Nucleating and Plasticization Effects in Drawn Poly(Lactic Acid) Fiber during Accelerated Weathering Degradation


Abstract
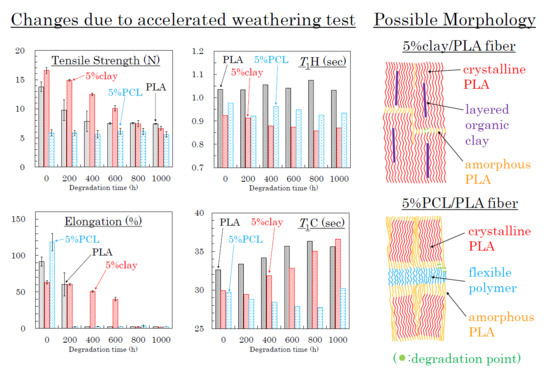
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Melt Spinning and Drawing of PLA Compounds
2.3. Accelerated Weathering Degradation
2.4. Fiber Chractrization
2.5. Solid-State NMR
2.6. Tensile Testing
3. Results and Discussion
3.1. Fiber Chatactrization
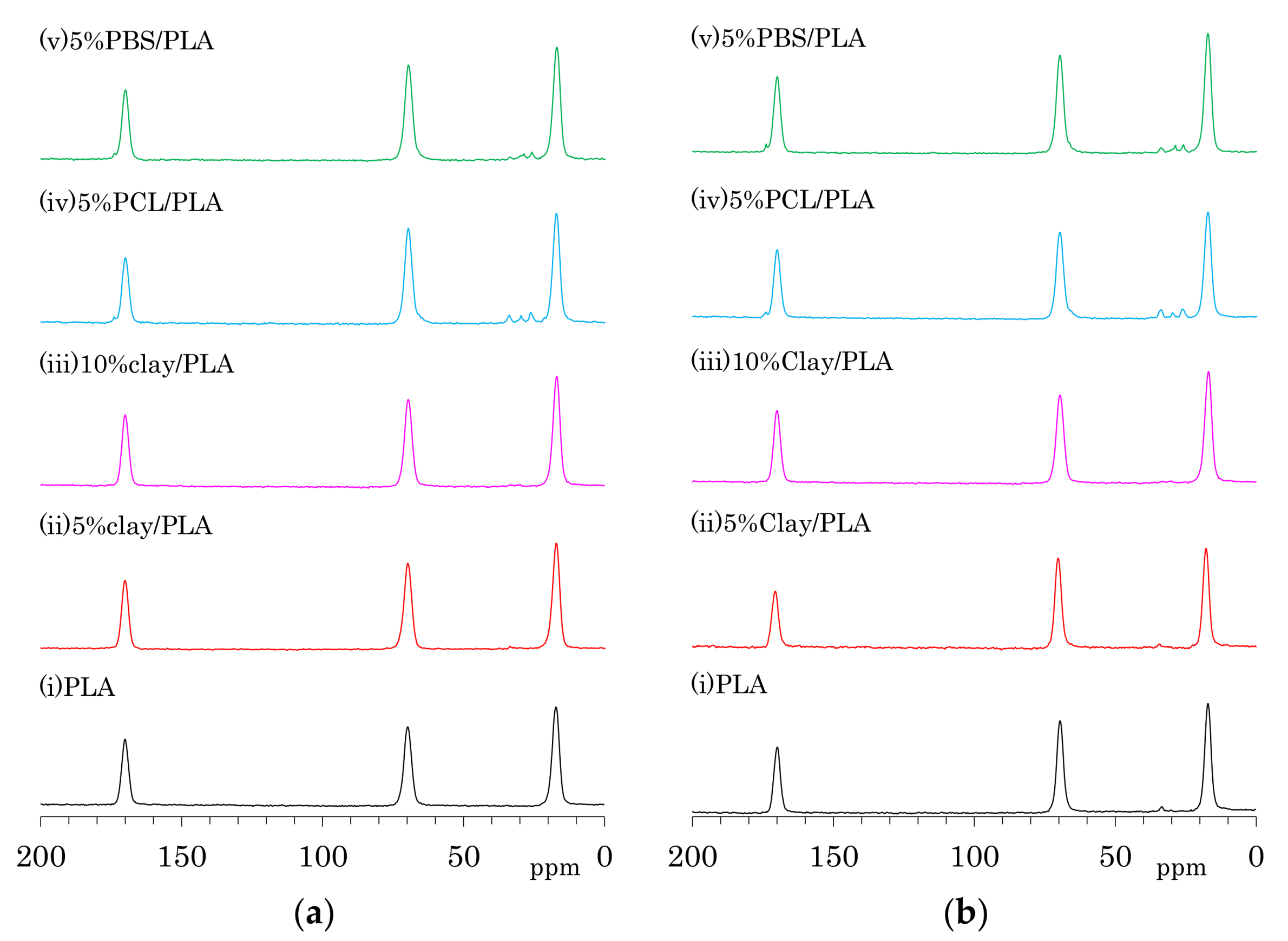
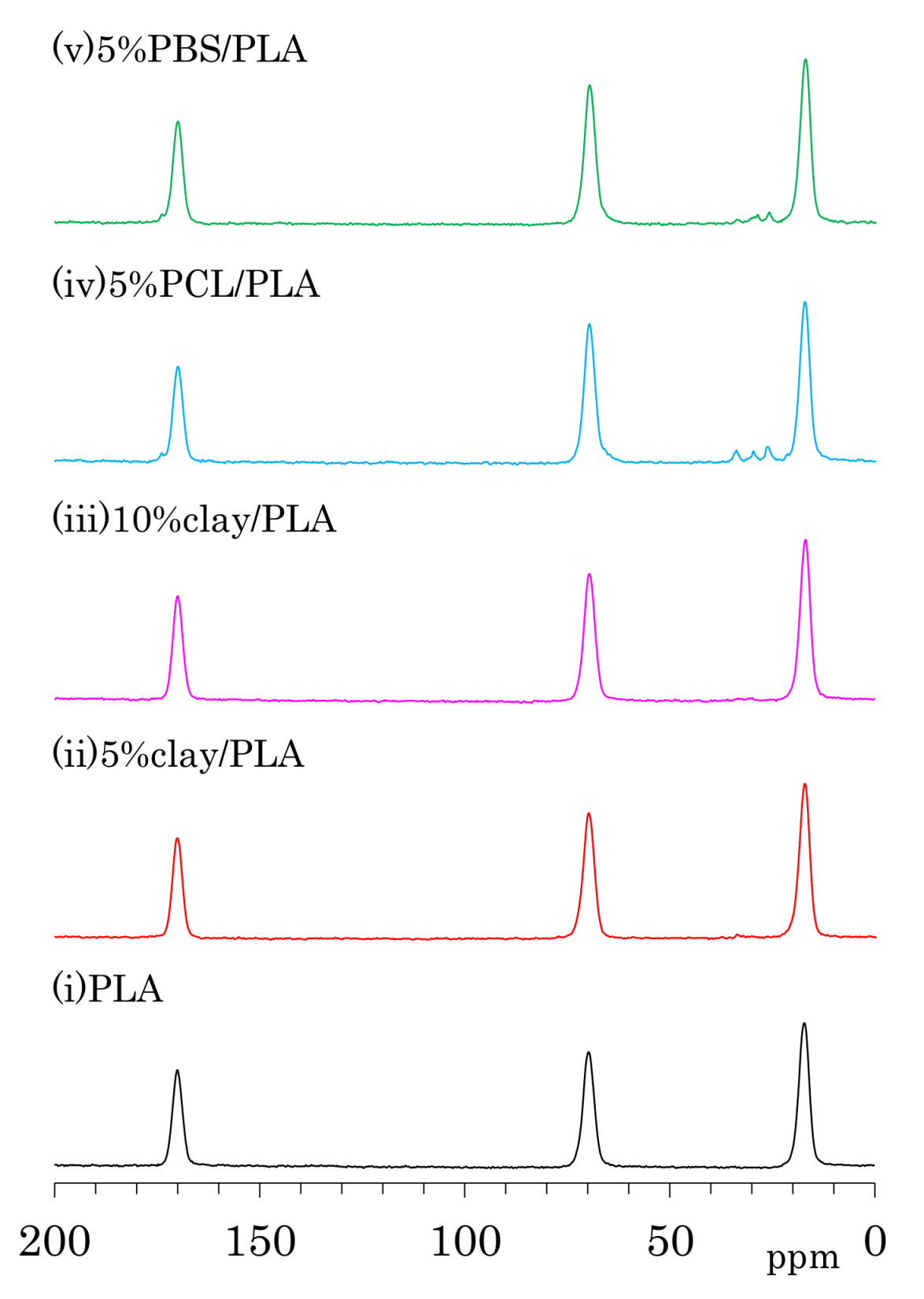
3.2. 13C CP-MAS NMR Spectra of PLA Fibers
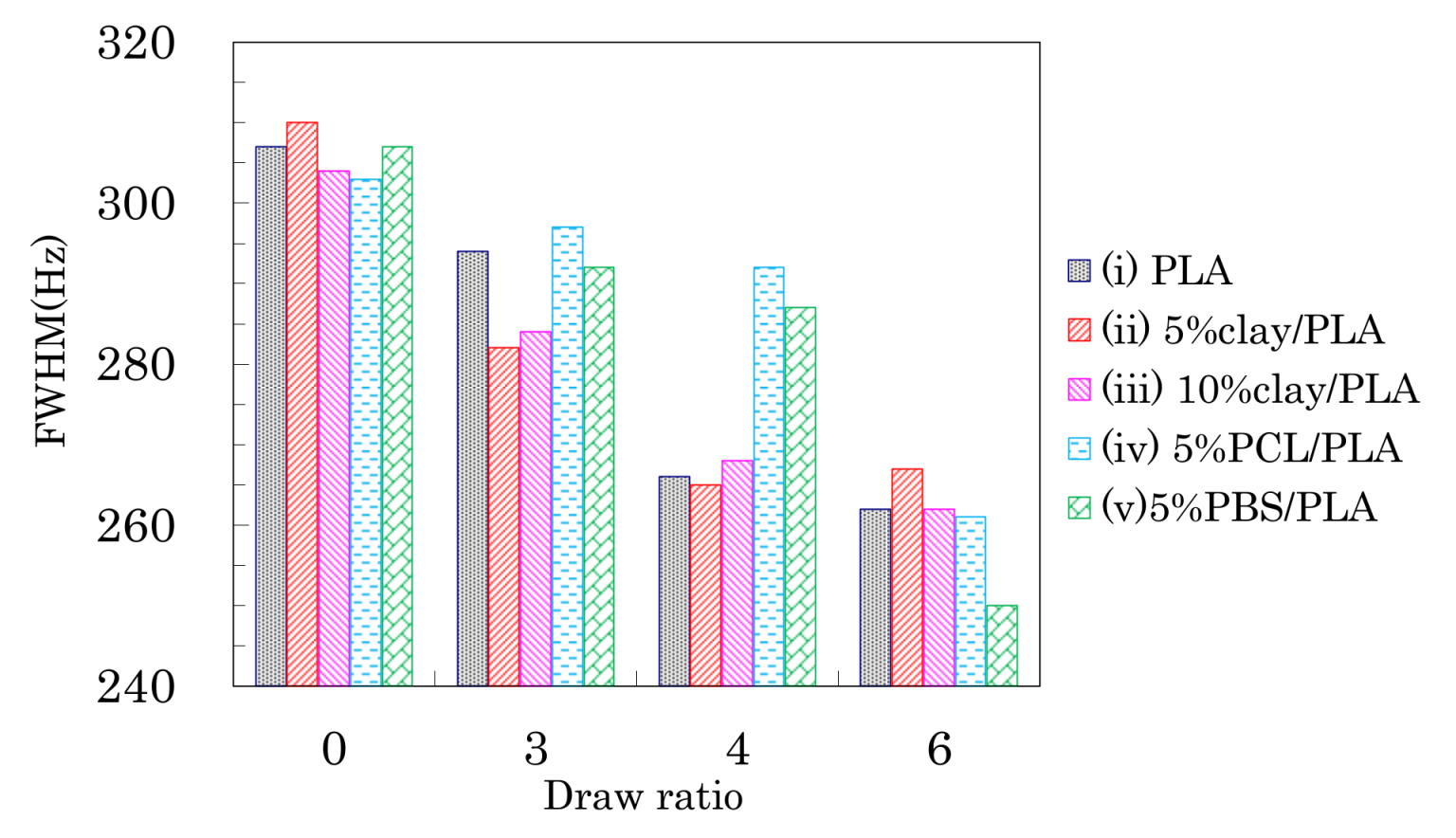
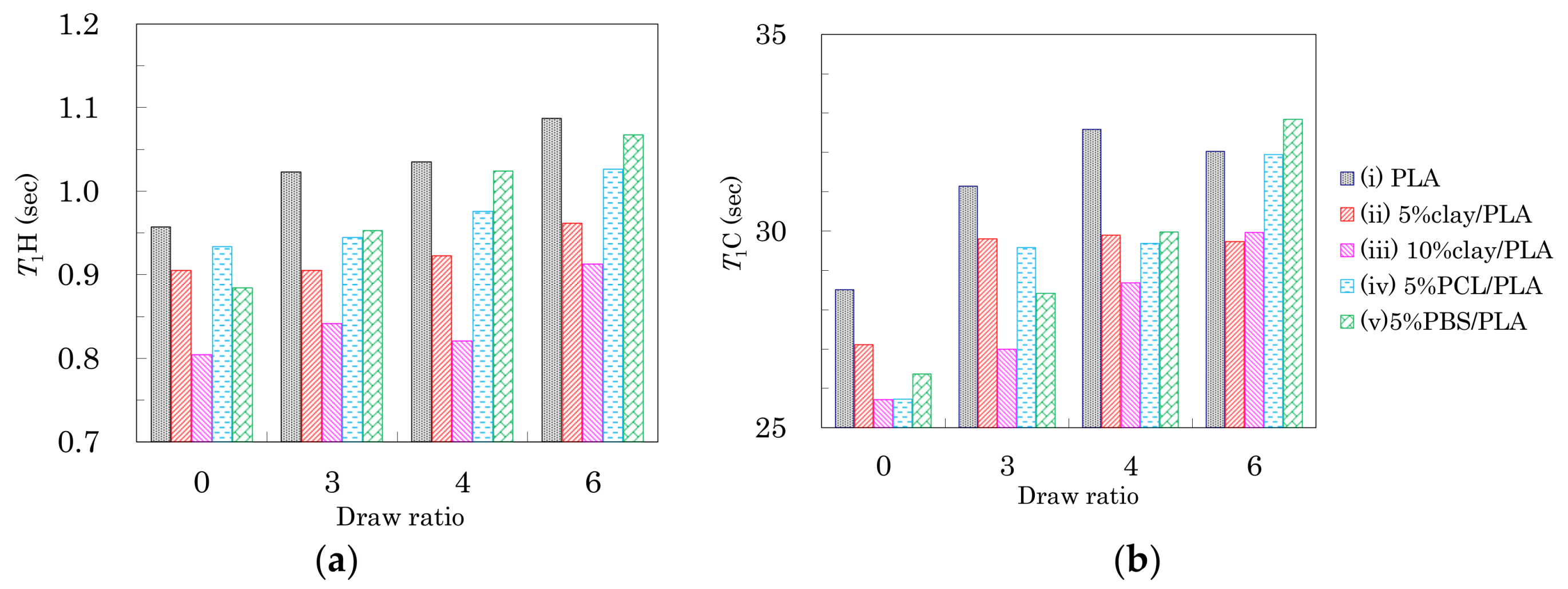
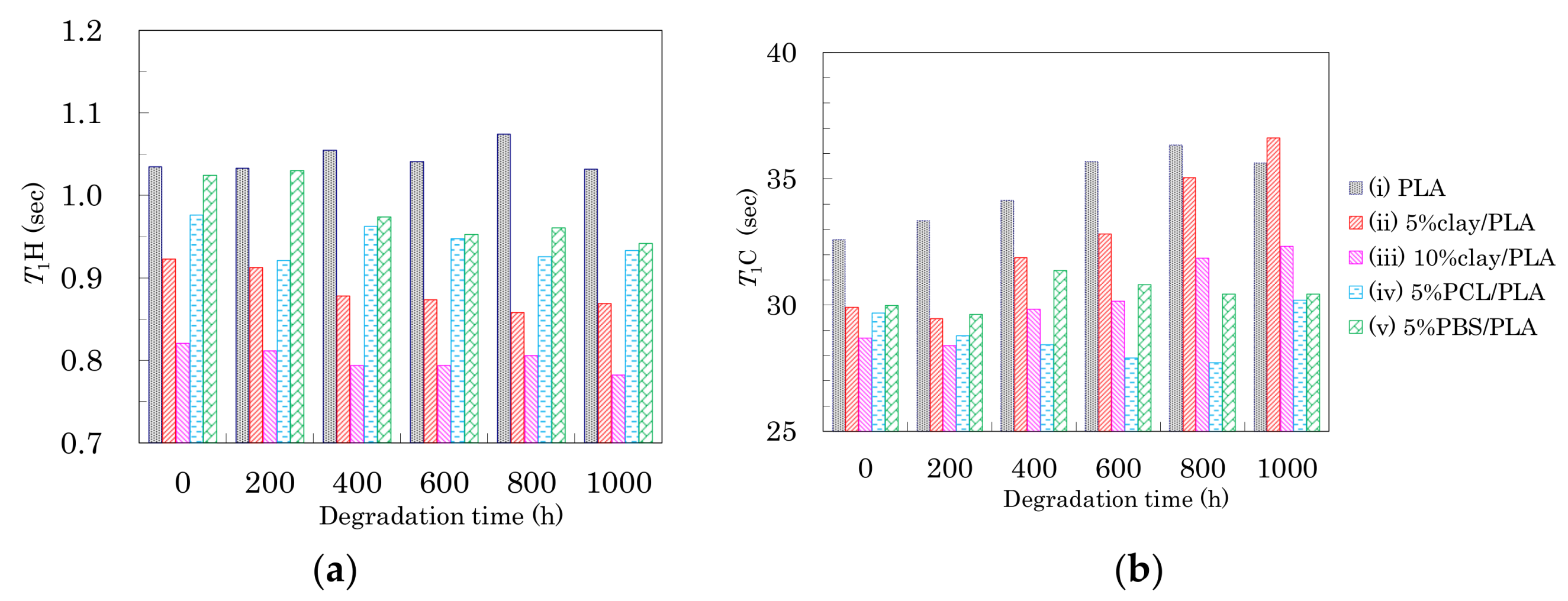
3.3. Nuclear Magnetic Relaxation Times of PLA Fibers
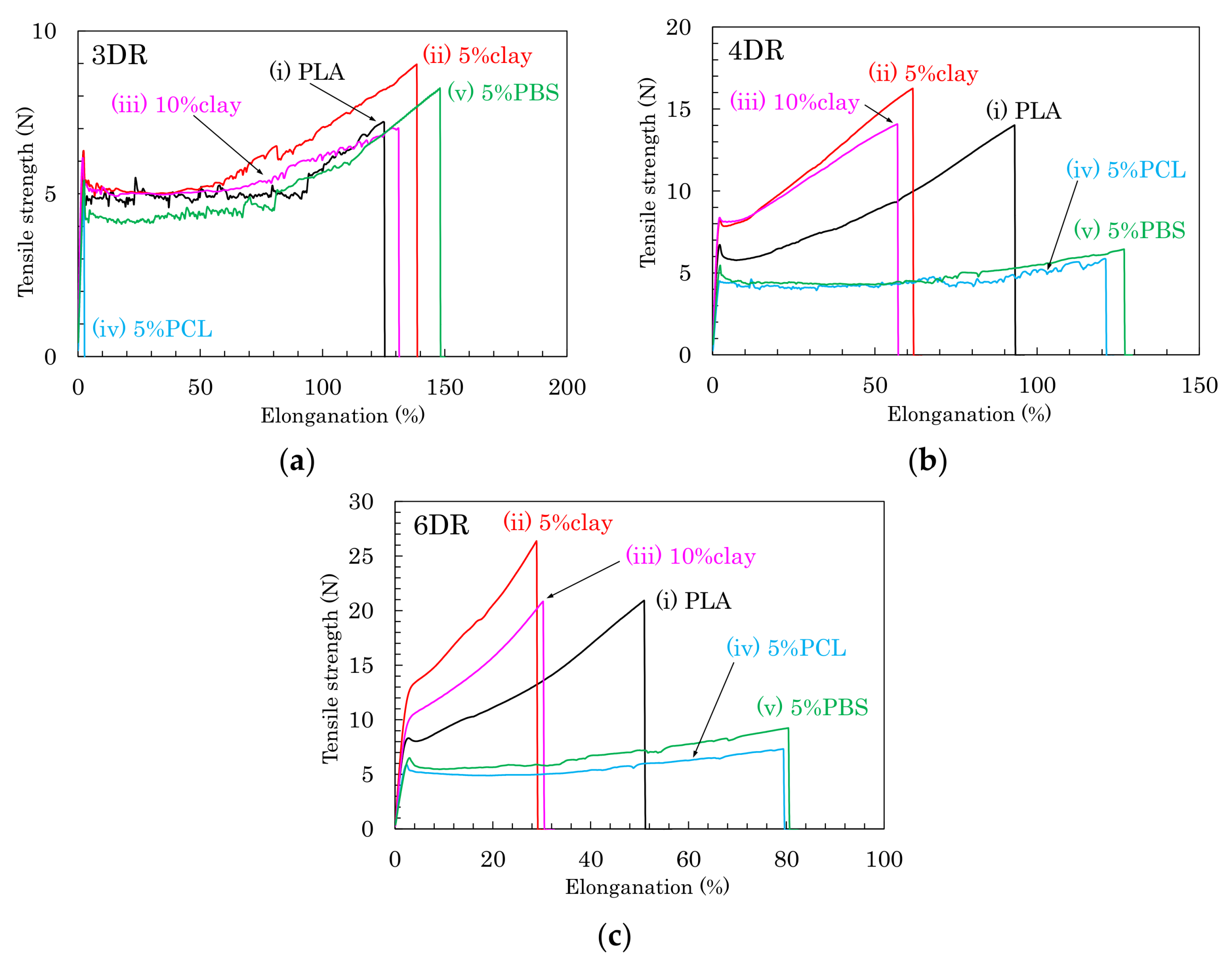
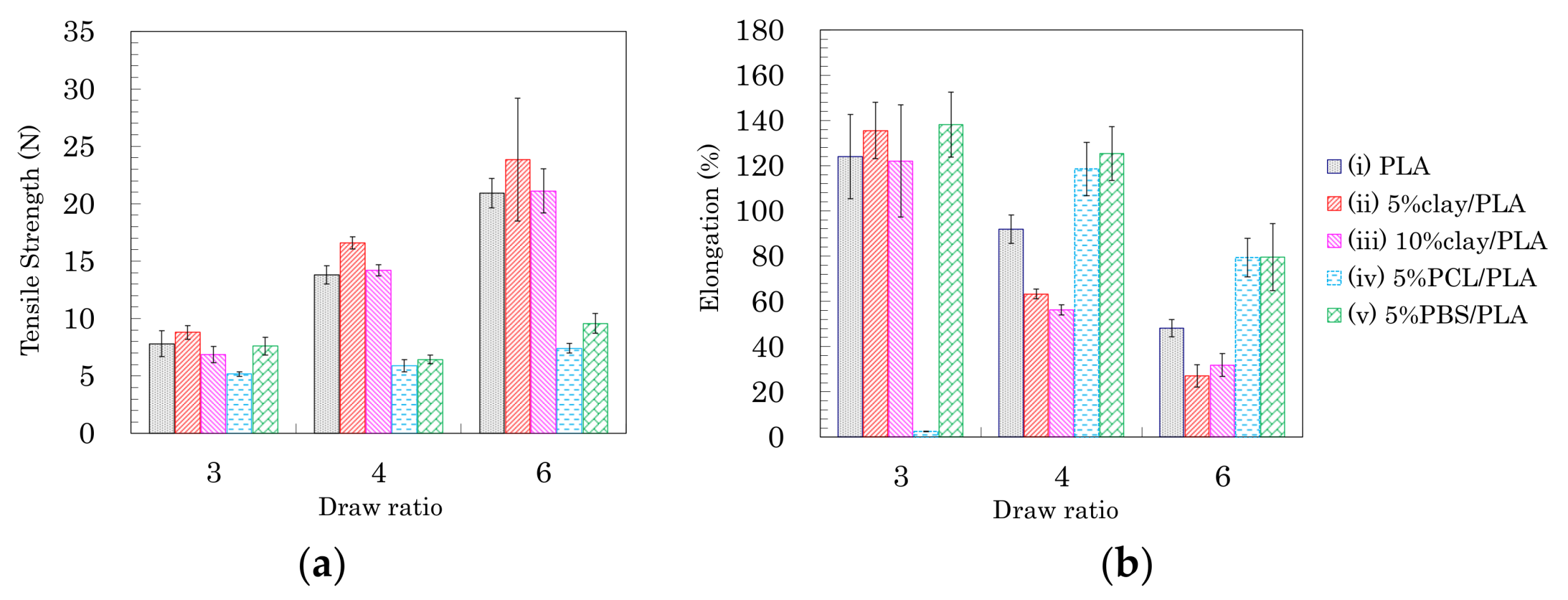
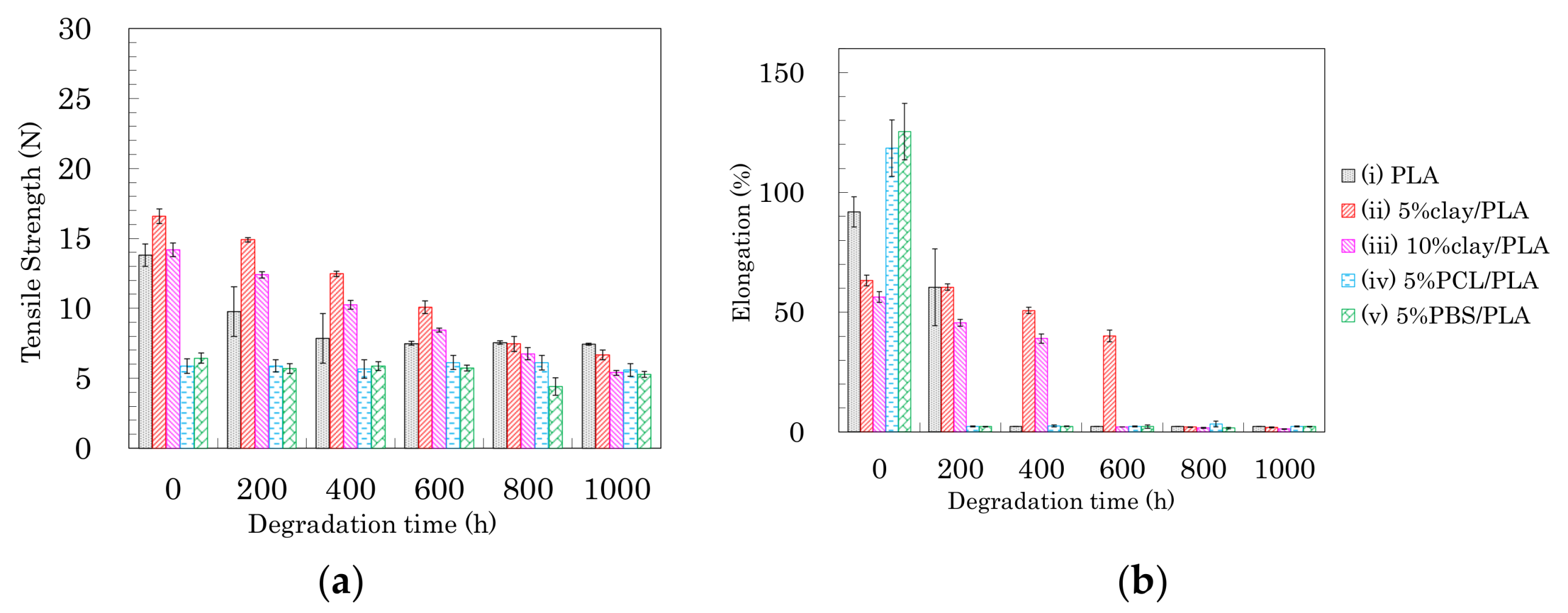
3.4. Tensile Properties of the Drawn PLA Fibers
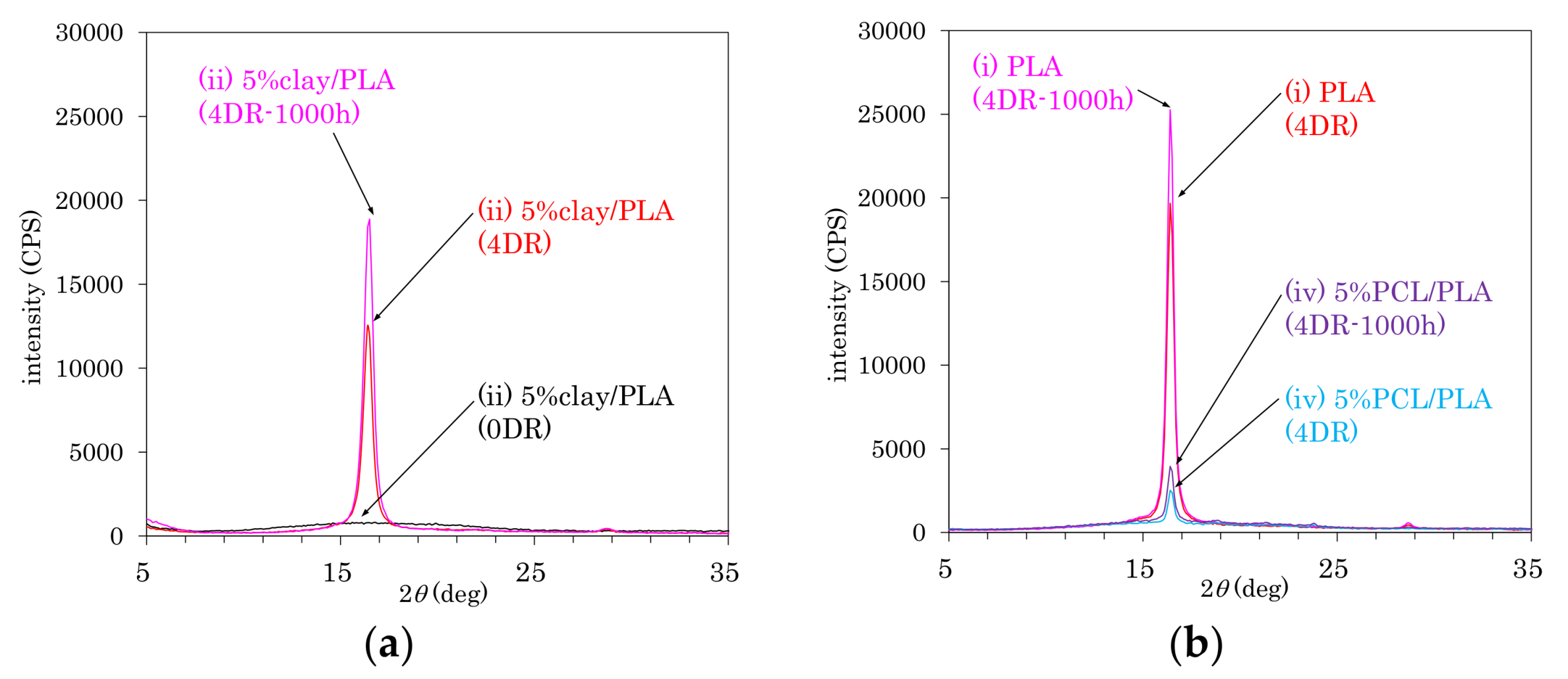
3.5. Accelerated Weathering Degradation—Changes of 13C CP-MAS NMR Spectra
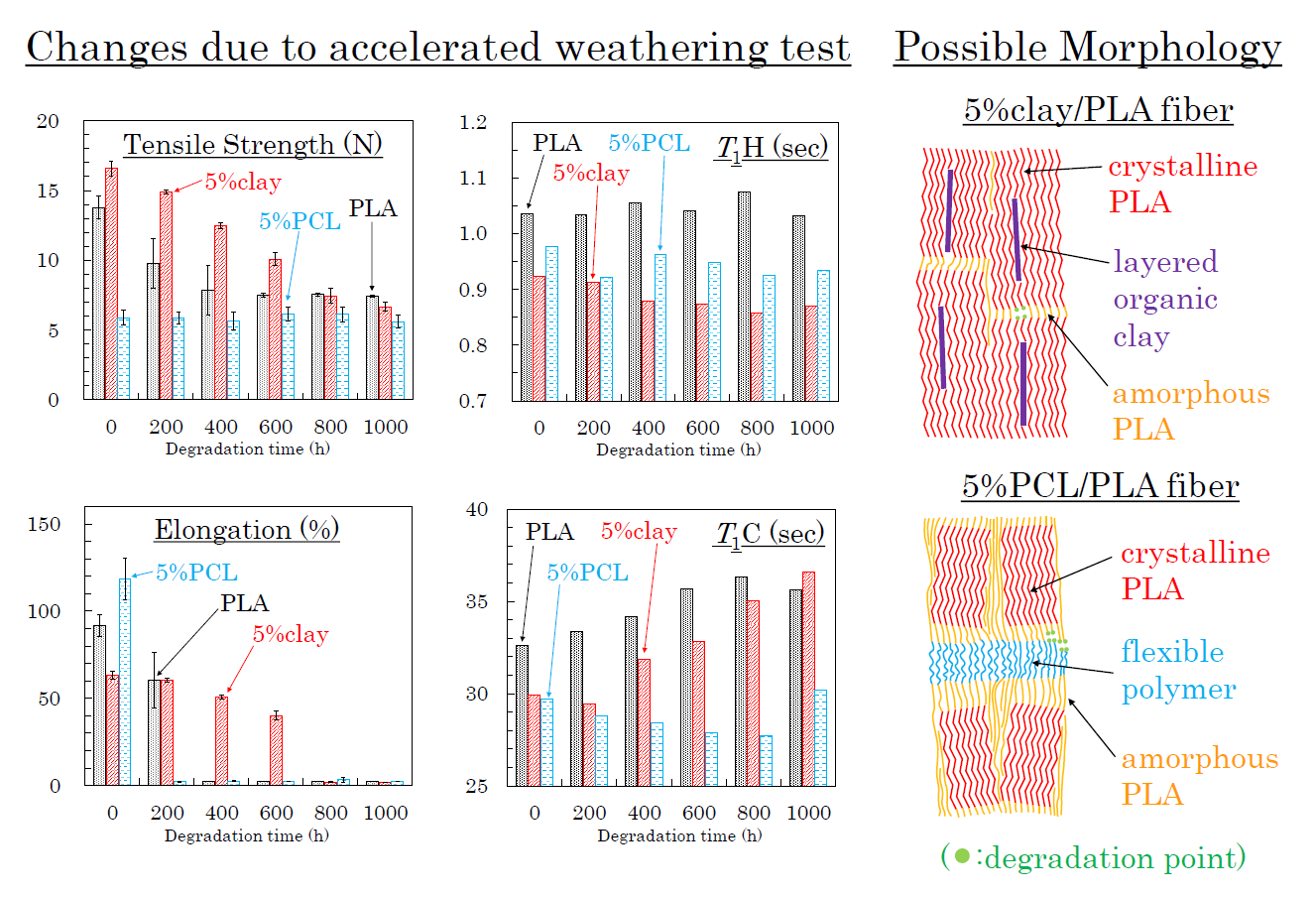
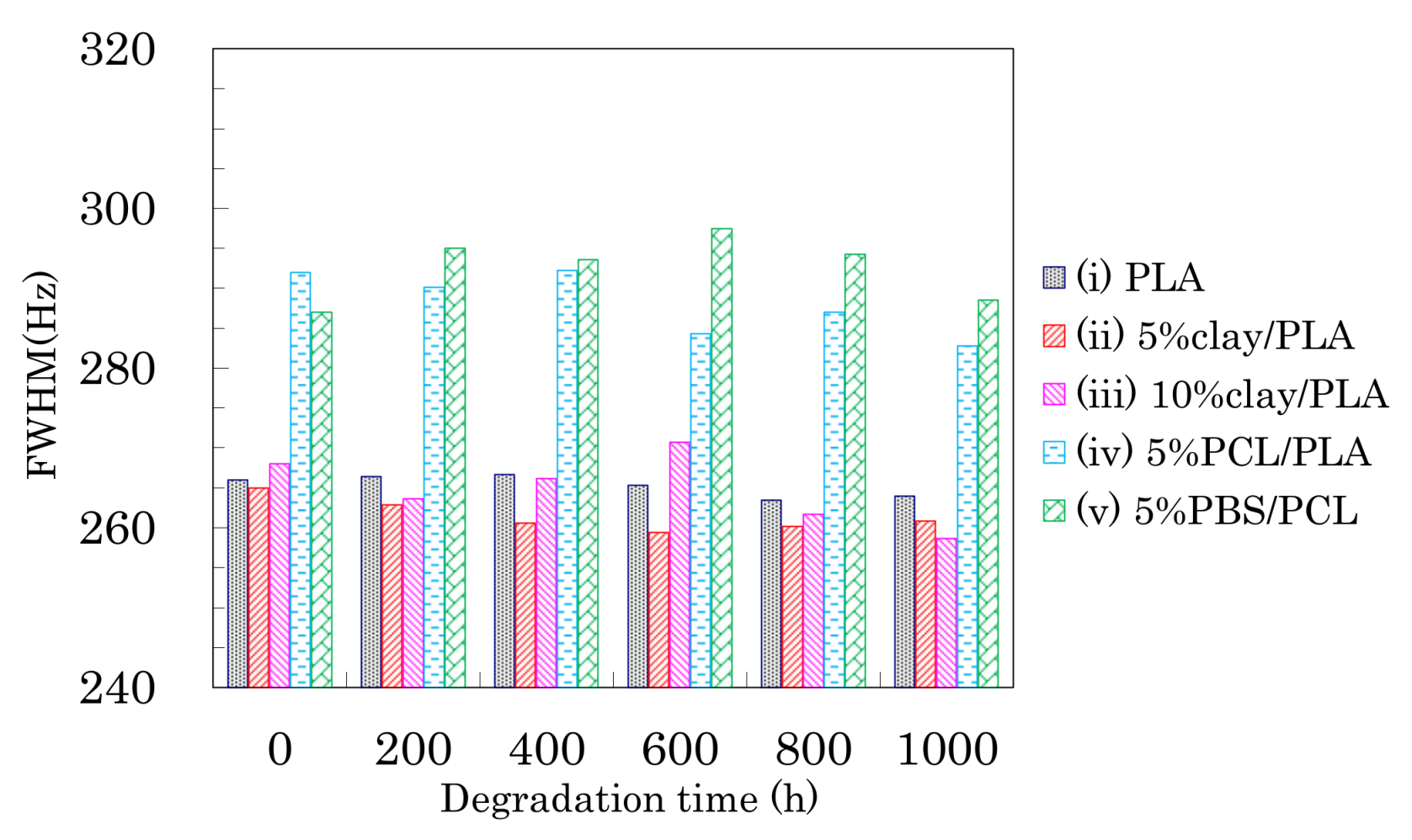
3.6. Accelerated Weathering Degradation—Changes of Nuclear Magnetic Relaxation Times
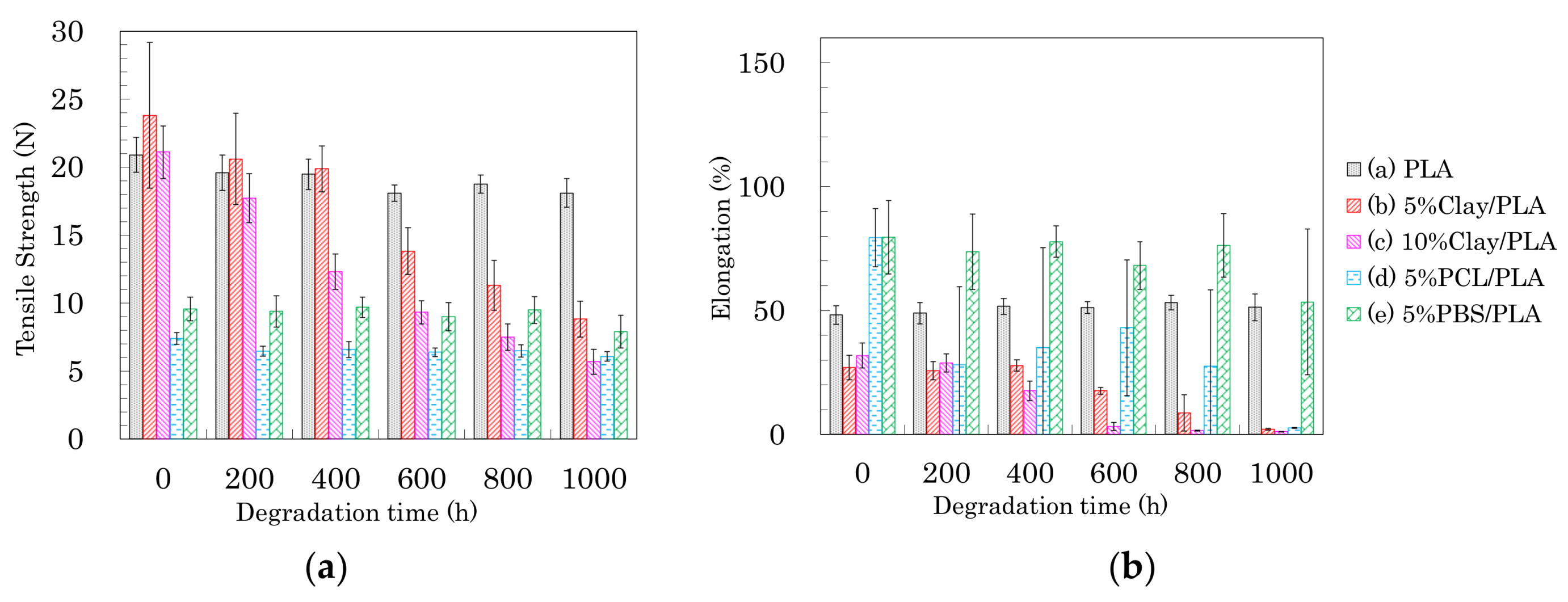
3.7. Accelerated Weathering Degradation—Changes of Tensile Properties
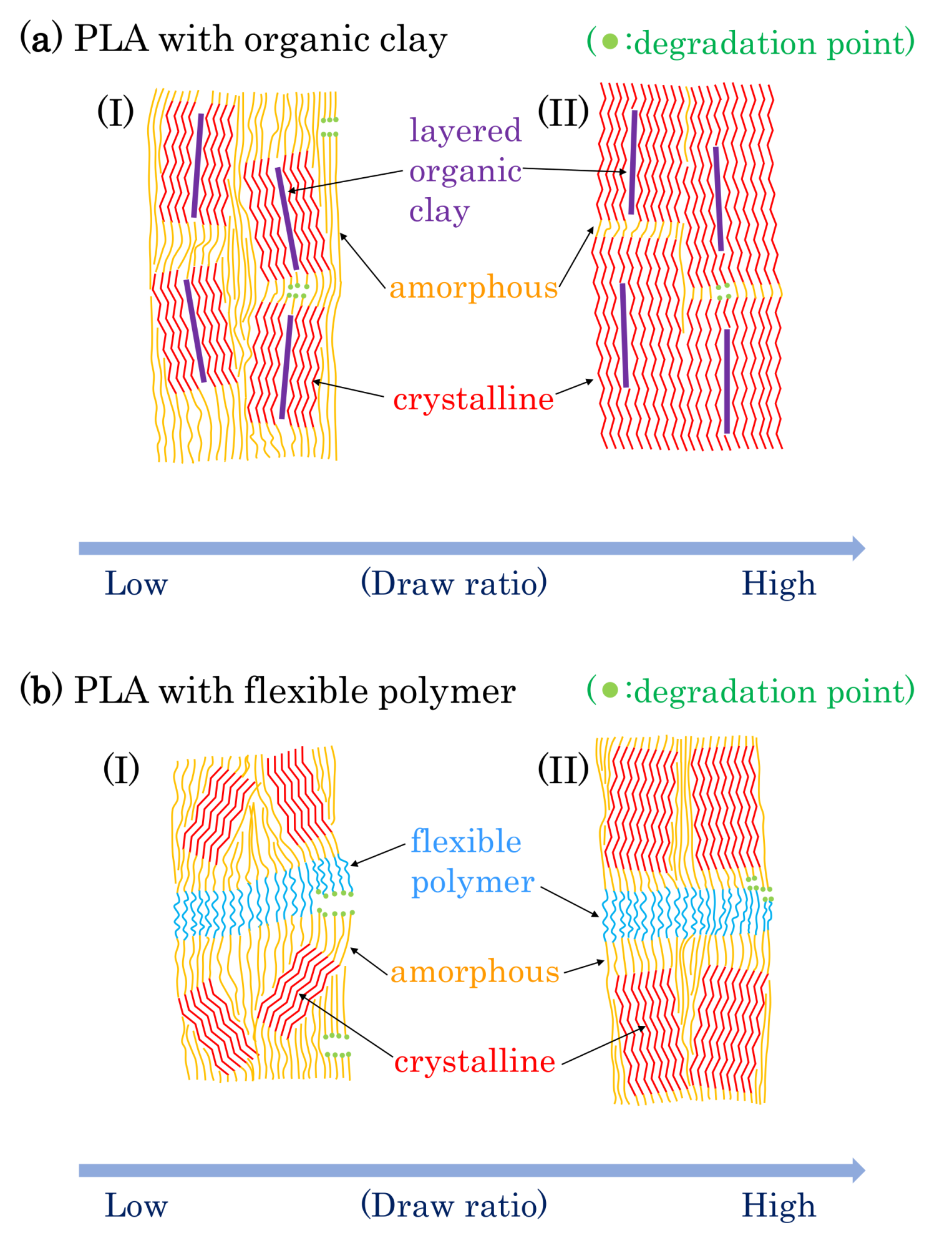
