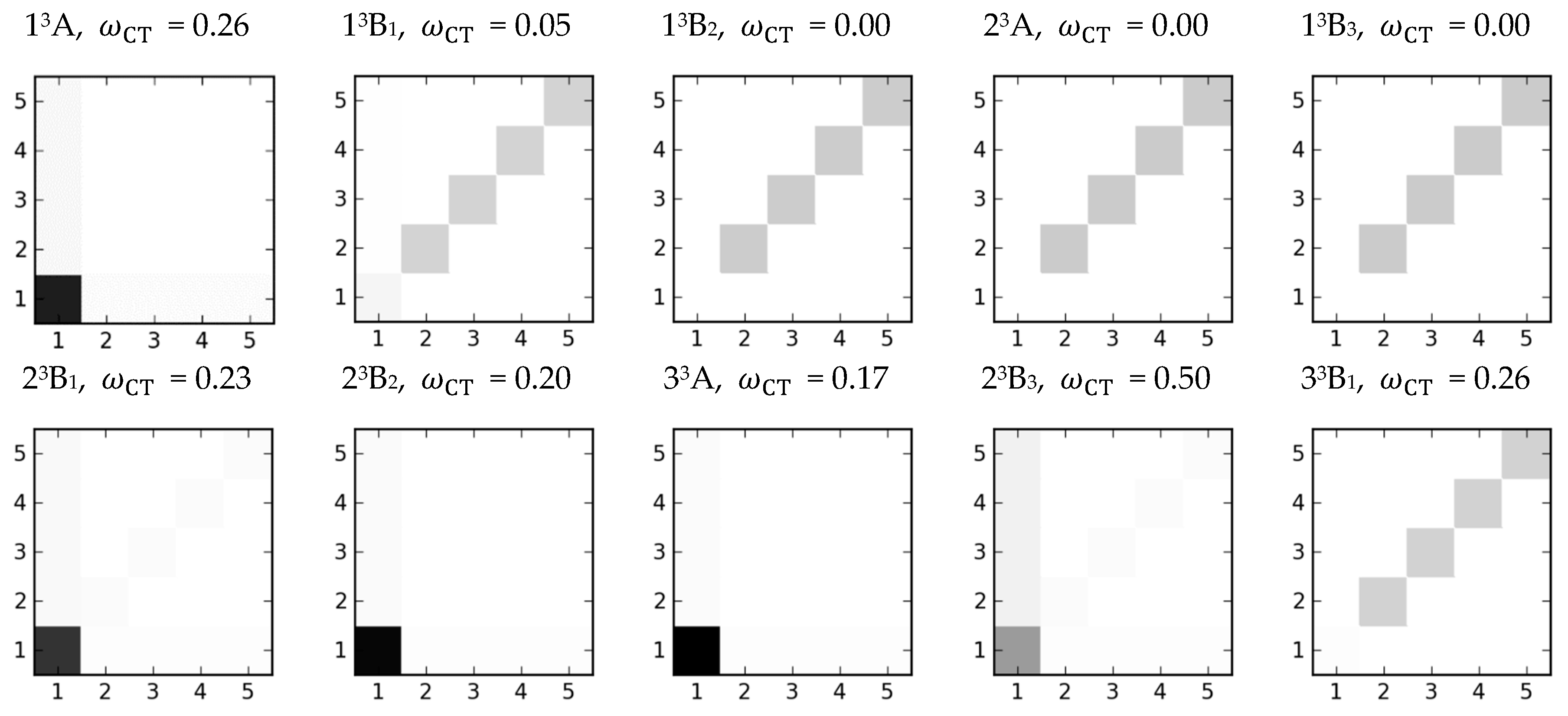
normalization factor corresponds to the squared norm of 1-TDM. The matrix can be visualized in a direct manner yielding the so-called electron-hole correlation plots (
-matrix plots) [
49]. The
index may be partitioned into metal centred (MC), ligand centred (LC), metal-to-ligand (MLCT), ligand-to-metal (LMCT), and ligand-to-ligand (LLCT) charge-transfer contributions [
32]. Consequently, the total CT index can be expressed as follows:
. For the purpose of the described analysis, a single
Cu4 molecule was divided into five fragments, i.e., the
Cu4 metal core and four benzoic acid ligands. In
Figure 7 the obtained electron-hole correlation plots for the first ten triplet excited states of the gas-phase-optimised
Cu4 complex are presented, while
Table S3 contains the corresponding excitation energies and the 1-TDM analysis results.
