1. Introduction
Due to popular awareness of sustainability, polymers obtained from renewable sources have been developed in the last decade, with the purpose of replacing those obtained from fossil resources [
1,
2,
3,
4,
5].
One renewable monomer that has attracted much attention is 2,5-furandicarboxylic acid (FDCA), an aromatic building block obtained from C5 and C6 sugars, that is able to replace terephthalic acid, a petrochemical compound widely used for the preparation of aromatic polyesters such as PET or PBT [
6,
7,
8]. Poly(ethylene furanoate) (PEF) has been extensively studied because it has not only similar properties to PET, but also improved gas barrier properties, which make it a serious alternative for applications in soft drink bottles. In contrast, poly(butylene furanoate) (PBF) has been much less studied; as such, the knowledge available on this polyester is relatively scarce. PBF is a semicrystalline polymer with a melting temperature of 172 °C and a glass transition temperature of 39 °C [
9]. As with PBT, the presence of the butylene segment in the repeating unit of PBF confers to this polymer a strong propensity for rapid crystallization, which is inconvenient for some injection molding processes due to the excessive mold shrinkage. In order to overcome these problems, one solution is copolymerization. The insertion of either a diol or diacid comonomeric unit in small quantities in the PBF chain decreases both the melting temperature and enthalpy, therefore reducing processing costs. Copolymerization has been applied to various technical polyesters in order to tune their thermal properties, such as crystallizability, melting or glass transition temperatures [
10]. PBF copolyesters with enhanced biodegradability have already been prepared by either melt polycondensation [
11,
12,
13,
14] or ring opening polymerization (ROP) of cyclic oligomers [
15,
16]. This last method, which uses cyclic oligomers for the synthesis, has some advantages because it does not require by-product removal during reaction, implies small or no heat exchange, and attains very high-molecular-weight polymers in reaction times of minutes. ROP for these systems was first examined in detail by Brunelle [
17] and recently reviewed by Hodge [
18] and Strandman et al. [
19]. The technique has been successfully used by us to prepare various PEF and PBF copolyesters with enhanced properties [
20,
21].
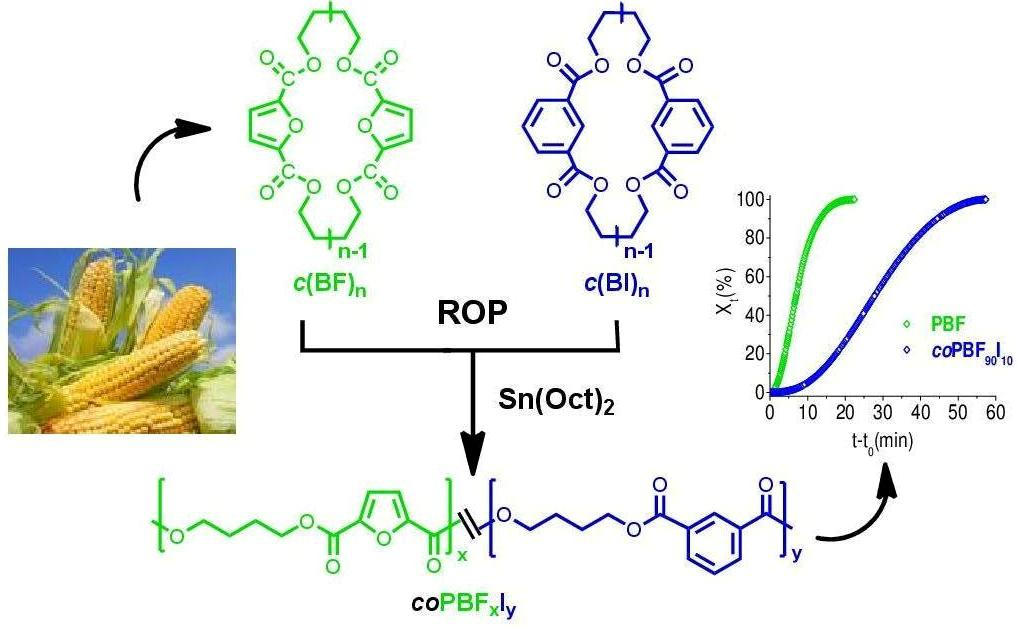
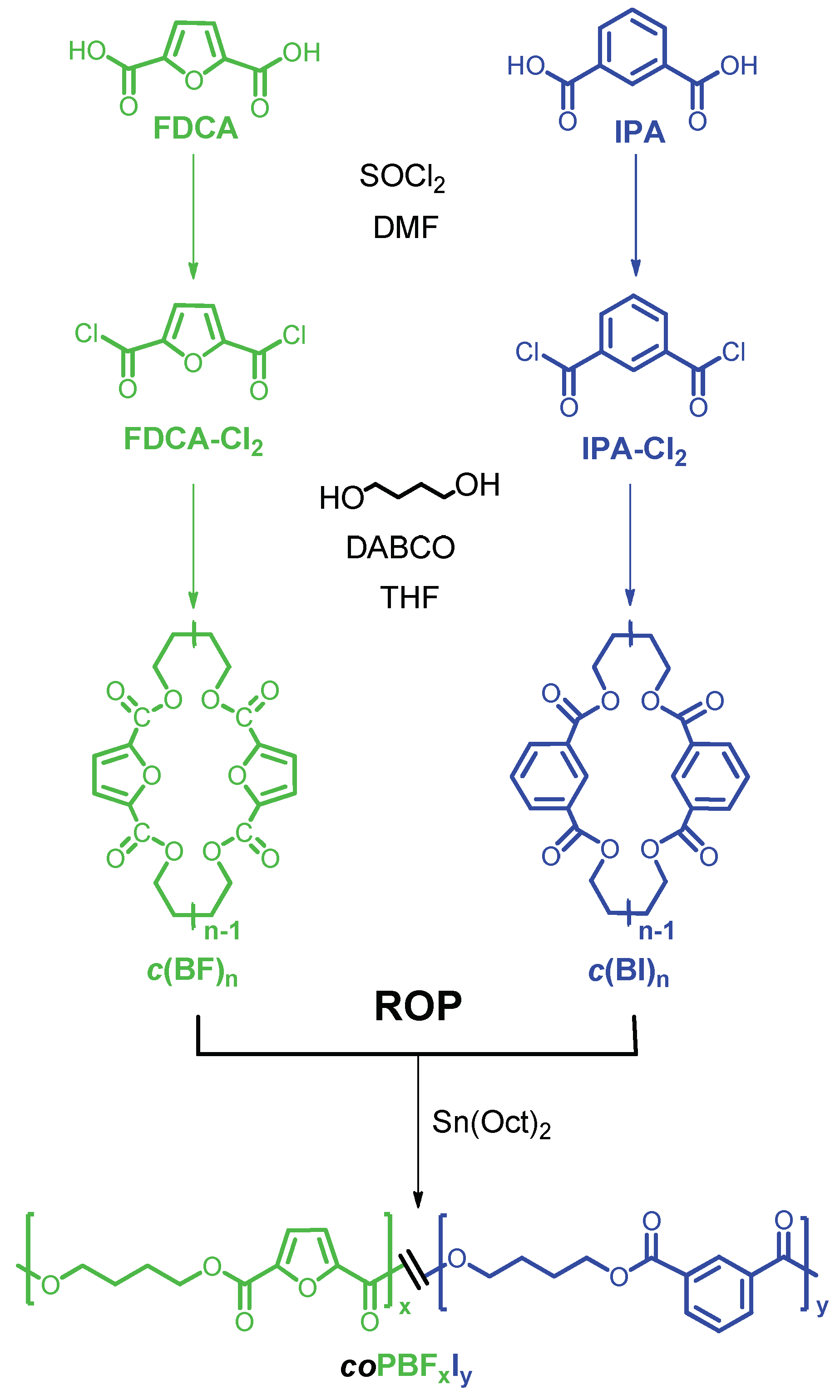
In this work we would like to report on the synthesis and characterization, evaluation of thermal properties, and crystallization behavior of new, partially renewable PBF copolyesters containing isophthalate units that are prepared by ROP of mixtures of cyclic butylene furanoate and butylene isophthalate oligomers. The 3D chemical structures of FDCA and isophthalic acid (IPA) are depicted in
Scheme 1.