3.8. Molpholiogical Changes Due to Accelerated Weathering Degradation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nampoothiri, K.M.; Nair, N.P.; John, R.H. An overview of the recent developments in polylactide (PLA) research. Bioresour. Technol. 2010, 101, 8493–8501. [Google Scholar] [CrossRef] [PubMed]
- Rasal, R.M.; Janorkar, A.V.; Hirt, D.E. Poly(lactic acid) modification. Prog. Polym. Sci. 2010, 35, 338–356. [Google Scholar] [CrossRef]
- Lim, L.-T.; Auras, R.; Rubino, M. Processing technologies for poly(lactic acid). Prog. Polym. Sci. 2008, 33, 820–852. [Google Scholar] [CrossRef]
- Saeidlou, S.; Huneault, M.A.; Li, H.; Park, C.B. Poly(lactic acid) crystallization. Prog. Polym. Sci. 2012, 37, 1657–1677. [Google Scholar] [CrossRef]
- Pan, P.; Kai, W.; Zhu, B.; Dong, T.; Inoue, Y. Polymorphous crystallization and multiple melting behavior of poly(l-lactide): Molecular weight dependence. Macromolecules 2007, 40, 6898–6905. [Google Scholar] [CrossRef]
- Zhang, J.; Tashiro, K.; Tsuji, H.; Domb, A.J. Disorder-to-Order Phase Transition and Multiple Melting Behavior of Poly(l-lactide) Investigated by Simultaneous Measurements of WAXD and DSC. Macromolecules 2008, 41, 1352–1357. [Google Scholar] [CrossRef]
- Raquez, J.-M.; Habibi, Y.; Murariu, M.; Dubois, P. Polylactide (PLA)-based nanocomposites. Prog. Polym. Sci. 2013, 38, 1504–1542. [Google Scholar] [CrossRef]
- Graupner, N.; Herrmann, A.S.; Müssig, J. Natural and man-made cellulose fibre-reinforced poly(lactic acid) (PLA) composites: An overview about mechanical characteristics and application areas. Composites A 2009, 81, 810–821. [Google Scholar] [CrossRef]