Based on the information from 1-TDM, it can be concluded that two types of triplet states are distinguishable. The first type gathers the core-centred states with significant contributions from the metal-to-ligand charge-transfer excitations. The second type includes states of Frenkel exciton nature involving
excitation localised on the ligands (
Figure 7).
In this view, the lowest-lying
3A triplet together with states from 6th to 9
th can be classified as the first type, i.e., they all share the dominant core-centred character, which is reflected in the notable
values ranging from 0.7 to 0.8, and in the MLCT contributions being above 0.1. The mixed charge transfer character is also supported by the number of participating fragments (referred to as PR) ranging between 1.21 and 2.56. The largest CT index, indicated by grey off-diagonal elements in the
-matrix plot (
Figure 7), is observed for the 9th state. In turn, deviation from the
symmetry results in a stronger CT character of all these states calculated for the QM/MM geometries, manifested by larger MLCT and LMCT contributions (for details see
Table S3). In particular, in the first excited state
amounts to 0.202, whereas it equals 0.165 in the
case. The delocalisation of the excitation from the complex’s core to one of the ligands affects also the PR value, which increases from 1.3 to 2.0.
The second type of triplet states encompasses states from the 2nd to 5th, as well as, the 10th triplet state. The local character of these excitations can be deduced from the electron-hole correlation plots dominated by diagonal elements, the
values being close to zero, and the number of participating fragments around 3.5 (
Figure 7,
Table S3).
The nature of excited states may also be analysed by inspection of natural transition orbitals (NTOs). NTOs are obtained from the singular value decomposition of 1-TDM:
where matrices
and
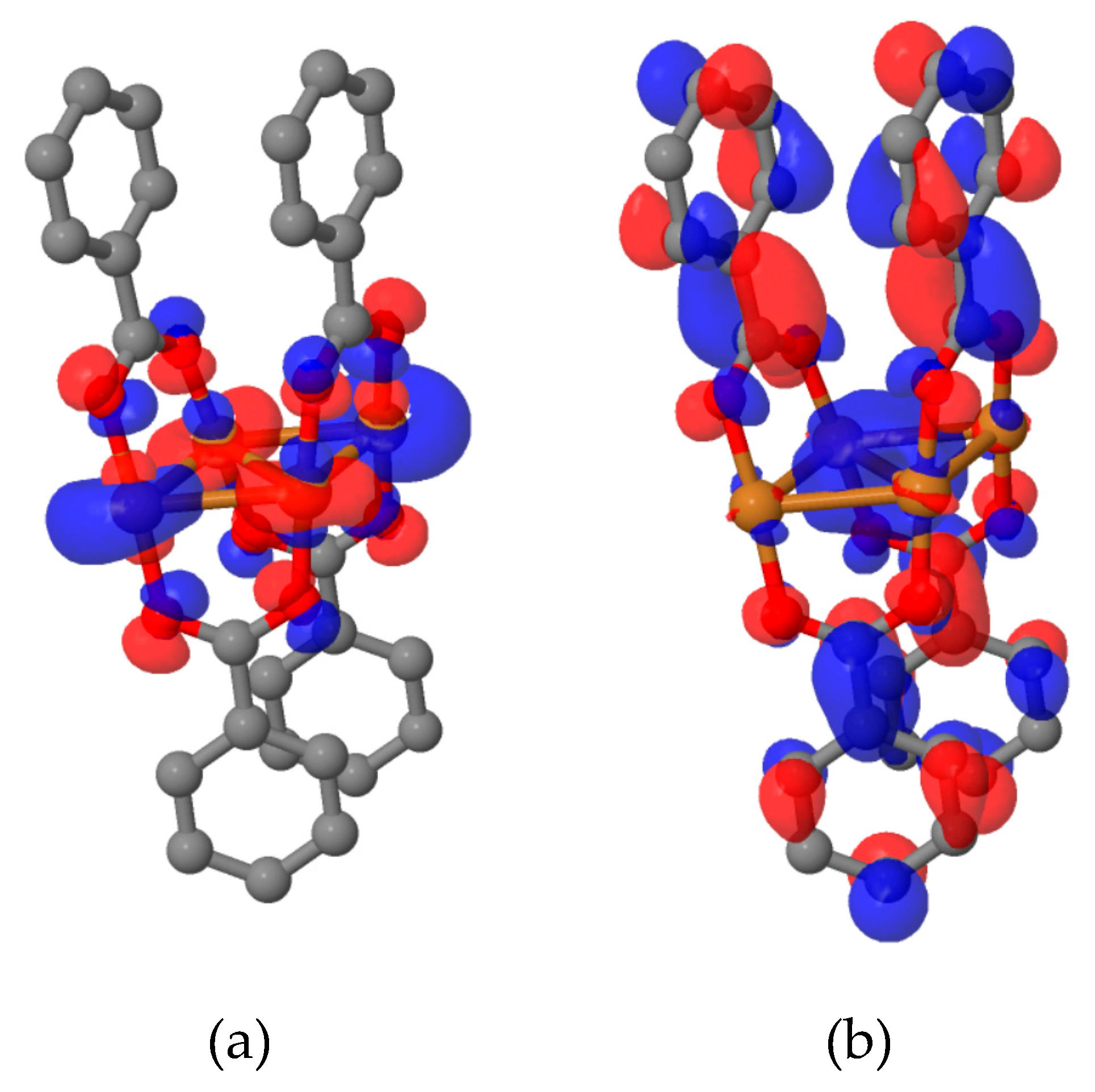
contain hole (initial) and particle (final) orbitals, respectively. The advantage of NTOs over canonical orbitals is that the former provide a more compact representation of the excitation, i.e., typically only a few configurations are needed to describe the electronic transition. In
Figure 8 we present the dominant NTO pairs for the 1
3A and 1
3B
1 states. Examination of NTOs corroborates the character of triplet excitations as deduced from the
-matrix plots. In the
3CC states the electron and hole are localised primarily in the core region, and the antibonding-to-bonding transition is clearly visible. The contributions from 2p orbitals of oxygen atoms correspond to the MLCT transitions. In turn, visual inspection of NTOs computed for the 1
3B
1 state confirms the
character of excitations localised on the benzoic acid fragments.
Finally, to model the most probable structural changes occurring during the excitation, two lowest-lying triplets were optimised using the analytic gradients from the TDDFT calculations. The selected optimised geometrical parameters are presented in
