Structural, Mechanical, and Transport Properties of Electron Beam-Irradiated Chitosan Membranes at Different Doses




Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Electron Beam Irradiation
2.3. Membrane Preparation
2.4. Characterization Methods
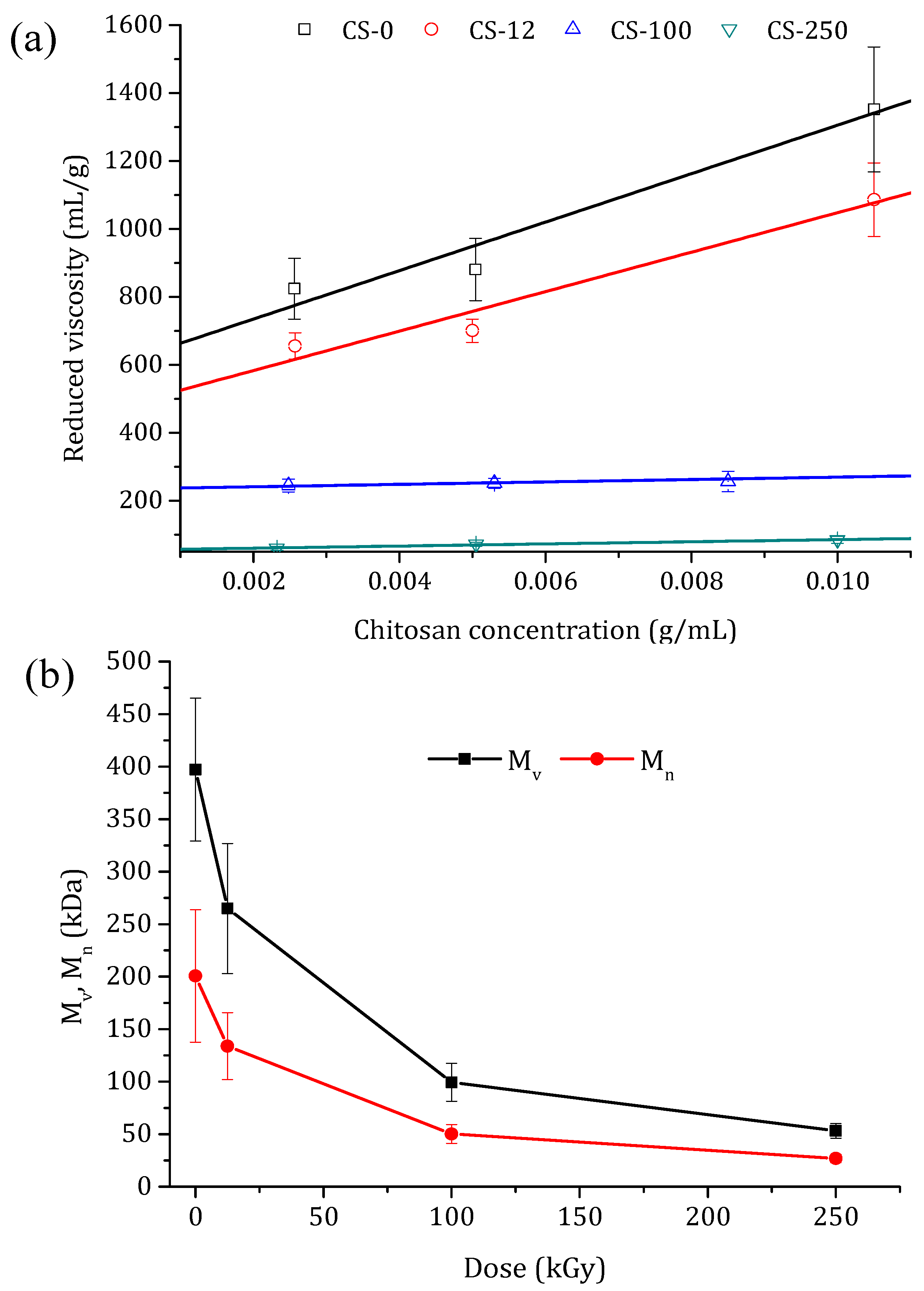
2.4.1. Viscosity, Molecular Weight, and the Irradiation-Induced Chemical Degradation of Chitosan Powder
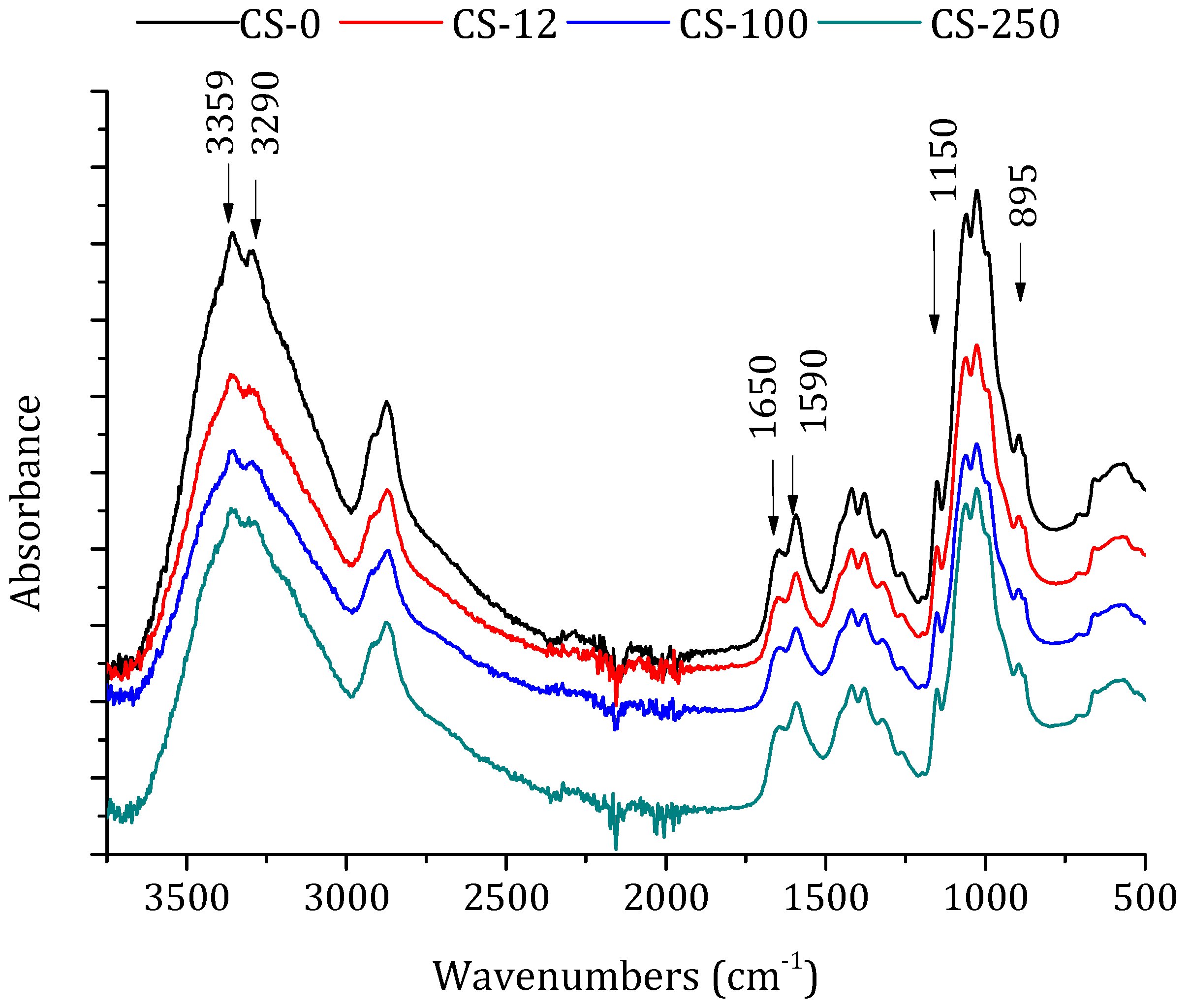
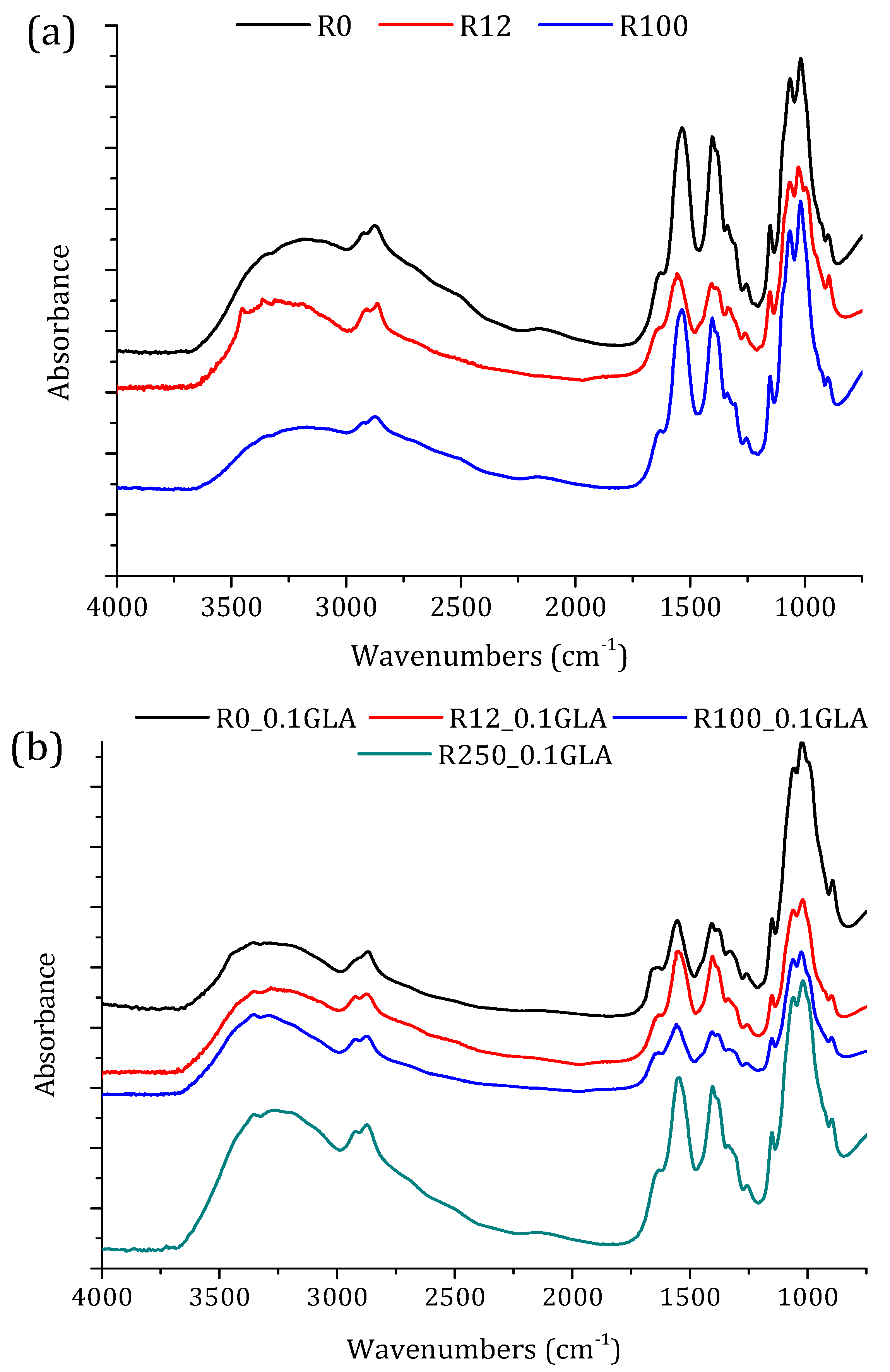
2.4.2. Attenuated Total Reflectance-Fourier Transforms Infrared (ATR-FTIR) Spectroscopy and Degree of Deacetylation (DD)
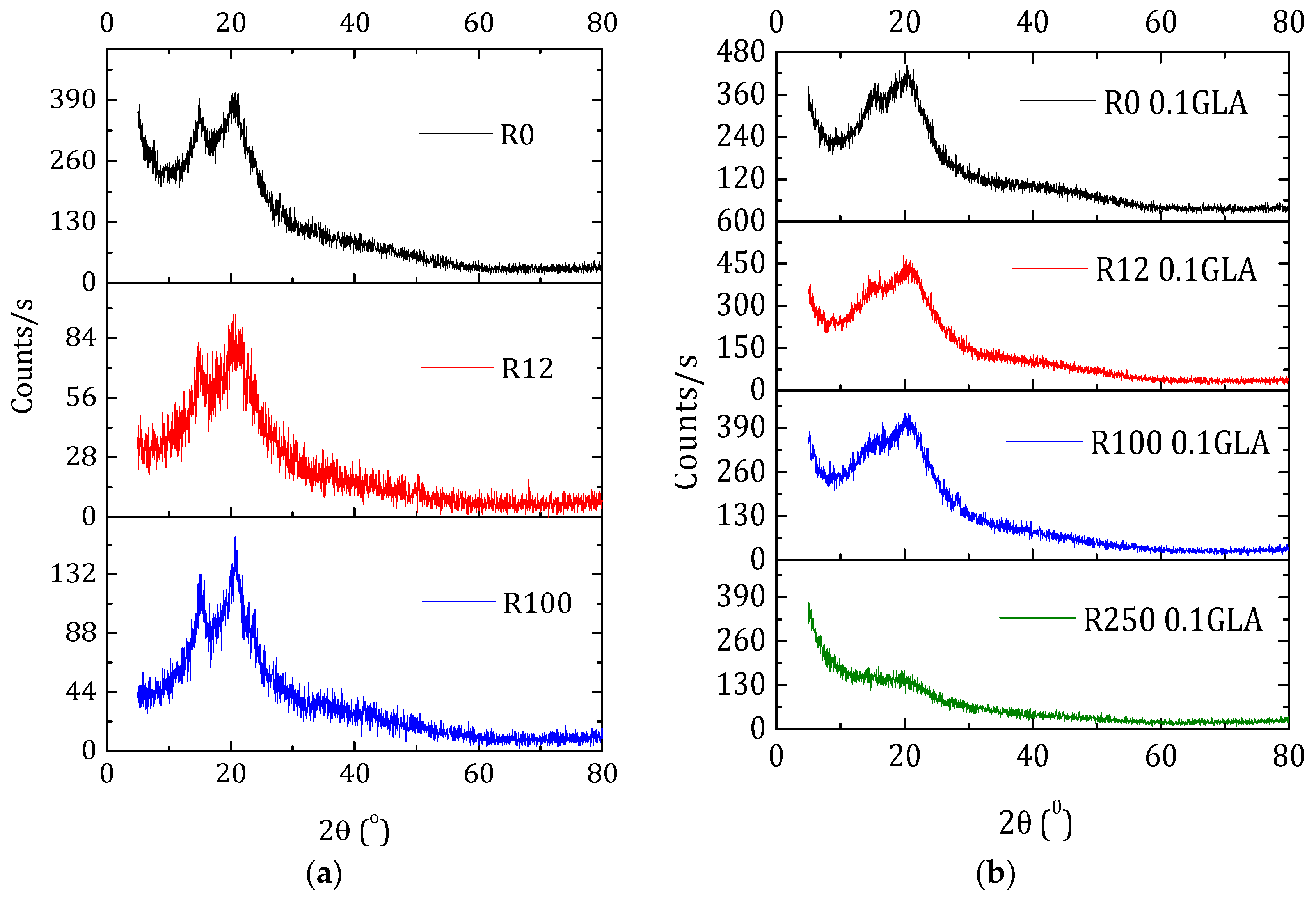
2.4.3. X-ray Diffraction Spectroscopy
2.4.4. Other Characterization Methods for Chitosan Membranes
2.5. Drug Permeability Experiments
3. Results and Discussion
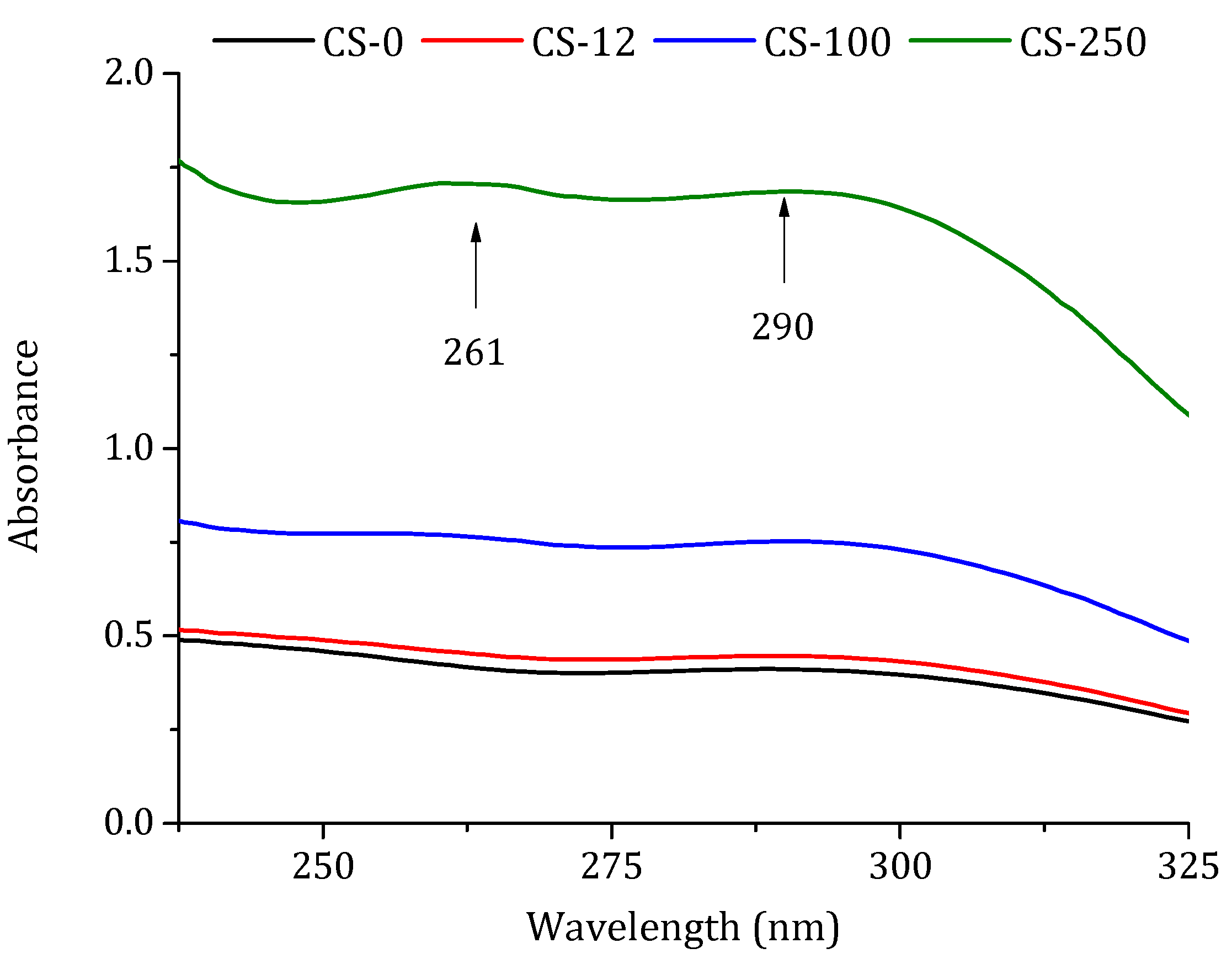
3.1. Chitosan Powders
3.2. Characterization of Chitosan Membranes
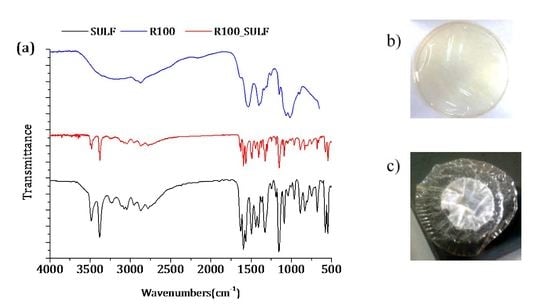
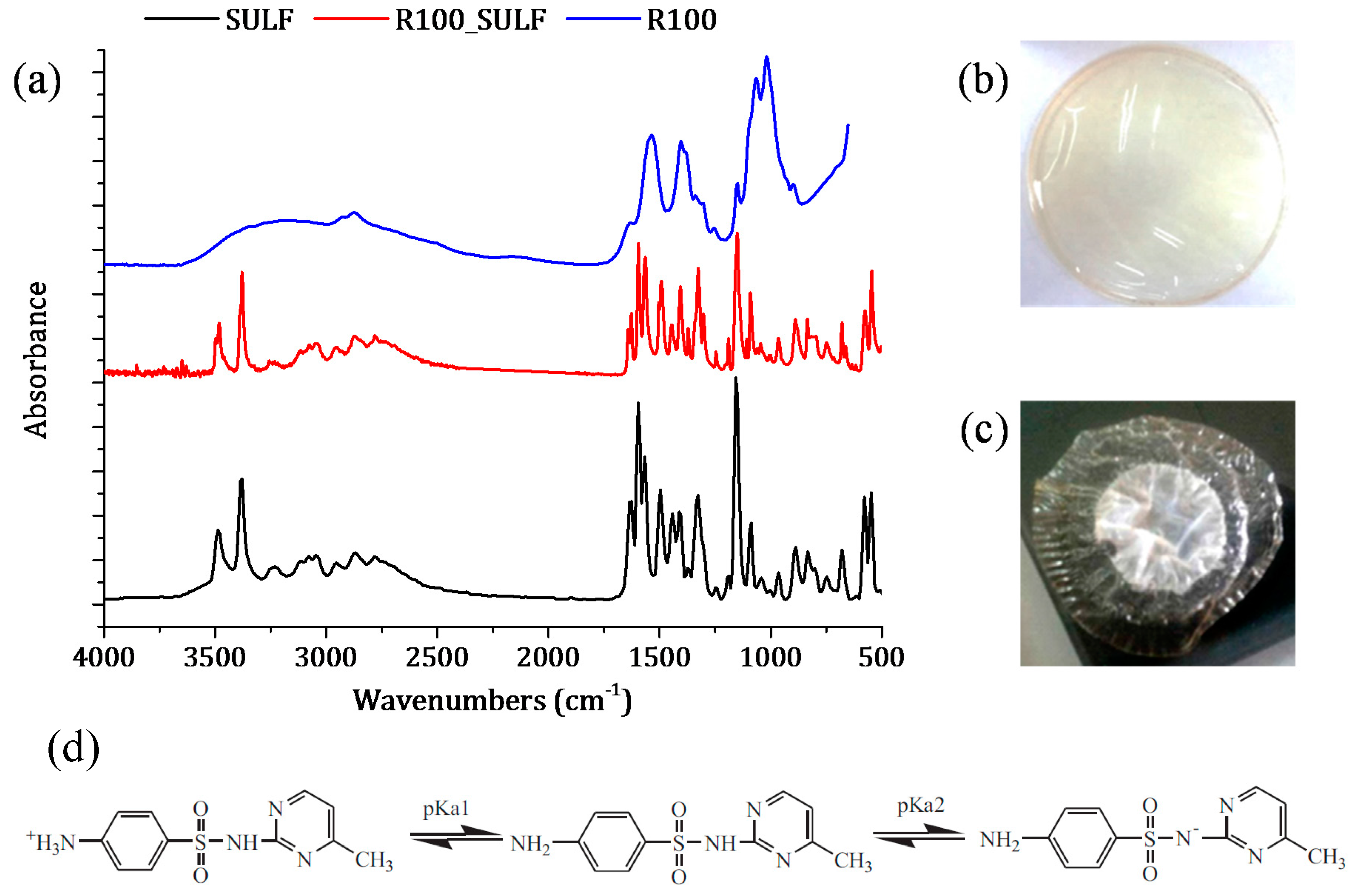
3.2.1. ATR-FTIR Spectroscopy and X-ray Diffraction
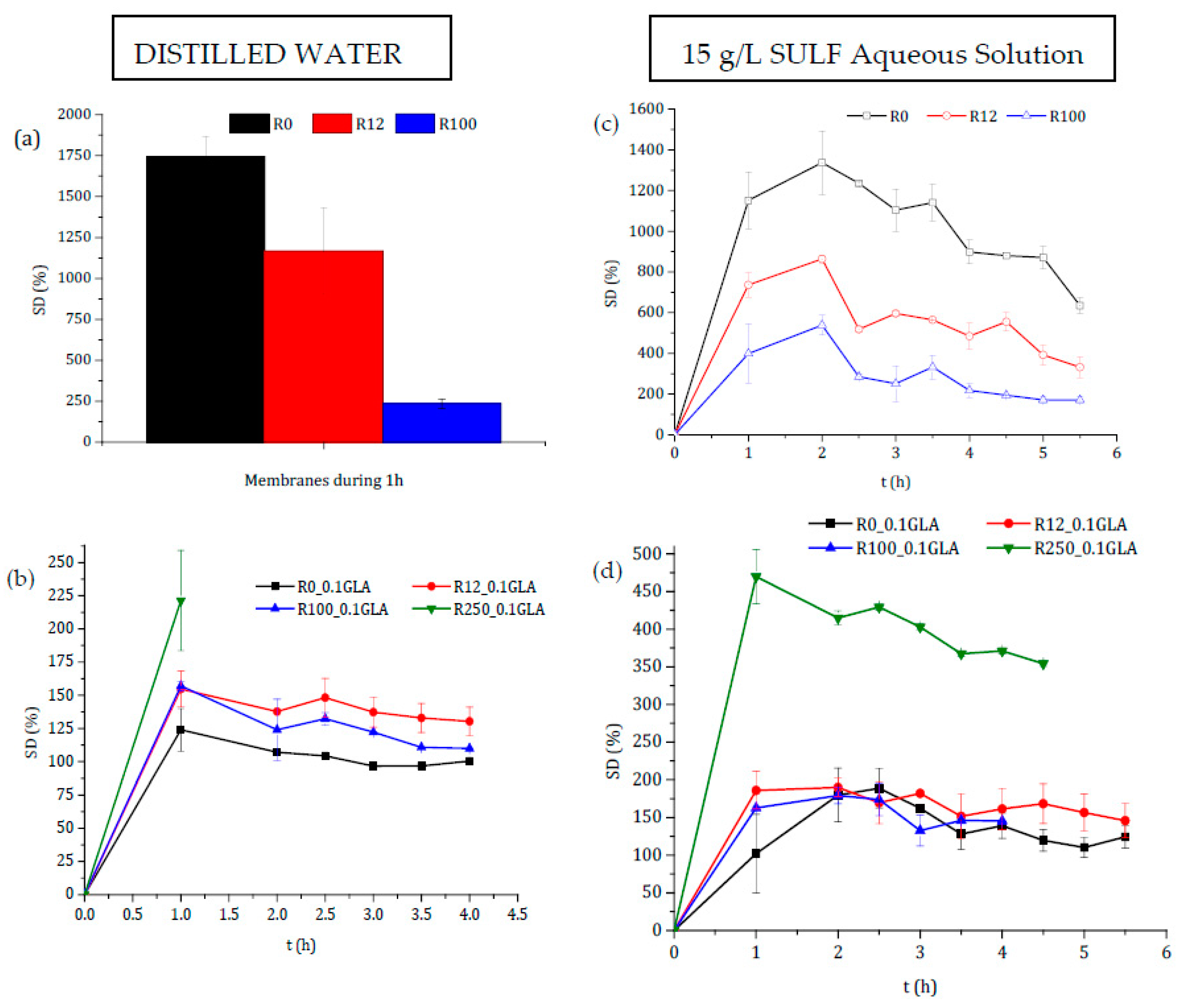
3.2.2. Swelling
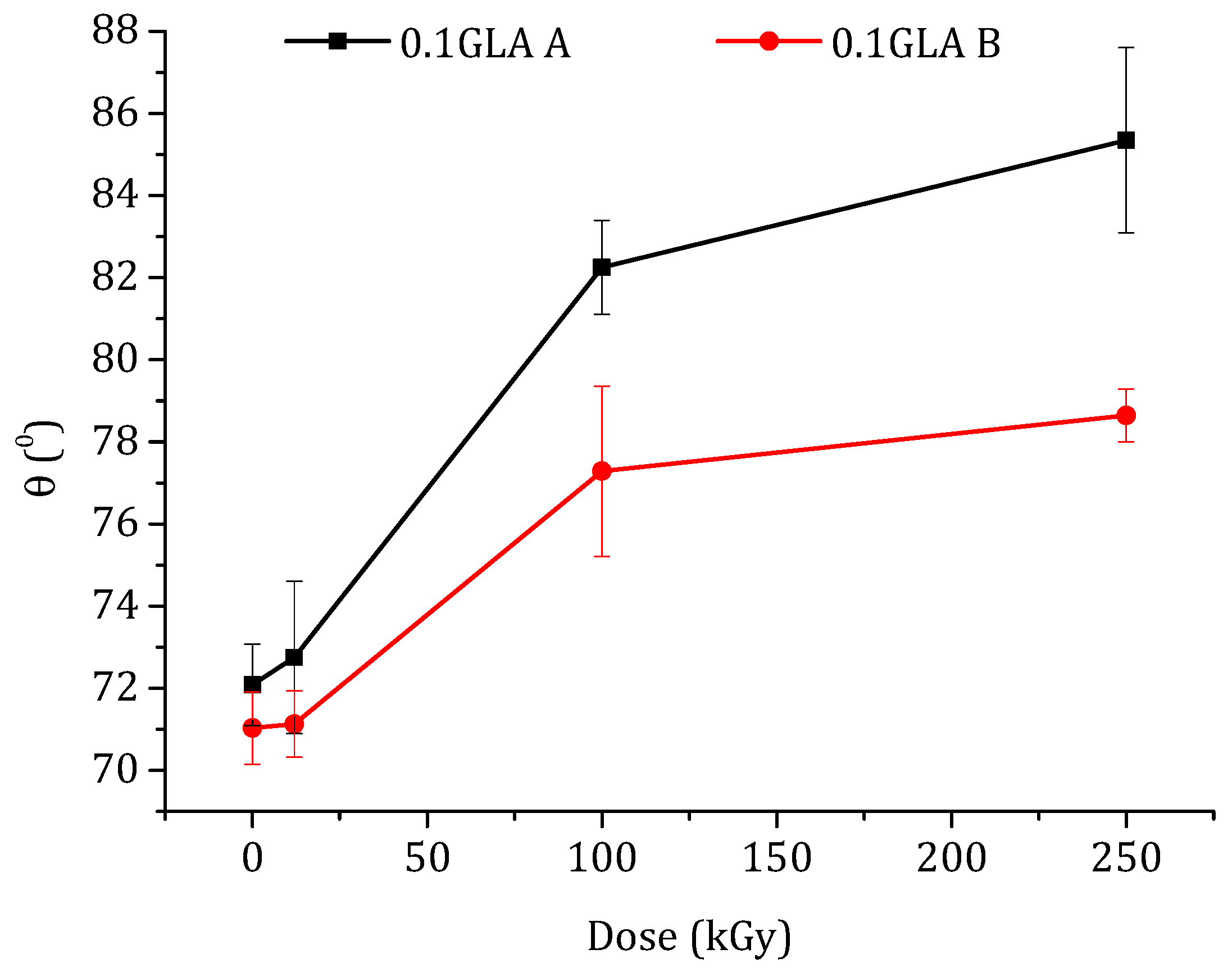
3.2.3. Contact Angle
3.2.4. Mechanical Properties
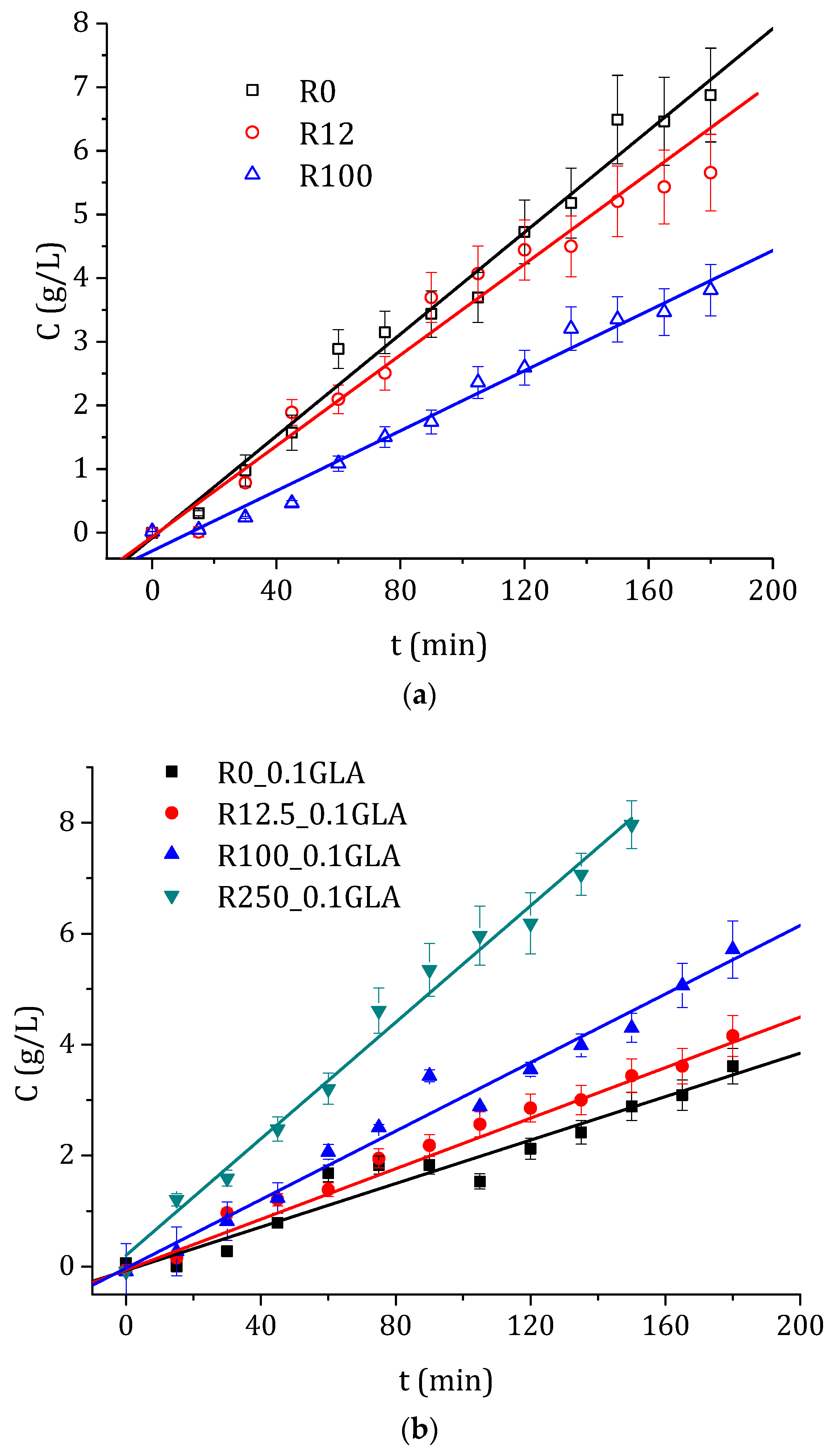
3.3. Drug Permeability Experiments
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Aranaz, I.; Mengíbar, M.; Harris, R.; Paños, I.; Miralles, B.; Acosta, N.; Galed, G.; Heras, Á. Functional characterization of chitin and chitosan. Curr. Chem. Biol. 2009, 3, 203–230. [Google Scholar]
- Berger, J.; Reist, M.; Mayer, J.M.; Felt, O.; Peppas, N.A.; Gurny, R. Structure and interactions in covalently and ionically crosslinked chitosan hydrogels for biomedical applications. Eur. J. Pharm. Biopharm. 2004, 57, 19–34. [Google Scholar] [CrossRef]
- Rinaudo, M.; Pavlov, G.; Desbrie, J. Influence of acetic acid concentration on the solubilization of chitosan. Polymer 1999, 40, 7029–7032. [Google Scholar] [CrossRef]
- Nilsen-Nygaard, J.; Strand, S.; Vårum, K.; Draget, K.; Nordgård, C. Chitosan: Gels and Interfacial Properties. Polymers 2015, 7, 552–579. [Google Scholar] [CrossRef]
