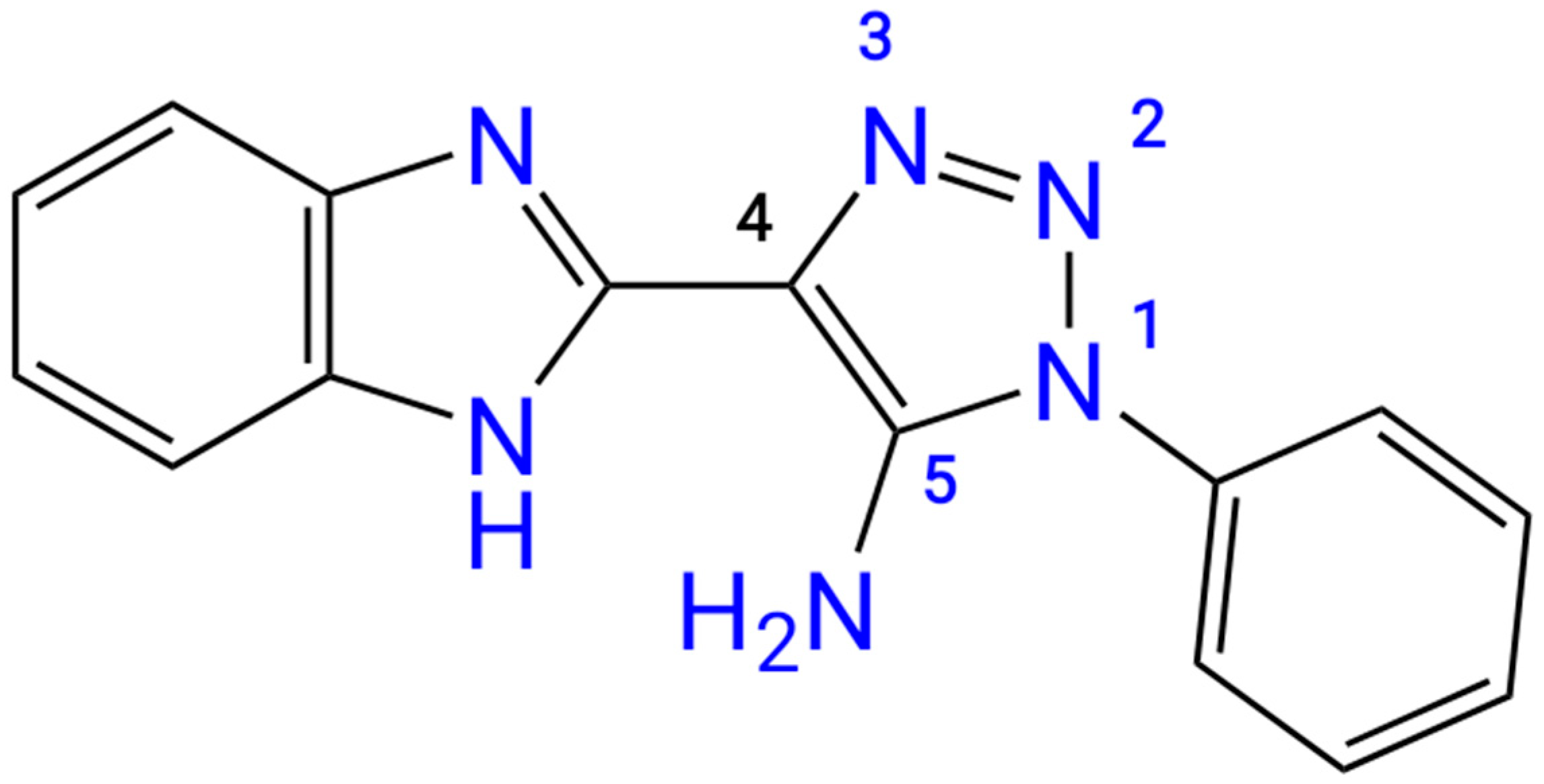
The initial structure of the studied “benzimidazole – 1,2,3-triazole” dual moiety molecules, 4-(1
H-benzo[d]imidazol-2-yl)-1-phenyl-1
H-1,2,3-triazol-5-amine
3, has not yet been described in the literature. There is only information on the synthesis of its 4-nitro- and 4-bromo-derivatives [
25]. We synthesized the title compound
3 and studied it using UV-, IR-,
1H NMR, and
13C NMR spectroscopy (
Supplementary Materials, Figures S1–S4) as well as LC/MS data for structural determination (
Figure S5). Finally, the structure of the title compound was confirmed by X-ray analysis (
Figure 1).