Table 7. The calculations were carried out both in the gas phase and in the crystal environment, where possible. In each case, frequency calculations confirmed the presence of the minima. The first excited
state structures obtained in different approaches remain in close agreement. The second excited state, denoted as
, was determined only for the
symmetry (gas phase)
Cu4 complex. The
optimisation was conducted using two approaches, which led to identical results, i.e., either by converging the 6th root in TDDFT, or by optimisation of the unrestricted Kohn-Sham (UKS) determinant with a deformed
Cu4-core as a starting point. It was not possible to reach the excited state minima corresponding to the
excitations, for which geometrical changes should occur primarily on the organic parts of the ligands.
It appeared that both
and
states involve significant changes in the
Cu4 core compared to
(
Table 7,
Figure S3), which is consistent with the
3CC character of the first and 6th excited states predicted from the 1-TDM analysis (
Figure 7 and
Figure 8). Among these, the core contraction was estimated to be notably larger for the
transition, for which the Cu−Cu distance is shortened by 0.240 Å upon excitation (for the gas-phase-optimised
Cu4 geometries; for
it is shortened by 0.073 Å;
Table 7). In the case of
, the changes in the metal core were predicted to be smaller, however, more significant elongation of the Cu−O bonds with respect to the
state was observed (1.93 Å vs. 1.85 Å, respectively;
Table 7). This finding supports the presence of non-negligible MLCT and/or LMCT contribution to the
3CC state.
The TDDFT excitation energies from the ground state to the first triplet state (referred to as 1st
) calculated for the optimised geometries of the
,
, and
states are gathered in
Table 8. Additionally, the
values, i.e., the difference between the electronic energy of the triplet state and the energy of respective singlet states (both energies calculated at the triplet minimum-energy geometry), are reported. In the case of the
geometry, the lowest triplet excited state occurs at 1.952 eV (635 nm), while the corresponding
value is equal to 592 nm. The QM/MM calculations predicted this state at 2.077 eV (597 nm) for the A molecule, while at 2.001 eV (618 nm) for B. The emission from the
state computed using the TDDFT approach matches the experimental low-energy band observed at 660−715 nm. The identification of the second emitting state is less straightforward. In the case of the
geometry the lowest triplet state was placed by TDDFT at 2.149 eV (578 nm), while the
amounted to 523 nm. Thus, the anticipated emission from
in the
symmetry is close to the experimental value of 546 nm. It should be noted, however, that upon distortion of the
symmetry, which is observed in crystals, as well as in the QM/MM-optimized structures, the two types of states gain significantly more MLCT and LMCT contributions. Furthermore, it was not possible to identify a minimum on the excited potential energy surfaces complying with the states dominated by the
excitations.