- Beppu, M.M.; Vieira, R.S.; Aimoli, C.G.; Santana, C.C. Crosslinking of chitosan membranes using glutaraldehyde: Effect on ion permeability and water absorption. J. Membr. Sci. 2007, 301, 126–130. [Google Scholar] [CrossRef]
- Kamari, A.; Ngah, W.S.W.; Chong, M.Y.; Cheah, M.L. Sorption of acid dyes onto GLA and H2SO4 cross-linked chitosan beads. Desalination 2009, 249, 1180–1189. [Google Scholar] [CrossRef]
- Salehi, E.; Daraei, P.; Arabi Shamsabadi, A. A review on chitosan-based adsorptive membranes. Carbohydr. Polym. 2016, 152, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Bui, V.; Park, D.; Lee, Y.-C. Chitosan Combined with ZnO, TiO2 and Ag Nanoparticles for Antimicrobial Wound Healing Applications: A Mini Review of the Research Trends. Polymers 2017, 9, 21. [Google Scholar] [CrossRef]
- Falk, B.; Garramone, S.; Shivkumar, S. Diffusion coefficient of paracetamol in a chitosan hydrogel. Mater. Lett. 2004, 58, 3261–3265. [Google Scholar] [CrossRef]
- Langer, R.; Peppas, N. Chemical and Physical Structure of Polymers as Carriers for Controlled Release of Bioactive Agents: A Review. J. Macromol. Sci. 2006, 23, 61–126. [Google Scholar] [CrossRef]
- Gupta, K.C.; Jabrail, F.H. Glutaraldehyde cross-linked chitosan microspheres for controlled release of centchroman. Carbohydr. Res. 2007, 342, 2244–2252. [Google Scholar] [CrossRef] [PubMed]
- Ražem, D.; Katušin-Ražem, B. The effects of irradiation on controlled drug delivery/controlled drug release systems. Radiat. Phys. Chem. 2008, 77, 288–344. [Google Scholar] [CrossRef]
- Hennink, W.E.; van Nostrum, C.F. Novel crosslinking methods to design hydrogels. Adv. Drug Deliv. 2012, 64, 223–236. [Google Scholar] [CrossRef]
- Monteiro, O.A.C.; Airoldi, C. Some studies of crosslinking chitosan-glutaraldehyde interaction in a homogeneous system. Int. J. Biol. Macromol. 1999, 26, 119–128. [Google Scholar] [CrossRef]