- Oksman, K.; Skrifvars, M.; Selinc, J.-F. Natural fibres as reinforcement in polylactic acid (PLA) composites. Compos. Sci. Technol. 2003, 63, 1317–1324. [Google Scholar] [CrossRef]
- Fukushima, K.; Tabuani, D.; Arena, M.; Gennari, M.; Camino, G. Effect of clay type and loading on thermal, mechanical properties and biodegradation of poly(lactic acid) nanocomposites. React. Funct. Polym. 2013, 73, 540–549. [Google Scholar] [CrossRef]
- Marra, S.I.; Zuburtikudis, I.; Panayiotou, C. Nanostructure vs. microstructure: Morphological and thermomechanical characterization of poly(l-lactic acid)/layered silicate hybrids. Eur. Polym. J. 2007, 43, 2191–2206. [Google Scholar] [CrossRef]
- Navarro-Baena, I.; Sessini, V.; Dominici, F.; Torre, L.; Kenny, J.M.; Peponi, L. Design of biodegradable blends based on PLA and PCL: From morphological, thermal and mechanical studies to shape memory behavior. Polym. Degrad. Stab. 2016, 132, 97–108. [Google Scholar] [CrossRef]
- Harada, M.; Ohya, T.; Iida, K.; Hayashi, H.; Hirano, K.; Fukuda, H. Increased impact strength of biodegradable poly(lactic acid)/poly(butylene succinate) blend composites by using isocyanate as a reactive processing agent. J. Appl. Polym. Sci. 2007, 106, 1813–1820. [Google Scholar] [CrossRef]