3.4. Time-Resolved X-ray Laue Diffraction Results
Determination of geometry changes occurring in the
Cu4 complex in the crystal form at 90 K and 225 K upon excitation may provide us some indication about the nature of the dominant excited states at low and high temperatures. This goal can be realised via a time-resolved laser-pump/X-ray-probe Laue technique, especially for heavier atoms rich in electrons [
50]. Nevertheless, even some information on the copper core deformation would be of help, as the changes in the cluster-centre geometry confronted with the theoretical results could give us premises about the emission origins.
In the studied case, the full response-ratio refinement [
51] was unfortunately not possible, most probably due to the rather low completeness of the collected data (35% and 17% for 90 K and 225 K data, respectively), as a result of low conversion, low space-group symmetry of the sample and the examined crystals’ shape. Nevertheless, some meaningful qualitative conclusions can still be drawn. The obtained photodifference maps, computed for the copper cores, are presented in
Figure 9. The negative peaks, as expected, are localised on copper atoms’ positions, which shows that these atoms shifted/moved upon excitation. The similarity of photodifference maps obtained from two independent measurements at both 90 and 225 K is striking. This suggests the same excited state was captured in both experiments and also indicates good reproducibility of the results. Since the pump-probe delay was set to several ns, while the studied excited state lifetimes are in the μs regime (
Table 3), such explanation is rational. The measurement could have been faster than the conversion to the final emissive state. In such a case, either some singlet excited state was captured, or the high-energy (“green”) triplet excited state. It also seems that most of the peaks are located closer to the centre of the cluster core, especially for molecule B (see, e.g., the large peak next to the Cu7 atom), which suggests its contraction. For this molecule, more peaks are visible than for A when the same isocontour values are applied. This suggests that the molecule B, exhibiting longer “short” diagonal Cu⋯Cu contact, should undergo more pronounced core contraction when excited, which is indeed observed in the
QM/MM results. Nevertheless, the experimental geometrical changes are not straightforward. Regarding the theoretical results, one may conclude that the low-energy excited state should be characterised by more significant changes of the copper core shape which becomes more quadratic (
Table 7), whereas the high-energy excited state is expected to exhibit more emphasized Cu−O bond elongation and less notable copper atoms’ distortions from the GS geometry (
Supplementary Materials). For both
and
states, the contraction of all Cu−Cu bonds is observed and also the shorter diagonal gets contracted (
Table 7).
3.5. Cu4 and Cu6 Comparison
As already mentioned, despite the similarity of
Cu4 and
Cu6, no luminescence thermochromism was observed in the latter case. In an attempt to explain such behaviour, the first few excited states of the
Cu6 complex were calculated and analysed. It appears that the nature of the first singlet and triplet states obtained for
Cu4 and
Cu6 is qualitatively very much alike. Namely, the triplet states have either a core-centred or ligand-centred character (see the discussion in
Section 3.3, and
Figures S6, S11 and S12 and
Tables S6 and S7), whereas the first singlets are core-centred with significant MLCT charge-transfer contributions (
Figures S7, S9 and S10 and
Tables S3 and S5). Clearly, without direct probing of the excited-state dynamics, it is not straightforward to establish whether thermochromism occurs for a single
Cu4 molecule. If this was the case, the lack of this phenomenon in
Cu6 could be attributed, for instance, to a lower energy barrier between the emissive triplet states. Another possibility is that the thermochromism arises due to crystal packing effects, which are markedly different in the studied structures.