2. Materials and Methods
2.1. Materials
2,5-Furandicarboxylic acid (FDCA, >98% purity) was purchased from Satachem (Shanghai, China). Isophthalic acid (IPA, 99%), 1,4-butanediol (BD), thionyl chloride (SOCl2, 99%), and 1,4-di-azabicyclo[2.2.2]octane (DABCO, 99%) and tin(II) ethylhexanoate (Sn(Oct)2, 99%) catalysts were purchased from Sigma-Aldrich Co. Triethylamine (Et3N, 98%) was purchased from Panreac. Solvents used for reaction, isolation and purification were of high-purity grade and used as received except tetrahydrofuran (THF) that was dried on 3 Å-molecular sieves. The DABCO catalyst was purified by sublimation.
2.2. Methods
1H- and 13C-NMR spectra were recorded on a Bruker AMX-300 spectrometer (Billerica, MA, USA) at 25 °C, operating at 300.1 and 75.5 MHz, respectively. For NMR analysis, monomers, cyclic oligomers and intermediate compounds were dissolved in deuterated chloroform (CDCl3) and polymers in pure CDCl3 or in a mixture of trifluoroacetic acid (TFA) and CDCl3 (1:8). About 10 and 50 mg of sample in 1 mL of solvent were used for 1H- and 13C-NMR, respectively. Sixty-four scans were recorded for 1H, and between 1000 and 10,000 scans for 13C-NMR. Spectra were internally referenced to tetramethylsilane (TMS).
High-performance liquid chromatography (HPLC) analysis was performed at 25 °C in a Waters apparatus equipped with a UV detector of Applied Biosystems operating at 254 nm wavelength, and a Scharlau Science column (Si60, 5 μm; 250 × 4.6 mm). Cyclic oligomers (1 mg) were dissolved in chloroform (1 mL) and eluted with hexane/1,4-dioxane 70/30 (v/v) at a flow rate of 1.0 mL·min−1. Molecular weight analysis was performed by GPC on a Waters equipment provided with RI and UV detectors. 100 μL of 0.1% (w/v) sample solution were injected and chromatographed with a flow of 0.5 mL·min−1 of 1,1,1,3,3,3-hexafluoroisopropanol (HFIP). HR5E and HR2 Waters linear Styragel columns (7.8 mm × 300 mm, pore size 103–104 Å) were packed with crosslinked polystyrene and protected with a precolumn. Molar mass average and distributions were calculated against PMMA standards.
Matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) mass spectra were recorded in a 4700 Proteomics Analyzer instrument (Applied Biosystems, Foster City, CA, USA) at the Proteomics Platform of Barcelona Science Park, University of Barcelona. Spectra acquisition was performed in the MS reflector positive-ion mode. About 0.1 mg of sample was dissolved in 50 μL of DCM and 2 μL of this solution was mixed with an equal volume of DCM solution of anthracene (10 mg·mL−1); the mixture was then left to evaporate to dryness onto the stainless steel plate of the analyzer. The residue was then covered with 2 μL of a solution of 2,5-dihydroxibenzoic acid in acetonitrile/H2O (1/1) containing 0.1% TFA, and the mixture was left to dry prior to exposition to the laser beam.
The thermal behavior of cyclic compounds and polymers were examined by differential scanning calorimetry (DSC), using a Perkin-Elmer Pyris 1 apparatus (Waltam, MA, USA). The thermograms were recorded from 3 to 6 mg samples at heating and cooling rates of 10 °C·min−1 under a nitrogen flow of 20 mL·min−1. Indium and zinc were used as standards for temperature and enthalpy calibration. The glass transition temperature (Tg) was taken as the inflection point of the heating DSC traces recorded at 20 °C·min−1 from melt-quenched samples, and the melting temperature (Tm) was taken as the maximum of the endothermic peak appearing on heating traces. Thermogravimetric analyses were performed on a Mettler-Toledo TGA/DSC 1 Star System under a nitrogen flow of 20 mL·min−1 at a heating rate of 10 °C·min−1 and within a temperature range of 30 to 600 °C. X-ray diffraction patterns from powdered samples coming directly from synthesis were recorded on a PANalytical X’Pert PRO MPD θ/θ diffractometer using the CuKα radiation of wavelength 0.1542 nm.