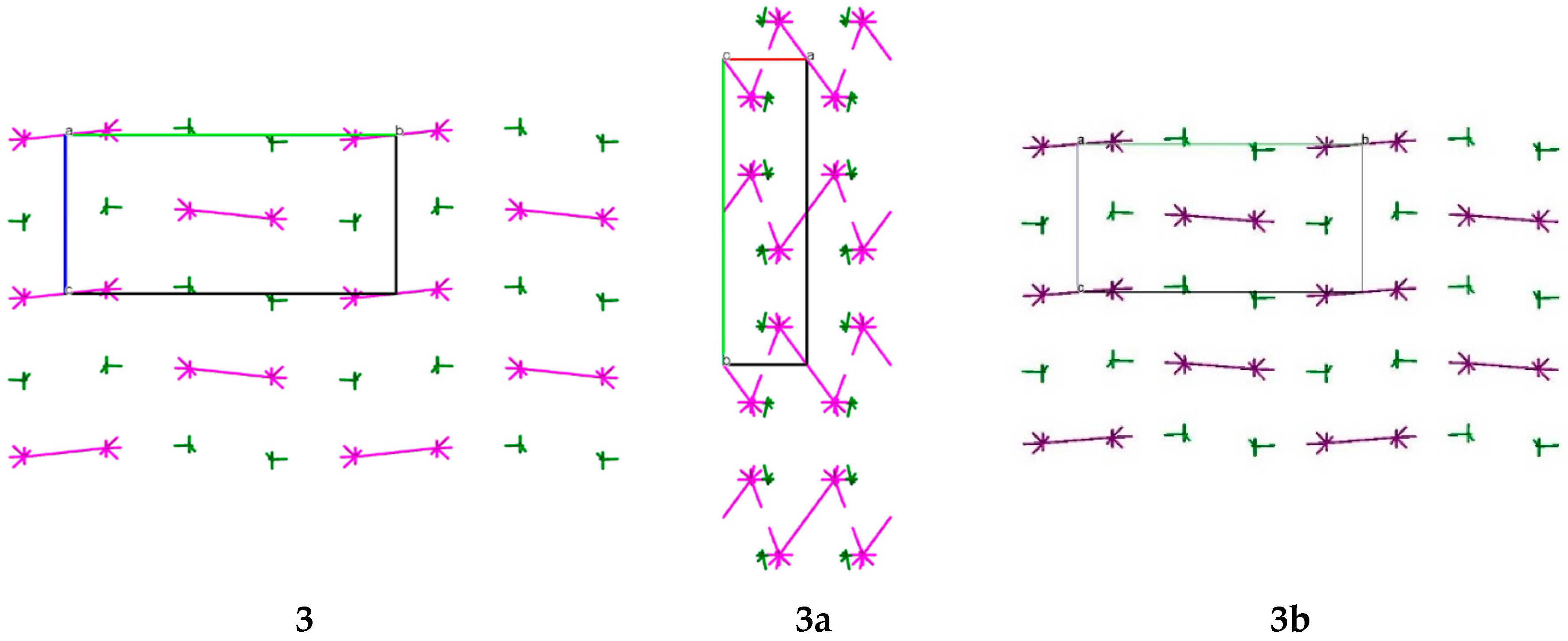
2.2. Molecular and Crystal Structure Analysis
The structures of compounds
3,
3a and
3b were studied using different spectral methods (see Materials and Methods section, and
Supplementary Materials) and finally confirmed using the single crystal X-ray diffraction analysis.
The crystallographic information and refinement data are presented in the Materials and Methods Section.
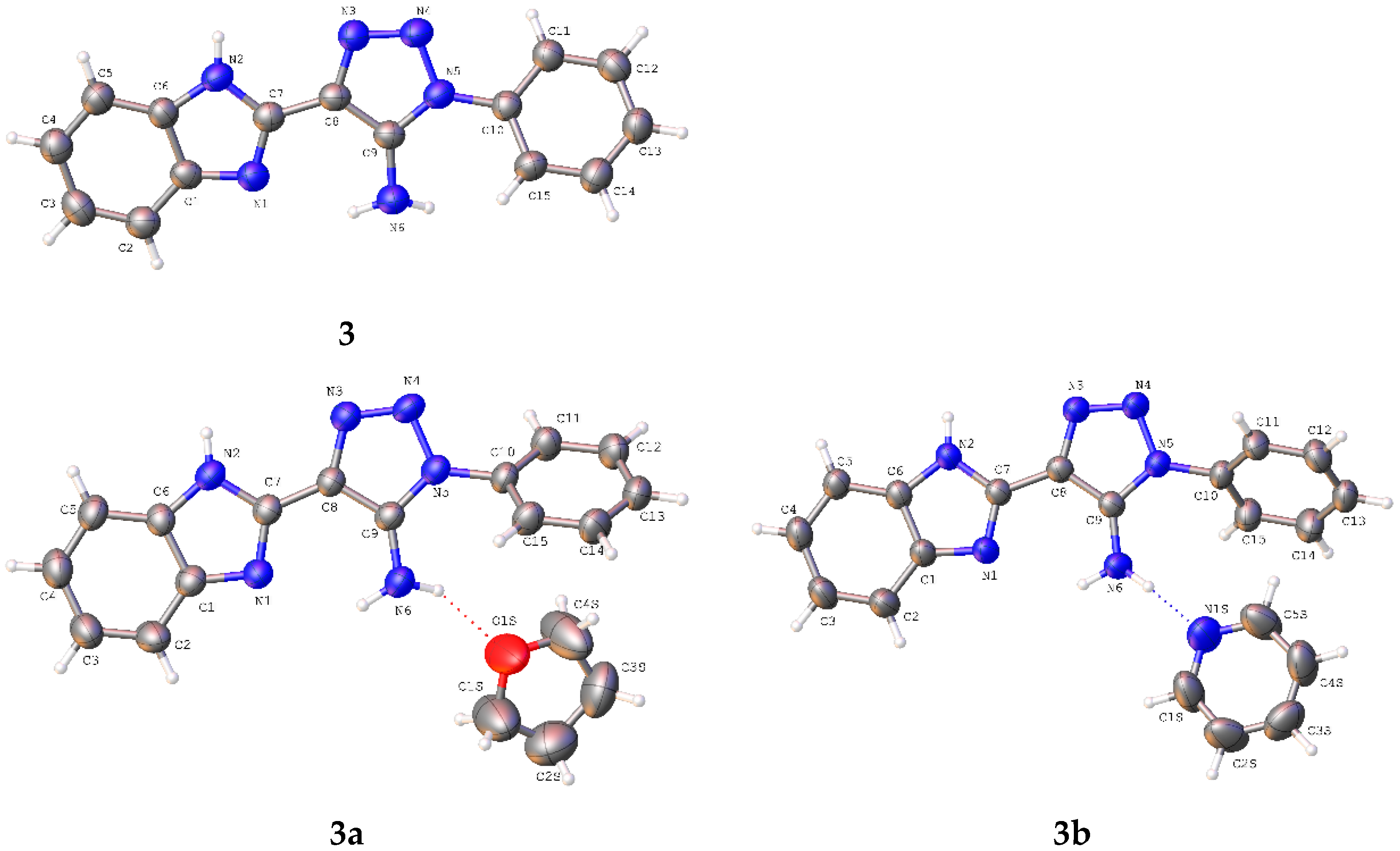
The compounds
3a and
3b have been obtained as solvates with THF and pyridine, respectively, at the stoichiometric ratio 1:1. The asymmetric part of the unit cell of the studied crystals contains one molecule of the title compounds (
Figure 2). The bond lengths are very similar in all basic molecules of the title compounds (
Table S1). The nitrogen atom of the amino group has a pyramidal configuration with different degrees of pyramidality in compounds
3,
3a and
3b (
Table 1). The highest degree of pyramidality is observed in crystal
3, while the nitrogen atoms of amino group in crystals
3a and
3b have a smaller degree of pyramidality. This reflects considerable conjugation between the π-system of the triazole ring and the N6 atom lone pair which is confirmed by the shorter length of the C9–N6 bond (
Table 1). It is caused by full involvement of two hydrogen atoms of amino group in formation of both intramolecular N6-H6NB…N1 and intermolecular hydrogen bonds with solvent molecules (
Table 2). The phenyl substituent is turned relative to the C9–N5 endocyclic bond (the C9–N5–C10–C15 torsion angles are shown in
Table 1) due to steric repulsion between vicinal substituents (the shortened intramolecular contacts are: H15…N6 2.55 Å (van der Waals radii sum [
26] is 2.66 Å) (
3), H6A…C10 2.74 Å (2.87 Å) (
3a), N6…C15 3.16 Å (3.21 Å) (
3b)).
The organic molecules such as the studied compound 3 have the ability to form solvates with different molecules which contain strong enough proton acceptors. Therefore, special attention should be paid to the intermolecular interactions and their role in the crystal structure formation.