- Baroni, P.; Vieira, R.S.; Meneghetti, E.; da Silva, M.G.; Beppu, M.M. Evaluation of batch adsorption of chromium ions on natural and crosslinked chitosan membranes. J. Hazard. Mater. 2008, 152, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, S.; Susheela, G.; Jayasimha, G.; Khan, A. Crosslinked chitosan membranes: Characterzation and study of dimethylhydrazine dehydration and pervaporation. Polym. Int. 2001, 5, 1156–1161. [Google Scholar] [CrossRef]
- Agnihotri, S.A.; Mallikarjuna, N.N.; Aminabhavi, T.M. Recent advances on chitosan-based micro- and nanoparticles in drug delivery. J. Control. Release 2004, 100, 5–28. [Google Scholar] [CrossRef] [PubMed]
- Vanichvattanadecha, C.; Supaphol, P.; Nagasawa, N.; Tamada, M.; Tokura, S.; Furuike, T.; Tamura, H.; Rujiravanit, R. Effect of gamma radiation on dilute aqueous solutions and thin films of N-succinyl chitosan. Polym. Degrad. Stab. 2010, 95, 234–244. [Google Scholar] [CrossRef]
- Franzén, H.; Draget, K.; Langebäck, J.; Nilsen-Nygaard, J. Characterization and Properties of Hydrogels Made from Neutral Soluble Chitosans. Polymers 2015, 7, 373–389. [Google Scholar] [CrossRef]
- Nunthanid, J.; Puttipipatkhachorn, S.; Yamamoto, K.; Peck, G.E. Physical properties and molecular behavior of chitosan films. Drug Dev. Ind. Pharm. 2001, 27, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Jaworska, M.; Sakurai, K.; Gaudon, P.; Guibal, E. Influence of chitosan characteristics on polymer properties. I: Crystallographic properties. Polym. Int. 2003, 52, 198–205. [Google Scholar] [CrossRef]
- Chen, R.H.; Hwa, H.D. Effect of molecular weight of chitosan with the same degree of deacetylation on the thermal, mechanical, and permeability properties of the prepared membrane. Carbohydr. Polym. 1996, 29, 353–358. [Google Scholar]
- Garcia, M.A.; Perez, L.; de la Paz, N.; Gonzalez, J.; Rapado, M.; Casariego, A. Effect of molecular weight reduction by gamma irradiation on chitosan film properties. Mater. Sci. Eng. C 2015, 55, 174–180. [Google Scholar] [CrossRef] [PubMed]
- BenBettaïeb, N.; Karbowiak, T.; Bornaz, S.; Debeaufort, F. Spectroscopic analyses of the influence of electron beam irradiation doses on mechanical, transport properties and microstructure of chitosan-fish gelatin blend films. Food Hydrocoll. 2015, 46, 37–51. [Google Scholar] [CrossRef]