2.3. Synthesis
2.3.1. Synthesis of Cyclic Oligomers
Cyclic oligomers of butylene 2,5-furandicarboxylate
c(BF)
n and butylene isophthalate
c(BI)
n were synthesized by high dilution condensation (HDC) from equimolar mixtures of BD and furandicarboxylic dichloride (FDCA-Cl
2) and isophthaloyl chloride (IPA-Cl
2), respectively, as previously reported Brunelle et al. [
22], and more recently by us [
23]. Briefly, a three necked round bottom flask charged with 250 mL of THF was cooled to 0 °C; 12.5 mmol (1.40 g) of DABCO was then added under stirring. 5 mmol (0.96 g) of FDCA-Cl
2 or 5 mmol (1.01 g) of IPA-Cl
2 in 10 mL and 5 mmol (0.46 g) of BD in THF were drop-wise added simultaneously for 40 min using two addition funnels, in order to maintain the reagents equimolarity in the reaction mixture. The reaction was finished by adding 1 mL of water, followed by 5 mL of 1M HCl; after stirring for 5 min, the mixture was diluted with DCM and filtered. The filtrate was washed with 0.1M HCl, dried on MgSO
4, and evaporated to dryness to render a mixture of linear and cyclic oligomers. Linear oligomers were removed by chromatography through a short column of silica gel using a cold mixture of DCM/diethyl ether 90/10 (
v/
v) as eluent.
c(BF)
n:
1H-NMR (
δ ppm, CDCl
3, 300 MHz): 7.23, 7.24, 7.25 (3s, 2H), 4.40 (m, 4H), 1.92, 1.99 (2m, 4H),
13C-NMR (
δ ppm, CDCl
3, 75.5 MHz): 158.1, 157.9, 146.7, 146.5, 118.7, 118.6, 118.5, 65.0, 64.8, 25.4.
c(BI)
n:
1H-NMR (
δ ppm, CDCl
3, 300 MHz): 8.62, 8.60 (2m, 1H), 8.26, 8.21 (2m, 2H), 7.56, 7.50 (2m, 1H), 4.45 (m, 4H), 2.01, 1.97 (2m, 4H).
13C-NMR (
δ ppm, CDCl
3, 75.5 MHz): 165.6, 165.5, 134.2, 133.8, 130.6, 130.5, 130.3 129.7, 128.8, 128.6, 64.8, 64.7, 64.6, 25.5, 25.4.
2.3.2. Synthesis of Polymers
Mixtures of cyclic oligoesters (
c(BF)
n and
c(BI)
n) at different molar ratios were polymerized following the procedure used by us for the synthesis of other PBF copolyesters [
15,
21]. A total of 47 mmol of the mixture of cyclic oligomers with the selected composition together with 0.5 mol % of Sn(Oct)
2 were dissolved in 10 mL of CHCl
3, the solution evaporated, and the remaining solid dried under vacuum at room temperature for 24 h. Subsequently, the mixture was left to react in a three necked round bottom flask for 6 h at 200 °C under a flow of N
2. For optimization of the reaction, the 50:50 mixture was polymerized at 180, 200 and 230 °C. The evolution in the molecular weight of all reactions was monitored by drawing aliquots at different times and analyzing them by GPC. The resulting polymers—without further treatment—were analyzed by NMR, GPC, TGA, DSC and WAXS.
3. Results and Discussion
The synthetic route used for the preparation of
c(BF)
n and
c(BI)
n cyclic oligomers and their ROP is represented in
Figure 1. In a first step, the cyclic oligomers were obtained by HDC of BD and either FDCA-Cl
2 or IPA-Cl
2. The mixture of cyclic oligomers and linear species were separated by column chromatography, and the purity of cyclic oligomers was ascertained by HPLC, NMR and MALDI-TOF mass spectra (
Figures S2 and S3).
The 1H-NMR spectra show the absence of any peaks at around 3.8 ppm due to the presence of CH2OH groups, which indicates that only cyclic oligomers are present in the purified fractions. Some signals in both 1H- and 13C-NMR spectra are split due to the sensitivity of these nuclei to the size of the oligomeric cycle. On the other hand, MALDI-TOF MS spectra allowed determining the molar mass of the different cyclic species.