- Gupta, B.; Revagade, N.; Hilborn, J. Poly(lactic acid) fiber: An overview. Prog. Polym. Sci. 2007, 32, 455–482. [Google Scholar] [CrossRef]
- Schmack, G.; Tändler, B.; Vogel, R.; Beyreuther, R.; Jacobsen, S.; Fritz, H.-G. Biodegradable fibers of poly(l-lactide) produced by high-speed melt spinning and spin drawing. J. Appl. Polym. Sci. 1999, 73, 2785–2797. [Google Scholar] [CrossRef]
- Cicero, J.A.; Dorgan, J.R.; Garrett, J.; Runt, J.; Lin, J.S. Effects of molecular architecture on two-step, melt-spun poly(lactic acid) fibers. J. Appl. Polym. Sci. 2002, 86, 2839–2846. [Google Scholar] [CrossRef]
- Yuan, X.; Mak, A.F.T.; Kwok, K.W.; Yung, B.K.O.; Yao, K. Characterization of poly(l-lactic acid) fibers produced by melt spinning. J. Appl. Polym. Sci. 2001, 81, 251–260. [Google Scholar] [CrossRef]
- Hossain, K.M.Z.; Parsons, A.J.; Rudd, C.D.; Ahmed, I.; Thielemans, W. Mechanical, crystallisation and moisture absorption properties of melt drawn polylactic acid fibres. Eur. Polym. J. 2014, 53, 270–281. [Google Scholar] [CrossRef]
- Solarski, S.; Ferreira, M.; Devaux, E. Characterization of the thermal properties of PLA fibers by modulated differential scanning calorimetry. Polymer 2005, 46, 11187–11192. [Google Scholar] [CrossRef]
- Solarski, S.; Ferreira, M.; Devaux, E.; Fontaine, G.; Bachelet, P.; Bourbigot, S.; Delobel, R.; Coszach, P.; Murariu, M.; Ferreira, A.D.; et al. Designing polylactide/clay nanocomposites for textile applications: Effect of processing conditions, spinning, and characterization. J. Appl. Polym. Sci. 2007, 109, 841–851. [Google Scholar] [CrossRef]