- Antony, R.; Theodore David, S.; Karuppasamy, K.; Sanjeev, G.; Balakumar, S. Influence of electron beam irradiation on spectral, thermal, morphological and catalytic properties of Co(II) complex immobilized on chitosan’s Schiff base. Spectrochim. Acta 2014, 124, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Choi, Y.-J.; Park, H.S.; Noh, I. Analysis of chitosan irradiated with high-energy cyclotron ion beams. J. Phys. Chem. Solids 2008, 69, 1569–1572. [Google Scholar] [CrossRef]
- Ramaprasad, A.T.; Rao, V.; Praveena, M.; Sanjeev, G.; Ramanani, S.P.; Sabharwal, S. Preparation of crosslinked chitosan by electron beam irradiation in the presence of CCl4. J. Appl. Polym. Sci. 2009, 111, 1063–1068. [Google Scholar] [CrossRef]
- Zainol, I.; Akil, H.M.; Mastor, A. Effect of γ-irradiation on the physical and mechanical properties of chitosan powder. Mater. Sci. Eng. C 2009, 29, 292–297. [Google Scholar] [CrossRef]
- Chmielewski, A.G.; Migdal, W.; Swietoslawski, J.; Swietoslawski, J.; Jakubaszek, U.; Tarnowski, T. Chemical-radiation degradation of natural oligoamino-polysaccharides for agricultural application. Radiat. Phys. Chem. 2007, 76, 1840–1842. [Google Scholar] [CrossRef]
- Gryczka, U.; Dondi, D.; Chmielewski, A.G.; Migdal, W.; Buttafava, A.; Faucitano, A. The mechanism of chitosan degradation by gamma and e-beam irradiation. Radiat. Phys. Chem. 2009, 78, 543–548. [Google Scholar] [CrossRef]
- Hien, N.Q.; Phu, D.V.; Duy, N.N.; Lan, N.T.K. Degradation of chitosan in solution by gamma irradiation in the presence of hydrogen peroxide. Carbohydr. Polym. 2012, 87, 935–938. [Google Scholar] [CrossRef]
- Tahtat, D.; Mahlous, M.; Benamer, S.; Nacer Khodja, A.; Larbi Youcef, S. Effect of molecular weight on radiation chemical degradation yield of chain scission of γ-irradiated chitosan in solid state and in aqueous solution. Radiat. Phys. Chem. 2012, 81, 659–665. [Google Scholar] [CrossRef]