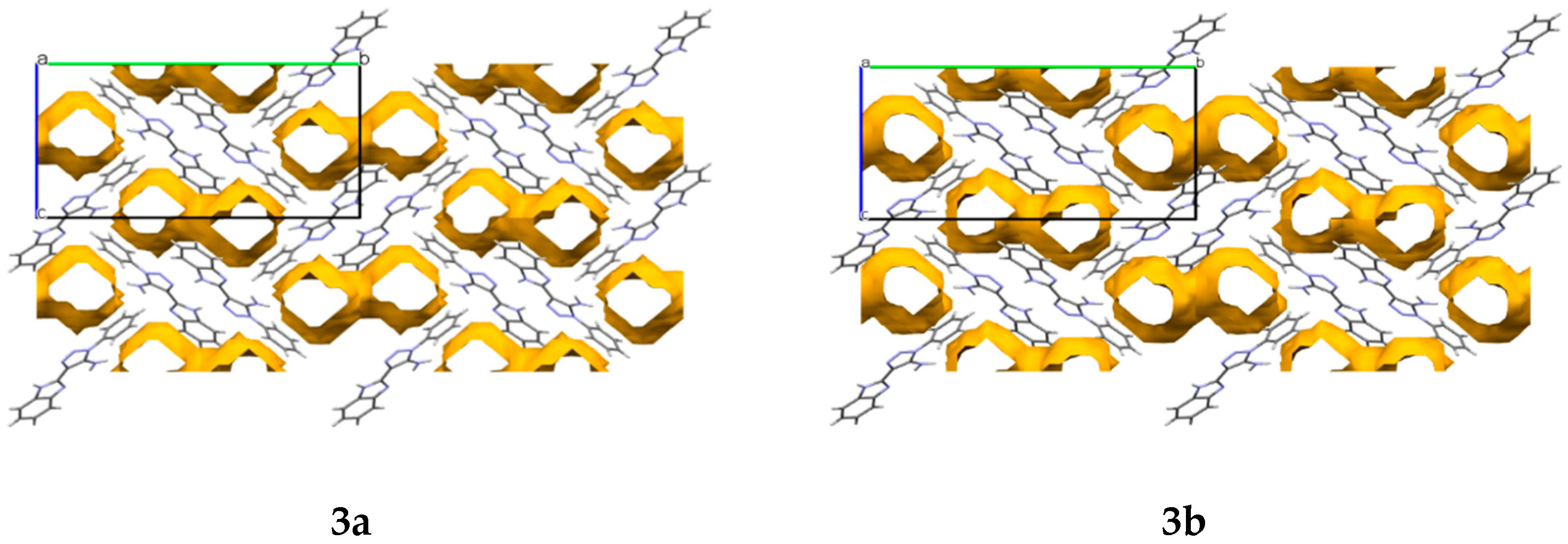
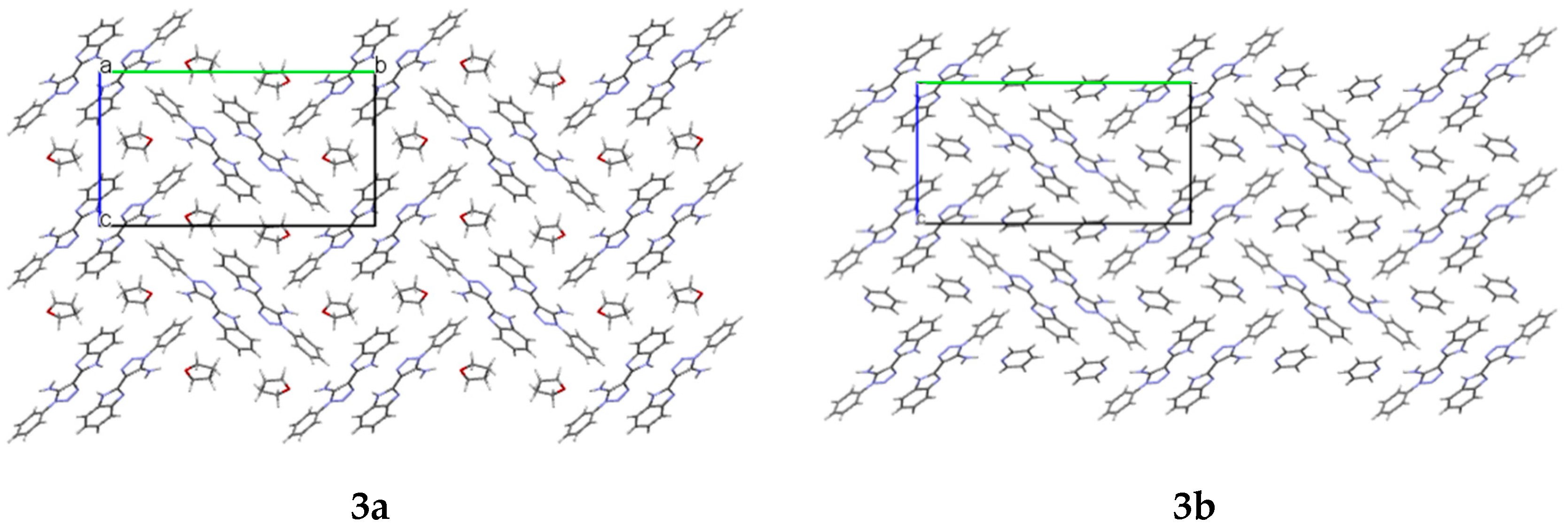
In the crystals
3a and
3b, cavities that contain solvent molecules are formed. The cavities have a volume of 487.52 Å
3 in compound
3a and 471.76 Å
3 in compound
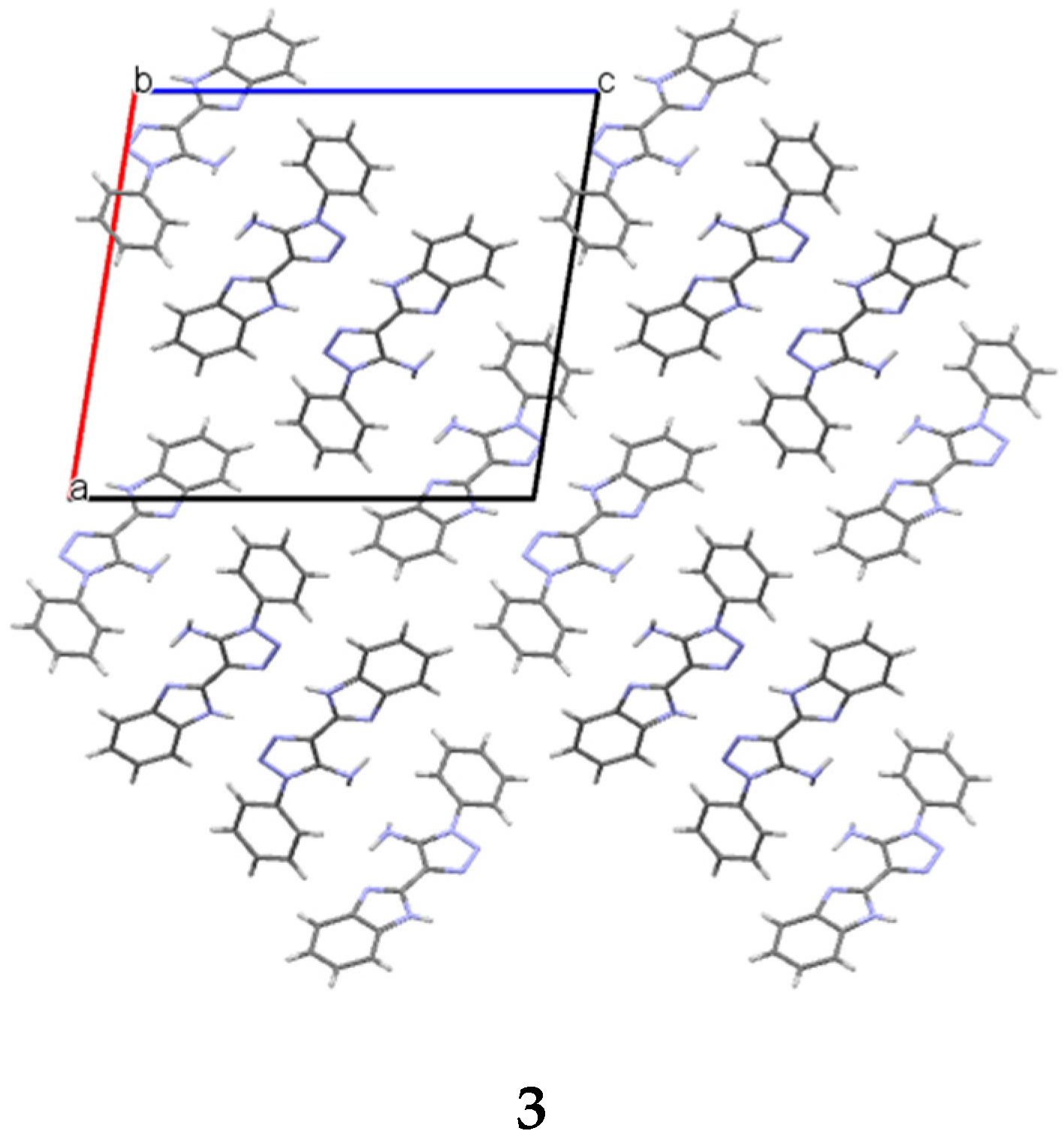
3b, which account for 28% and 27% of their unit cell size, respectively. They are formed along the
a crystallographic axis (
Figure 3).
Modern crystal engineering is based on various approaches to the crystal structure analysis, including the close packing principles of Kitaigorodskii [
27], the Popelier’s principles [
28], the set of graphs applied to the crystal structure analysis [
29], the Etter’s rules [
30], or the Desiraju’s concept of supramolecular synthons [
31,
32,
33]. All these approaches are based mainly on the study and geometric parameters comparison of intermolecular interactions. The modern basic method is the concept of supramolecular synthons, which allows us to describe a crystal as a packing of molecular assemblies bound by any specific intermolecular interactions (hydrogen or halogen bonds, stacking interaction, etc. [
31,
32,
33]). This concept turned out to be very useful and has made significant progress in studying and predicting the crystal structure [
32]. Nevertheless, the release of the basic supramolecular synthon is reliable in the presence of only one strong intermolecular interaction. This task becomes very equivocal in the presence of two or more strong hydrogen bonds, or weak interactions with close geometric parameters, or when specific intermolecular interactions are absent at all. For example, as shown earlier, the application of the geometric approach has proved to be unsuccessful in determining the strongest intermolecular interaction in crystals of diaminobenzenes and nitro-diaminobenzenes [
34,
35].