- Jompang, L.; Thumsorn, S.; On, J.W.; Surin, P.; Apawet, C.; Chaichalermwong, T.; Kaabbuathong, N.; O-Charoen, N.; Srisawat, N. Poly(lactic acid) and poly(butylene succinate) blend fibers prepared by melt spinning technique. Energy Procedia 2013, 34, 493–499. [Google Scholar] [CrossRef]
- Nishimura, Y.; Takasu, A.; Inai, Y.; Hirabayashi, T. Melt spinning of poly(l-lactic acid) and its biodegradability. J. Appl. Polym. Sci. 2005, 97, 2118–2124. [Google Scholar] [CrossRef]
- Shinzawa, H.; Nishida, M.; Tanaka, T.; Kanematsu, W. Accelerated weathering-induced degradation of poly(lactic acid) fiber studied by near-infrared (NIR) hyperspectral imaging. Appl. Spectrosc. 2012, 66, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Thakur, K.A.M.; Kean, R.T.; Zupfer, J.M.; Buehler, N.U.; Doscotch, M.A.; Munson, E.J. Solid state 13C CP-MAS NMR studies of the crystallinity and morphology of poly(l-lactide). Macromolecules 1996, 29, 8844–8851. [Google Scholar] [CrossRef]
- Zell, M.T.; Padden, B.E.; Paterick, A.J.; Hillmyer, M.A.; Kean, R.T.; Thakur, K.A.M.; Munson, E.J. Direct observation of stereodefect sites in semicrystalline poly(lactide) using 13C solid-state NMR. J. Am. Chem. Soc. 1998, 120, 12672–12673. [Google Scholar] [CrossRef]