- Desai, K.G.; Park, H.J. Study of gamma-irradiation effects on chitosan microparticles. Drug Deliv. 2006, 13, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Al-Assaf, S.; Coqueret, X.; Khairul, Z.; Haji, M.D.; Sen, M.; Ulanski, P. (Eds.) The Radiation Chemistry of Polysaccharides; International Atomic Energy Agency: Vienna, Austria, 2016. [Google Scholar]
- Ulański, P.; Rosiak, J. Preliminary studies on radiation-induced changes in chitosan. Int. J. Radiat. Appl. Instrum. 1992, 39, 53–57. [Google Scholar] [CrossRef]
- Tahtat, D.; Uzun, C.; Mahlous, M.; Güven, O. Beneficial effect of gamma irradiation on the N-deacetylation of chitin to form chitosan. Nucl. Instrum. Methods Phys. Res. 2007, 265, 425–428. [Google Scholar] [CrossRef]
- Shen, K.; Hu, Q.; Wang, Z.; Qu, J. Effect of 60Co irradiation on the properties of chitosan rod. Mater. Sci. Eng. C 2011, 31, 866–872. [Google Scholar] [CrossRef]
- Chmielewski, A.G.; Haji-Saeid, M.; Ahmed, S. Progress in radiation processing of polymers. Nucl. Instrum. Methods Phys. Res. 2005, 236, 44–54. [Google Scholar] [CrossRef]
- Maciszewski, W.; MigdaI, W.; Owczarczyk, B.; Chmielewski, A. Application of “Electronica 10-10” Electron Linac for Food Processing. In Proceedings of the Materials: 4th European Particle Accelerator Conference, London, UK, 27 June–1 July 1994; Volume 24, pp. 2644–2646. [Google Scholar]
- Brugnerotto, J.; Lizardi, J.; Goycoolea, F.M.; Argüelles-Monal, W.; Desbrières, J.; Rinaudo, M. An infrared investigation in relation with chitin and chitosan characterization. Polymer 2001, 42, 3569–3580. [Google Scholar] [CrossRef]
- Kasaai, M. A review of several reported procedures to determine the degree of N-acetylation for chitin and chitosan using infrared spectroscopy. Carbohydr. Polym. 2008, 71, 497–508. [Google Scholar] [CrossRef]
- Hussain, P.R.; Wani, I.A.; Suradkar, P.P.; Dar, M.A. Gamma irradiation induced modification of bean polysaccharides: Impact on physicochemical, morphological and antioxidant properties. Carbohydr. Polym. 2014, 110, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Crank, J. The Mathematics of Diffusion, 2nd ed.; Clarendon Press: Oxford, UK, 1975. [Google Scholar]