The analysis of interatomic distances in crystal
3 indicates the presence of a set of intermolecular C–H…X H-bonds, one N-H…N hydrogen bond and stacking interaction with a “head-to-head” orientation (
Table 2). Based on their geometrical parameters, it is possible to suggest that all of these interactions are weak, except for N2-H2N…N3′ H-bond (
Table 2). In crystals
3a and
3b, the number of C-H...X interactions is reduced (
Table 3). In these crystals, the set of intermolecular interactions is almost identical. It should be noted that the N2-H2N…N3′ hydrogen bond and the hydrogen bond formed between the amino group and the solvate molecule are also stronger than others (
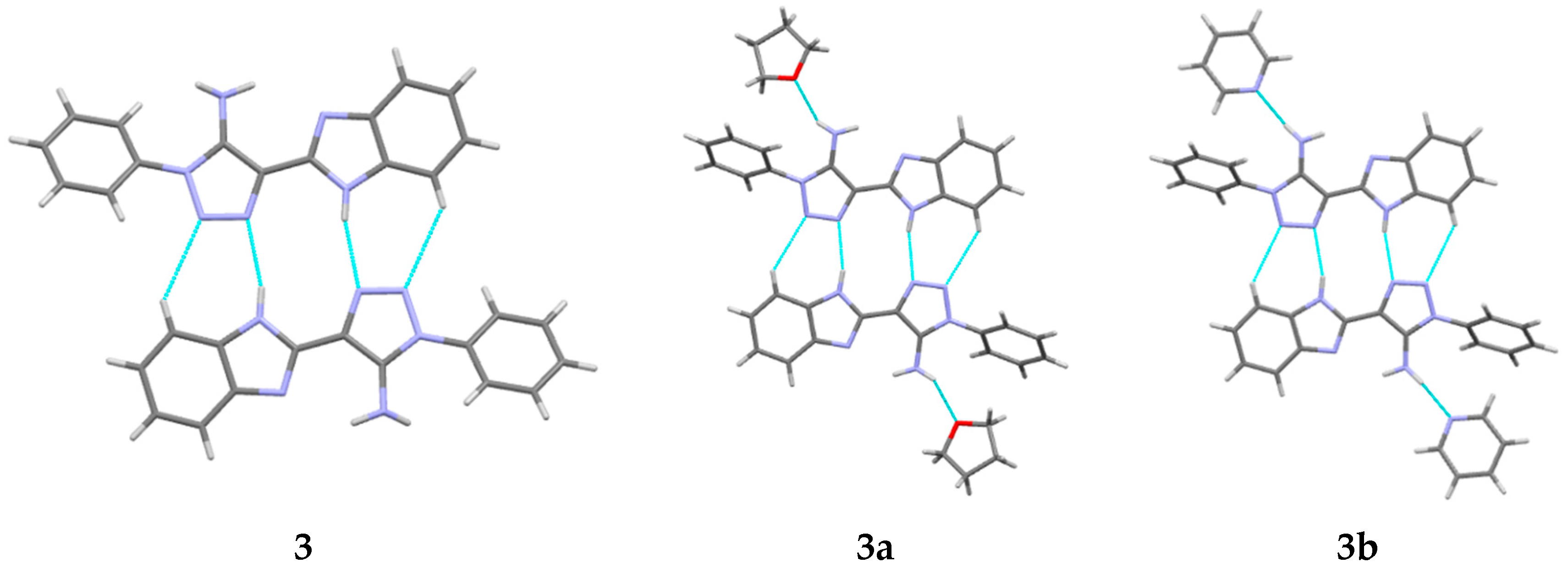
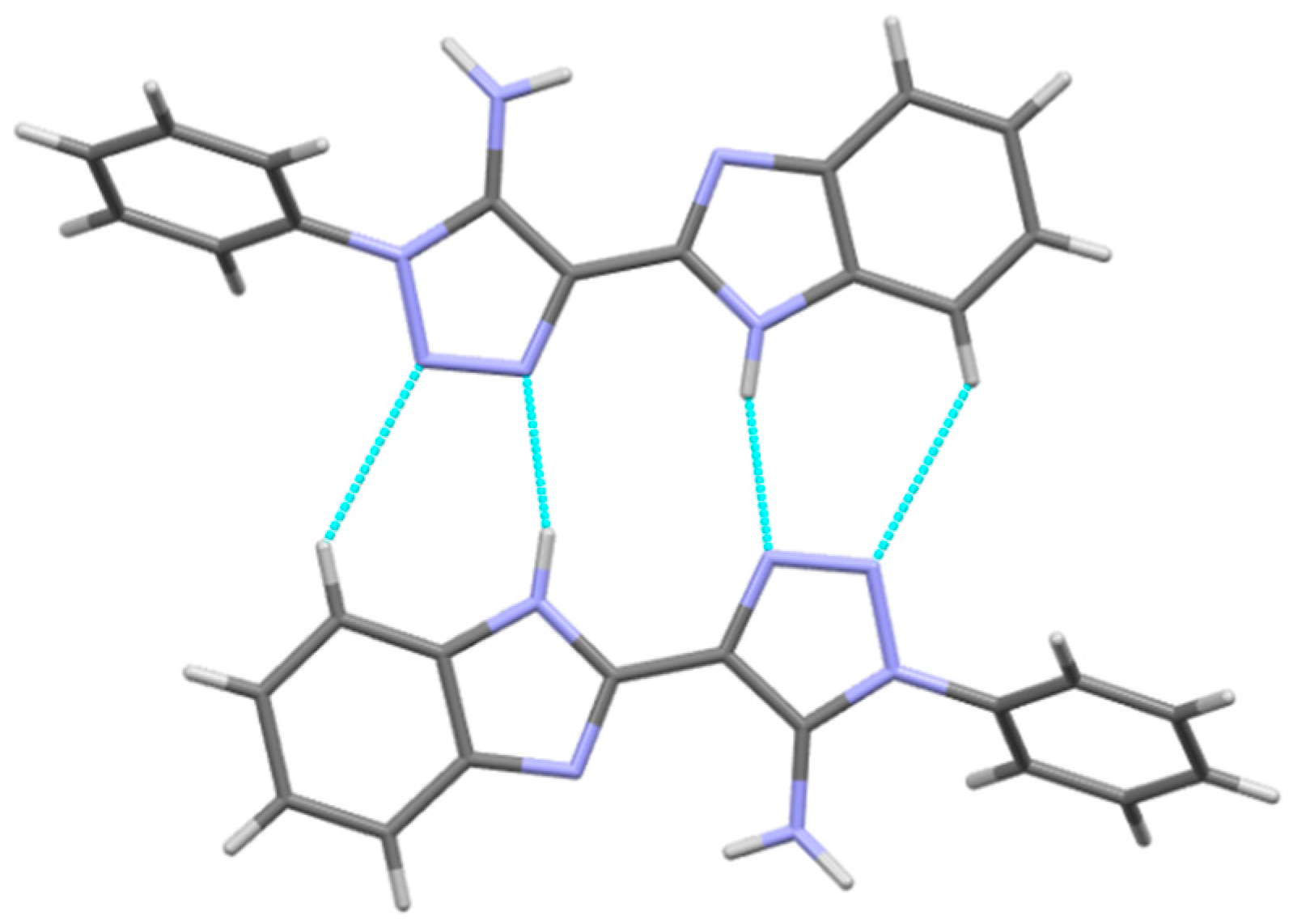
Table 3). Visual analysis of crystal structures
3,
3a and
3b allows us to recognize the centrosymmetric dimers bound by the N2-H2N…N3′ and C5-H5…N4′ hydrogen bonds (
Figure 4,
Table 2 and
Table 3). These dimers are packed in the layers due to the stacking interactions (
Figure 5). Ergo, routine analysis of specific intermolecular interactions in the crystal does not allow us to make definite conclusions about the preferred packing arrangement of molecules and the role of weak hydrogen bonds. This may be done only on the basis of consideration of topology of interactions between molecules in terms of their directionality and energies.
In terms of the energy of interactions between molecules, the study of the crystal structure was carried out using the method described previously [
36,
37] (calculation procedure is presented in detail in the Materials and Methods Section). This method does not depend on the nature of specific intermolecular interactions and their geometric parameters and takes into account all possible types of the intermolecular interactions. The first coordination sphere for each of the molecules located in the asymmetric part of the unit cell contains 16–27 molecules in the crystals of compounds
3,
3a and
3b. Data for dimers with interaction energies higher than 1% of the total interaction energy of the basic molecule with all the molecules belonging to its first coordination sphere are given in