- Chen, W.; Reichert, D.; Miyoshi, T. Helical jump motions of poly(l-lactic acid) chains in the α phase as revealed by solid-state NMR. J. Phys. Chem. B 2015, 119, 4552–4563. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, T.; Jaworska, M.; Potrzebowski, M.J. NMR crystallography of α-poly(l-lactide). Phys. Chem. Chem. Phys. 2013, 15, 3137–3145. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.; Han, L.; Shan, G.; Bao, Y. Heating and annealing induced structural reorganization and embrittlement of solution-crystallized poly(l-lactic acid). Macromolecules 2014, 47, 8126–8130. [Google Scholar] [CrossRef]
- Zhang, X.; Espiritu, M.; Bilyk, A.; Kurniawan, L. Morphological behaviour of poly(lactic acid) during hydrolytic degradation. Polym. Degrad. Stab. 2008, 93, 1964–1970. [Google Scholar] [CrossRef]
- Tsuji, H.; Kamo, S.; Horii, F. Solid-state 13C NMR analyses of the structures of crystallized and quenched poly(lactide)s: Effects of crystallinity, water absorption, hydrolytic degradation and tacticity. Polymer 2010, 51, 2215–2220. [Google Scholar] [CrossRef]
- Nishida, M.; Tanaka, T.; Yamaguchi, T.; Suzuki, K.; Kanematsu, W. Intercalative polymerization of l-lactide with organically modified clay by a reactive extrusion method and instrumental analyses of the poly(lactic acid)/clay nanocomposites. J. Appl. Polym. Sci. 2012, 125, E681–E690. [Google Scholar] [CrossRef]