- Osman, Z.; Arof, A.K. FTIR studies of chitosan acetate based polymer electrolytes. Electrochim. Acta 2003, 48, 993–999. [Google Scholar] [CrossRef]
- Pasanphan, W.; Rimdusit, P.; Choofong, S.; Piroonpan, T.; Nilsuwankosit, S. Systematic fabrication of chitosan nanoparticle by gamma irradiation. Radiat. Phys. Chem. 2010, 79, 1095–1102. [Google Scholar] [CrossRef]
- Nagasawa, N.; Mitomo, H.; Yoshii, F.; Kume, T. Radiation-induced degradation of sodium alginate. Polym. Degrad. Stab. 2000, 69, 279–285. [Google Scholar] [CrossRef]
- Bigi, A.; Bracci, B.; Cojazzi, G.; Panzavolta, S.; Roveri, N. Drawn gelatin films with improved mechanical properties. Biomaterials 1998, 19, 2335–2340. [Google Scholar] [CrossRef]
- Lavorgna, M.; Piscitelli, F.; Mangiacapra, P.; Buonocore, G.G. Study of the combined effect of both clay and glycerol plasticizer on the properties of chitosan films. Carbohydr. Polym. 2010, 82, 291–298. [Google Scholar] [CrossRef]
- Petrova, V.A.; Chernyakov, D.D.; Moskalenko, Y.E.; Gasilova, E.R.; Strelina, I.A.; Okatova, O.V.; Baklagina, Y.G.; Vlasova, E.N.; Skorik, Y.A. O,N-(2-sulfoethyl)chitosan: Synthesis and properties of solutions and films. Carbohydr. Polym. 2017, 157, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Mitomo, H.; Zhai, M.; Yoshii, F.; Nagasawa, N.; Kume, T. Synthesis of antibacterial PVA/CM-chitosan blend hydrogels with electron beam irradiation. Carbohydr. Polym. 2003, 53, 439–446. [Google Scholar] [CrossRef]
- Zhai, M.; Zhao, L.; Yoshii, F.; Kume, T. Study on antibacterial starch/chitosan blend film formed under the action of irradiation. Carbohydr. Polym. 2004, 57, 83–88. [Google Scholar] [CrossRef]
- Zhao, S.; Yao, Y.; Ba, C.; Zheng, W.; Economy, J.; Wang, P. Enhancing the performance of polyethylenimine modified nanofiltration membrane by coating a layer of sulfonated poly(ether ether ketone) for removing sulfamerazine. J. Membr. Sci. 2015, 492, 620–629. [Google Scholar] [CrossRef]