Table 4,
Table 5 and
Table 6.
The analysis of pairwise interaction energies in the crystal of
3 has revealed that the first coordination sphere of the basic molecule contains 16 neighboring molecules. The calculations of the pairwise interaction energies of the basic molecule with each of its neighbors have revealed one strong interaction and two weaker interactions (
Table 4). The strongest bonded dimer 3-d12 (
Figure 6) is formed due to classic N-H…N and C-H…N hydrogen bonds, while the molecules within two other strongly bonded dimers 3-d1 and 3-d2 are linked by the stacking interaction with the “head to head” orientation (
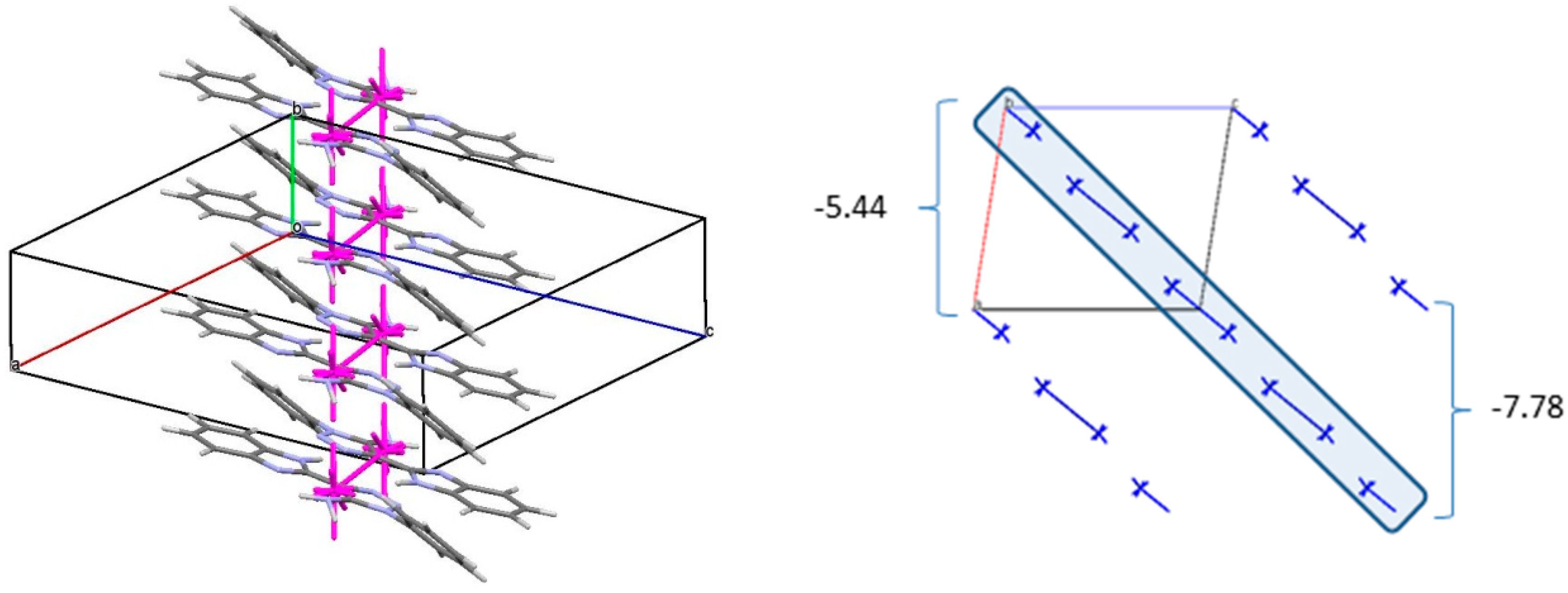
Table 4). As a result, the double column (
Figure 7) can be recognized as the primary basic structural motif (BSM1) in the crystal of
3. The interaction energy of each molecule with its neighbors within the column is −53.54 kcal/mol. The molecules belonging to the neighboring columns are bound by the C-H…π and N-H…N hydrogen bonds. The interaction energies of the primary BSM with neighboring columns are anisotropic enough. It allows to separate out the layer parallel to the (−1 0 1) crystallographic plane as the second basic structural motif (BSM2) (
Figure 7) where the interaction energy between neighboring layers is almost ten times smaller than the interaction energy within the layer (−72.10 kcal/mol). The neighboring layers are bound by the C-H…π hydrogen bonds and non-specific interactions. Therefore, we can conclude that crystal
3 has two levels of organization and can be classified as columnar-layered.
The introduction of the solvent molecule into the crystal causes some change in the crystal organization. The analysis of pairwise interaction energies has revealed that the first coordination sphere of the basic molecule
3 contains 16 neighboring molecules in both solvates while the solvate molecule is surrounded by 11 neighboring molecules in crystal
3a and 9 neighboring molecules in crystal
3b (
Table 5 and
Table 6). Similar to crystal