Table 1 shows the composition of the different cycles for the two oligomeric fractions as determined by HPLC. A mixture of cyclic oligomers, mainly from dimer to tetramer, were obtained for both
c(BF)
n and
c(BI)
n being the dimer the predominant cycle size. Both the flexibility of the butylene unit and the 1,3- or 2,5-substitution in benzene or furan respectively, favor the cyclization reaction due to the probable low ring strain of the cycles made of two repeating units.
The thermal properties of these cycles were evaluated by DSC and TGA (
Figure S4 and
Table 1). Both
c(BF)
n and
c(BI)
n showed melting peaks at around 150 °C, and it was observed that they were thermally stable up to 276 °C and 330 °C, respectively, which allowed their thermal polymerization at the temperature used for reaction (200 °C) without perceiving degradation.
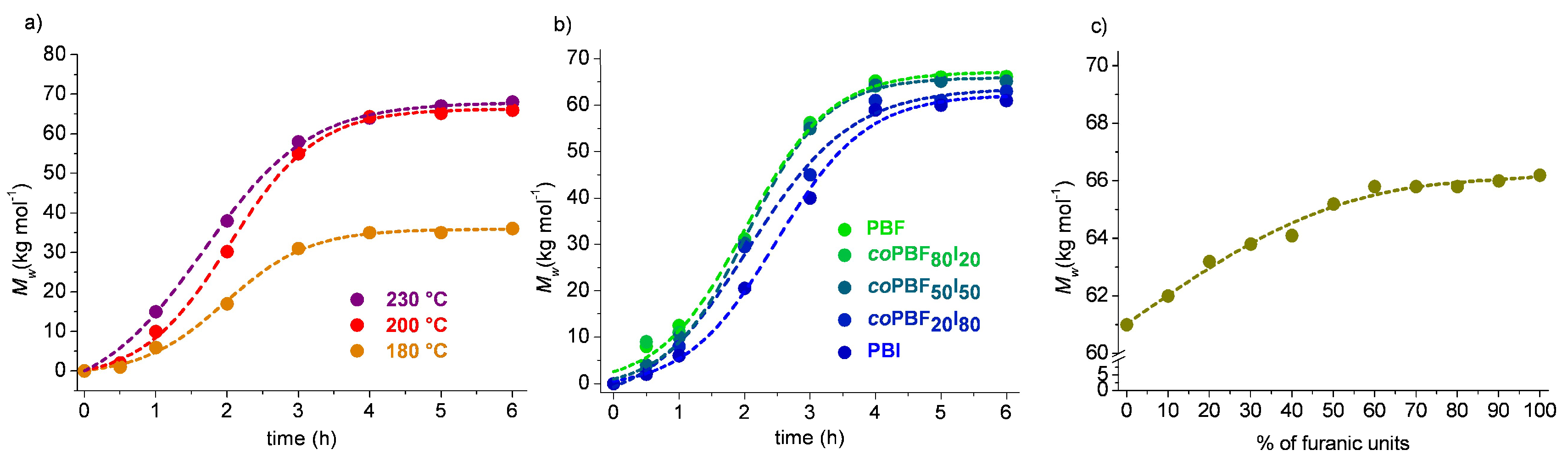
These cycles were then polymerized via ROP in bulk. First, an equimolar mixture of the two cyclic fractions was made to react in order to test the effect of time and temperature on the polymerization results. Three different temperatures above the melting point of the cycles were chosen, i.e., 180, 200 and 230 °C, and sample aliquots were drawn at scheduled periods to determine the evolution of the molar mass of the copolymer produced under different conditions. It was found that the molar mass of the polymer did not increase after six hours of reaction at above 200 °C, (
Figure 2a); such a temperature was then chosen for carrying out all copolyesters synthesis. The evolution of the molar mass of the copolyesters with time of reaction is depicted in
Figure 2b, where a similar tendency is observed for all the series. However the maximum molar mass attained was observed to increase slightly with the content in furanoate units in the copolyester (
Figure 2c and
Table 2), which can be due to the higher reactivity of the furanoate over isophthalate cyclic oligomers or the higher thermal stability of the former. The dispersities of the obtained copolyesters oscillated between 1.30 and 1.78 values, which are in accordance with those obtained by entropically driven ROP [
18,
19].