- Grundke, K.; Poschel, K.; Synytska, A.; Frenzel, R.; Drechsler, A.; Nitschke, M.; Cordeiro, A.L.; Uhlmann, P.; Welzel, P.B. Experimental studies of contact angle hysteresis phenomena on polymer surfaces—Toward the understanding and control of wettability for different applications. Adv. Colloid Interface Sci. 2015, 222, 350–376. [Google Scholar] [CrossRef] [PubMed]
- Kurek, M.; Galus, S.; Debeaufort, F. Surface, mechanical and barrier properties of bio-based composite films based on chitosan and whey protein. Food Packag. Shelf Life 2014, 1, 56–67. [Google Scholar] [CrossRef]
- Benbettaïeb, N.; Karbowiak, T.; Brachais, C.H.; Debeaufort, F. Impact of electron beam irradiation on fish gelatin film properties. Food Chem. 2016, 195, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, M.; Rajabzadeh, G.; Ahmadi, S.J. Characterization of nanocomposite film based on chitosan intercalated in clay platelets by electron beam irradiation. Carbohydr. Polym. 2017, 157, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, J.; Xia, J.; Kennedy, J.F.; Yie, X.; Liu, T.G. Effect of gamma irradiation on the condensed state structure and mechanical properties of konjac glucomannan/chitosan blend films. Carbohydr. Polym. 2011, 83, 44–51. [Google Scholar] [CrossRef]
- Benbettaïeb, N.; Chambin, O.; Assifaoui, A.; Al-Assaf, S.; Karbowiak, T.; Debeaufort, F. Release of coumarin incorporated into chitosan-gelatin irradiated films. Food Hydrocoll. 2016, 56, 266–276. [Google Scholar] [CrossRef]
- Nizam El-Din, H.M.; Abd Alla, S.G.; El-Naggar, A.W.M. Swelling and drug release properties of acrylamide/carboxymethyl cellulose networks formed by gamma irradiation. Radiat. Phys. Chem. 2010, 79, 725–730. [Google Scholar] [CrossRef]
- Oliveira, H.C.L.D.; Fonseca, J.L.C.; Pereira, M.R. Chitosan-poly(acrylic acid) polyelectrolyte complex membranes: Preparation, characterization and permeability studies. J. Biomater. Sci. Polym. Ed. 2008, 19, 143–160. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Nan, K.; Chen, H.; Xu, Y. Preparation and characterization of chitosan nanopores membranes for the transport of drugs. Int. J. Pharm. 2011, 420, 371–377. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dose (kGy) | DD (%) | (mL/g) | Mv (kDa) | Mn (kDa) | N |