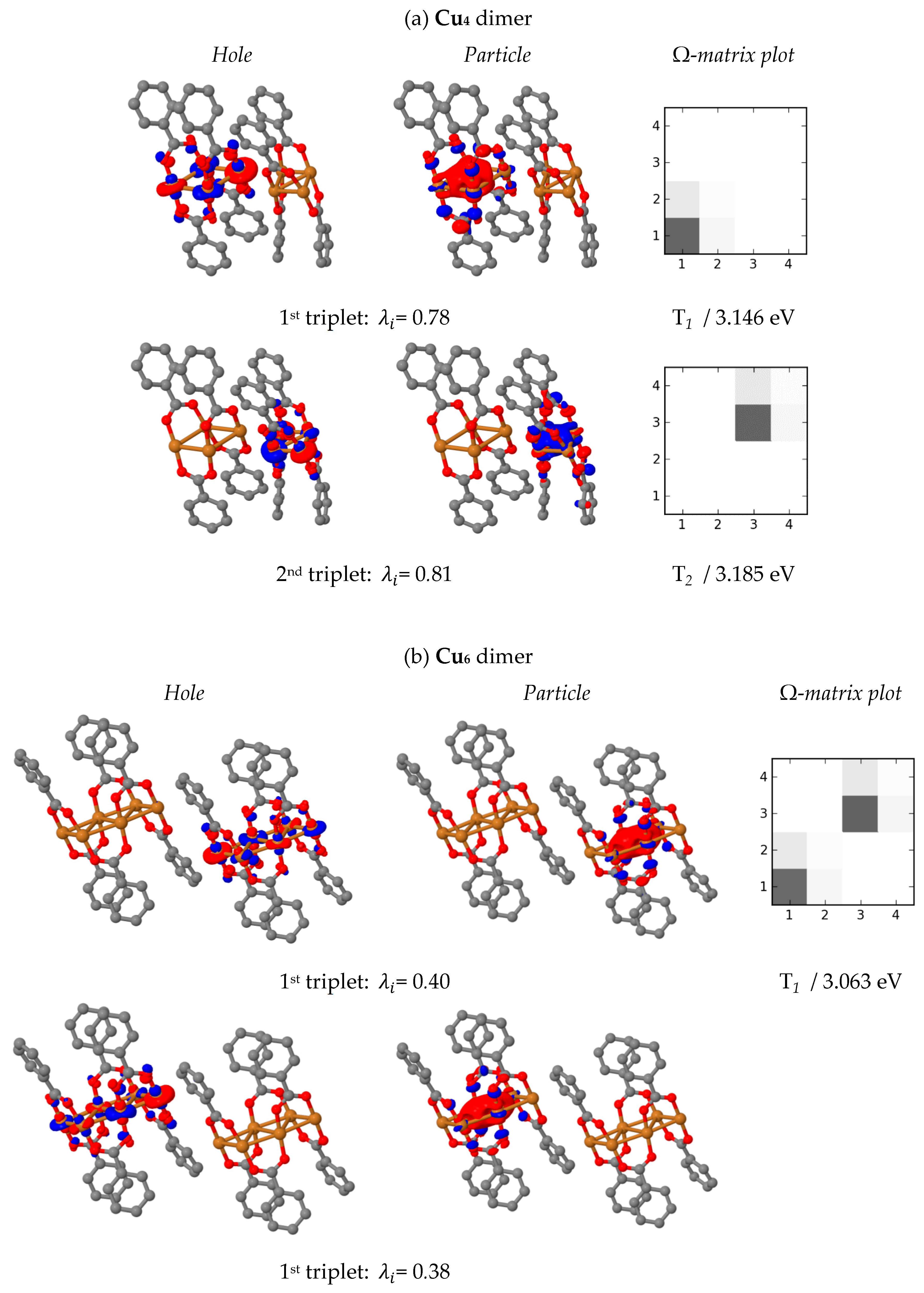
In order to explore the role of molecular packing in the
Cu4 and
Cu6 crystals, two closest-neighbour complex molecules were selected in both structures. The lowest-lying excited triplet states of such
Cu4 and
Cu6 dimers characterised in terms of natural transition orbitals and electron-hole correlation plots are shown in
Figure 10. The dimer geometries were taken directly from the crystal structures. Inspection of
Figure 10 reveals different characteristics of the excited states in the
Cu4 and
Cu6 dimers. In the former case excitations are localised on the separate monomers, whereas in the latter they are delocalised on both monomers forming exciton resonance states (no charge transfer between the monomers is observed, and each excited state of the
Cu6 dimer is doubly degenerate). Analogous behaviour was confirmed for singlet states (
Figures S8, S13 and S14 and
Tables S8 and S9).
The significant difference between the nature of excited states of Cu4 and Cu6 dimers indicates that the distinct spectroscopic behaviour of the two complexes in the solid state could be to some extent attributed to the mutual molecular arrangement in the crystal lattice. Nevertheless, the impact of such intermolecular interactions remains debatable due to the lack of unequivocal evidence on the matter.

 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}