The polyesters were obtained in good yields (85–93%). The chemical structure and composition of
coPBF
xI
y copolymers were determined by NMR.
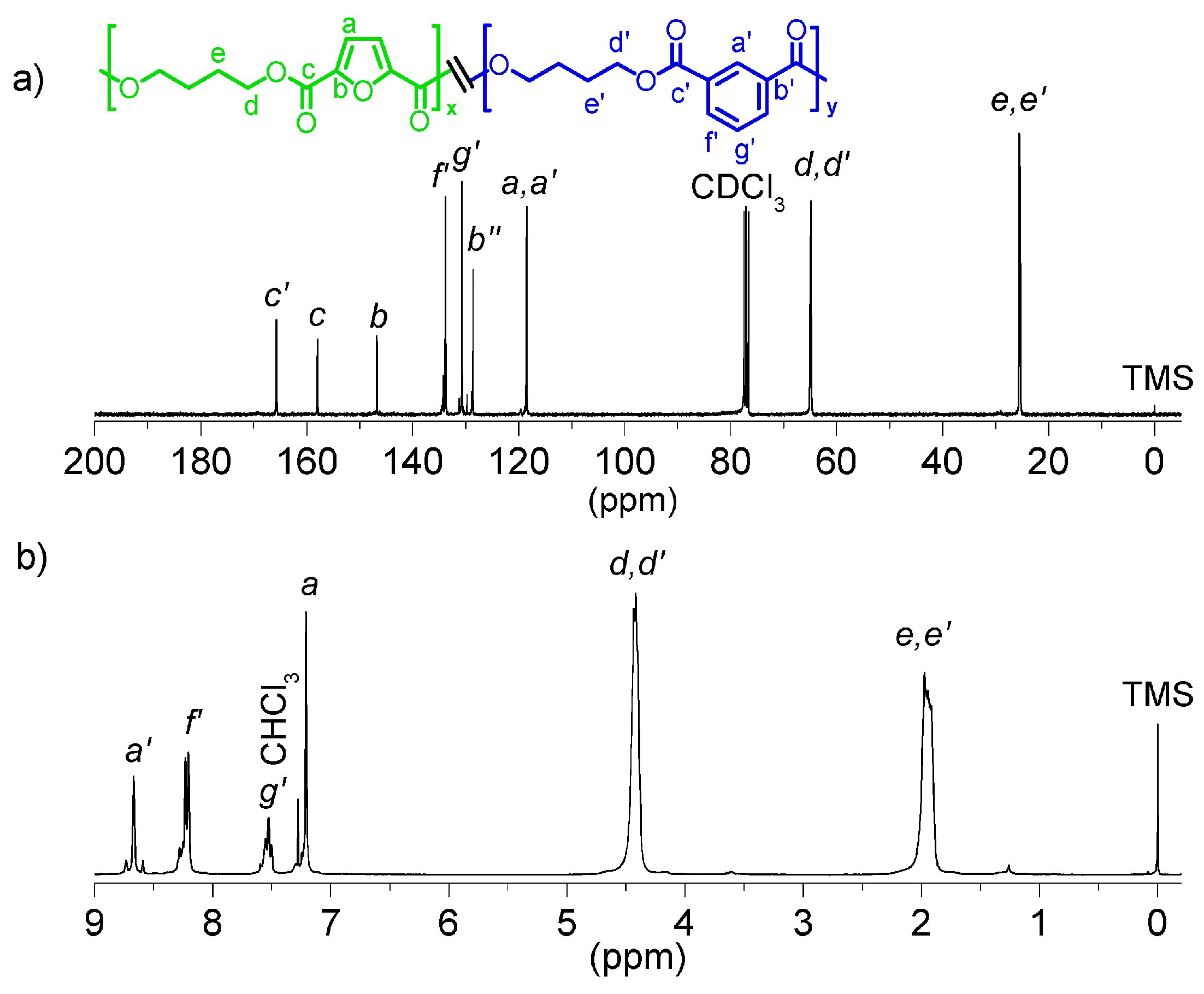
Figure 3 shows both
1H- and
13C-NMR spectra of
coPBF
50I
50 with peak assignments as a representative of the series. NMR spectra for all series are depicted in
Figure S5 of SI document. Signals due to the furanic proton
a and isophthalic protons
f’ were chosen for the determination of the copolyester composition. In general, a good correlation between the feed and the final copolyester composition determined by
1H-NMR was found, with slight fluctuations probably due to uncontrolled cycles volatilization (
Table 2).