- Nishida, M.; Nishimura, Y.; Tanaka, T.; Oonishi, M.; Kanematsu, W. Solid state NMR analysis of poly(l-lactide) random copolymer with poly(ε-caprolactone) and its reactive extrusion process. J. Appl. Polym. Sci. 2012, 123, 1865–1873. [Google Scholar] [CrossRef]
- Nishida, M.; Tanaka, T.; Miki, T.; Hayakawa, Y.; Kanayama, K. Integrated analysis of solid-state NMR spectra and nuclear magnetic relaxation times for the phenol formaldehyde (PF) resin impregnation process into soft wood. RSC Adv. 2017, 7, 54532–54541. [Google Scholar] [CrossRef]
- Nishida, M.; Tanaka, T.; Miki, T.; Hayakawa, Y.; Kanayama, K. Instrumental analyses of nanostructures and interactions with water molecules of biomass constituents of Japanese cypress. Cellulose 2017, 24, 5295–5312. [Google Scholar] [CrossRef]
- Nishida, M.; Tanaka, T.; Miki, T.; Ito, T.; Kanayama, K. Instrumental analyses of nanostructures and interactions with bound water of superheated steam treated plant materials. Ind. Crop. Prod. 2018, 114, 1–13. [Google Scholar] [CrossRef]
- Torchia, D.A. The Measurement of Proton-enhanced carbon-13 T1 values by a method which suppresses artifacts. J. Magn. Reson. 1978, 30, 613–616. [Google Scholar] [CrossRef]
- Testing Methods for Man-Made Filament Yarns, [JIS] Japanese Industrial Standards Committee, JIS L-1013; Japanese Standard Association: Tokyo, Japan, 2010. [CrossRef]