3, the most strongly bound dimer 3a-d8 and 3b-d8 is formed due to classic N-H…N and C-H…N hydrogen bonds, while the molecules within another strongly bound dimer 3a-d7 and 3b-d7 are linked by the stacking interaction with “head to tail” orientation (
Table 5 and
Table 6). As a result, the zig-zag columns can be recognized as the primary basic structural motif (BSM1) in crystals
3a and
3b (
Figure 8). The molecules within the columns are also bound by two strongest interactions, and the interaction energies of the basic molecule with its neighbors within the column are −42.15 kcal/mol (
3a) and −40.30 kcal/mol (
3b). The molecules belonging to the neighboring columns are bound by the N-H…O1S in
3a, N-H…N1S in
3b, C-H…π and C-H…N hydrogen bonds and non-specific interactions. The interactions of the primary BSM with neighboring columns are almost isotropic because of the interaction energies vary in the range of −4.24 ÷ −9.22 kcal/mol (
3a) and −6.10 ÷ −9.33 kcal/mol (
3b) (
Figure 8). The interaction energy between the neighboring columns is several times smaller than the interaction energy within the column. Thus we can conclude that the crystals
3a and
3b have only one level of organization and can be classified as columnar. It should be noted that despite different solvent molecules in crystals
3a and
3b, their crystal packings are almost isostructural (
Figure 8).
The comparison of the results of the crystal structure analysis performed by two ways shows clearly that the geometrical characteristics of intermolecular interactions are not informative enough to separate out the main structural motif and study the comparative role of different types of interactions. At the same time, the study of interaction energies between molecules allows us to recognize different levels of crystal structure organization and describe the interaction types causing their formation. Thus, the classic N-H…N and C-H…N hydrogen bonds play the main role in all the studied crystals forming the primary basic structural motif. Their role is comparable with the role of the stacking interactions. The inclusion of the solvent molecules in the crystals 3a and 3b lead to a more isotropic packing type.


 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}