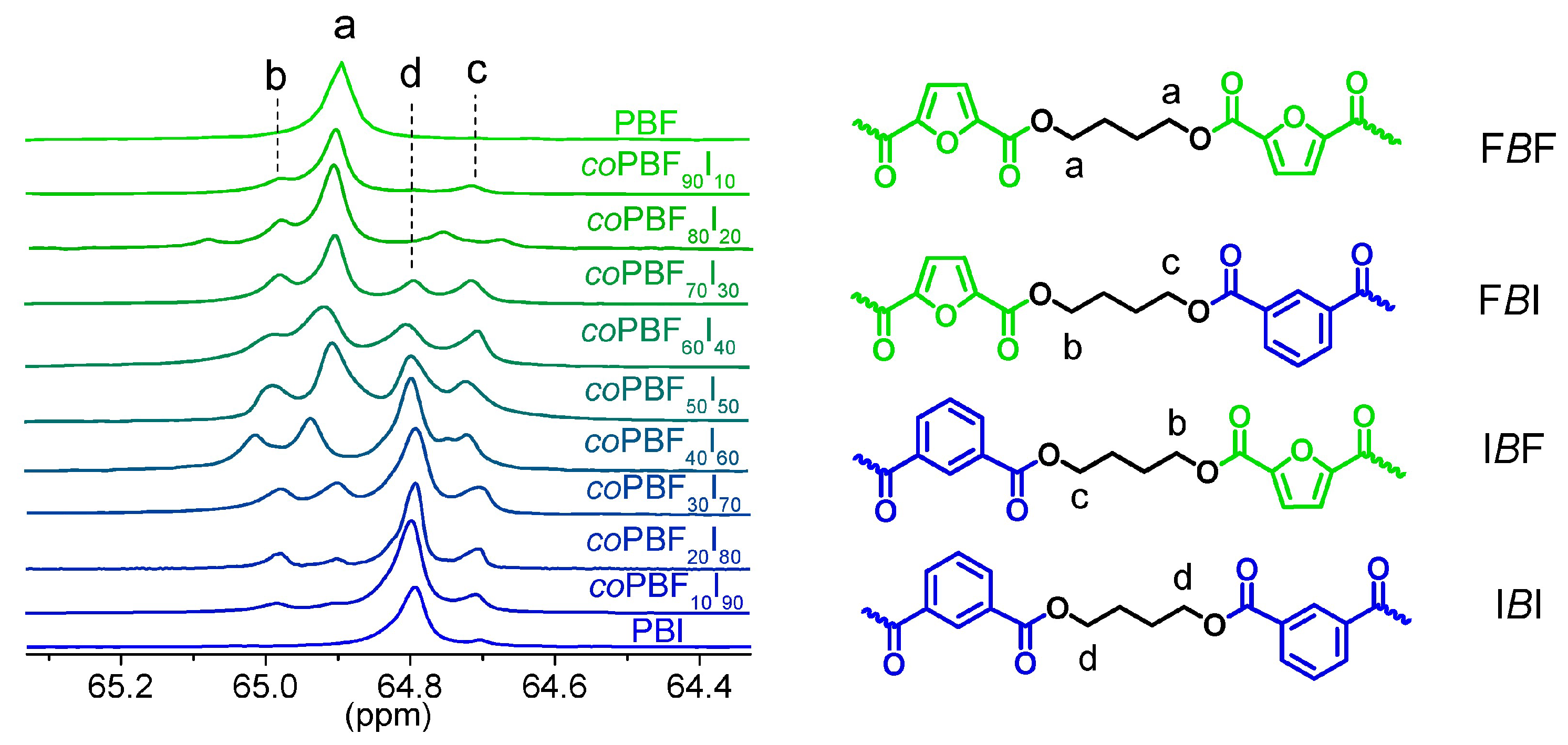
In contrast,
13C-NMR spectra were used for the determination of the copolymer microstructure. Each carbon signal of the butylene segment split into four peaks due to its sensitivity to sequence distribution at the level of dyads (
Figure 4). The assignment of the peaks contained in the different dyads was straightforward by comparison to those appearing in both PBF and PBI homopolyesters. By deconvolution of these signals, the dyad content (F
BF, F
BI + I
BF, I
BI) could be obtained and the number average sequence length and degree of randomness (
R) could be determined for each copolymer by applying the following expressions [
24]:
The degree of randomness was near to one with lower values for copolymers with contents in isophthalate units of between 50 mol % and 80 mol %. These values are higher than those that should be expected when only the ROP reaction takes place. In such cases, blocky copolymers and lower values of
R should be obtained [
25]. The observed values indicate that extensive transesterifications took place during polymerization, leading to nearly statistical copolymers (
Table 2).