|---|---|---|---|---|---|
| 0 | 88 ± 6 | 344 ± 172 | 367 ± 77 | 186 ± 39 | - |
| 12.5 | 87 ± 6 | 247 ± 104 | 256 ± 50 | 130 ± 26 | 0.2 ± 0.2 |
| 100 | 86 ± 6 | 87 ± 19 | 83 ± 13 | 42 ± 7 | 2.72 ± 0.04 |
| 250 | 85 ± 3 | 47 ± 5 | 43 ± 6 | 22 ± 3 | 6.11 ± 0.02 |
| Membrane | E (MPa) | τs (MPa) | εb (%) | D (10−12 cm2/s) | J (10−4 mol/m2s) | Tδ (min) |
|---|---|---|---|---|---|---|
| R0 | 2410 ± 650 | 62 ± 9 | 5.0 ± 0.4 | 1.74 ± 0.07 | 3.3 ± 0.1 | 1.9 ± 0.1 |
| R12 | 2270 ± 340 | 56 ± 9 | 4.1 ± 0.7 | 1.52 ± 0.09 | 3.0 ± 0.2 | 2.2 ± 0.2 |
| R100 | 1520 ± 180 | 20 ± 9 | 1.5 ± 0.8 | 0.3 ± 0.1 | 1.95 ± 0.08 | 12.1 ± 0.7 |
| R0_0.1GLA | 3140 ± 490 | 75 ± 5 | 11 ± 2 | 2.0 ± 0.2 | 1.6 ±0.5 | 1.7 ± 0.2 |
| R12_0.1GLA | 2670 ± 230 | 62 ± 15 | 8 ± 3 | 2.3 ± 0.1 | 1.9 ± 0.1 | 2.5 ± 0.2 |
| R100_0.1GLA | 2290 ± 150 | 47 ± 18 | 3 ± 1 | 3.8 ± 0.1 | 2.6 ± 0.1 | 0.9 ± 0.07 |
| R250_0.1GLA | 2140 ± 150 | 37 ± 8 | 2 ± 1 | 37 ± 1 | 4.7 ± 0.2 | 0.09 ± 0.04 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baroudi, A.; García-Payo, C.; Khayet, M. Structural, Mechanical, and Transport Properties of Electron Beam-Irradiated Chitosan Membranes at Different Doses. Polymers 2018, 10, 117. https://doi.org/10.3390/polym10020117
Baroudi A, García-Payo C, Khayet M. Structural, Mechanical, and Transport Properties of Electron Beam-Irradiated Chitosan Membranes at Different Doses. Polymers. 2018; 10(2):117. https://doi.org/10.3390/polym10020117
Chicago/Turabian StyleBaroudi, Alia, Carmen García-Payo, and Mohamed Khayet. 2018. "Structural, Mechanical, and Transport Properties of Electron Beam-Irradiated Chitosan Membranes at Different Doses" Polymers 10, no. 2: 117. https://doi.org/10.3390/polym10020117
APA StyleBaroudi, A., García-Payo, C., & Khayet, M. (2018). Structural, Mechanical, and Transport Properties of Electron Beam-Irradiated Chitosan Membranes at Different Doses. Polymers, 10(2), 117. https://doi.org/10.3390/polym10020117