- Shinzawa, H.; Nishida, M.; Kanematsu, W.; Tanaka, T.; Suzuki, K.; Noda, I. Parallel factor (PARAFAC) kernel analysis of temperature- and composition dependent NMR spectra of poly(lactic acid) nanocomposites. Analyst 2012, 137, 1913–1921. [Google Scholar] [CrossRef] [PubMed]
- Gupta, B.; Revagade, N.; Anjum, N.; Atthoff, B.; Hilborn, J. Preparation of poly(lactic acid) fiber by dry-jet-wet-spinning. I. Influence of draw ratio on fiber properties. J. Appl. Polym. Sci. 2006, 100, 1239–1246. [Google Scholar] [CrossRef]















© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishida, M.; Tanaka, T.; Tanaka, T.; Hayakawa, Y. Nucleating and Plasticization Effects in Drawn Poly(Lactic Acid) Fiber during Accelerated Weathering Degradation. Polymers 2018, 10, 365. https://doi.org/10.3390/polym10040365
Nishida M, Tanaka T, Tanaka T, Hayakawa Y. Nucleating and Plasticization Effects in Drawn Poly(Lactic Acid) Fiber during Accelerated Weathering Degradation. Polymers. 2018; 10(4):365. https://doi.org/10.3390/polym10040365
Chicago/Turabian StyleNishida, Masakazu, Tomoko Tanaka, Toshiyuki Tanaka, and Yoshio Hayakawa. 2018. "Nucleating and Plasticization Effects in Drawn Poly(Lactic Acid) Fiber during Accelerated Weathering Degradation" Polymers 10, no. 4: 365. https://doi.org/10.3390/polym10040365
APA StyleNishida, M., Tanaka, T., Tanaka, T., & Hayakawa, Y. (2018). Nucleating and Plasticization Effects in Drawn Poly(Lactic Acid) Fiber during Accelerated Weathering Degradation. Polymers, 10(4), 365. https://doi.org/10.3390/polym10040365