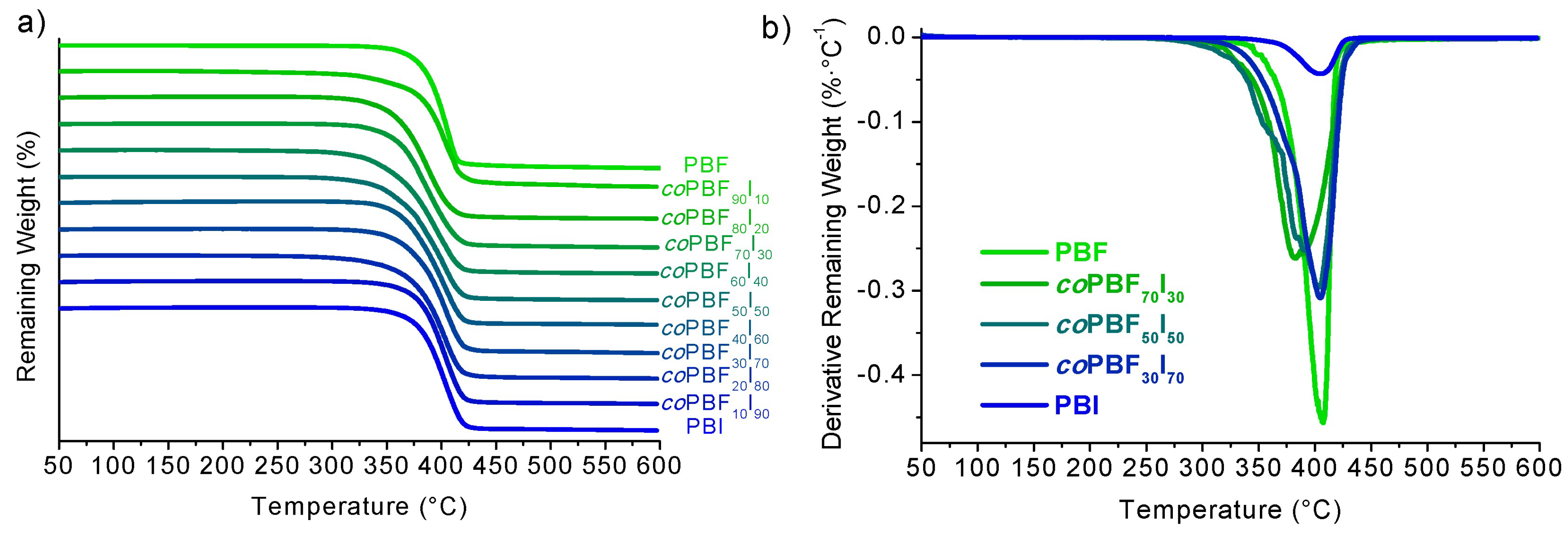
The thermal stability of PBF, PBI and their copolyesters was evaluated by TGA under an inert atmosphere. Both, polyesters, and copolyesters were observed to be thermally stable up to 300 °C with onset temperatures above 330 °C, a temperature of maximum decomposition rate close to 400 °C (
Figure 5 and
Table 3), and remaining weights at 600 °C between 7% and 11%. These values indicate that both homopolyesters and copolyesters have a good thermal stability, and may be processed above their melting temperature without suffering significant thermal degradation.
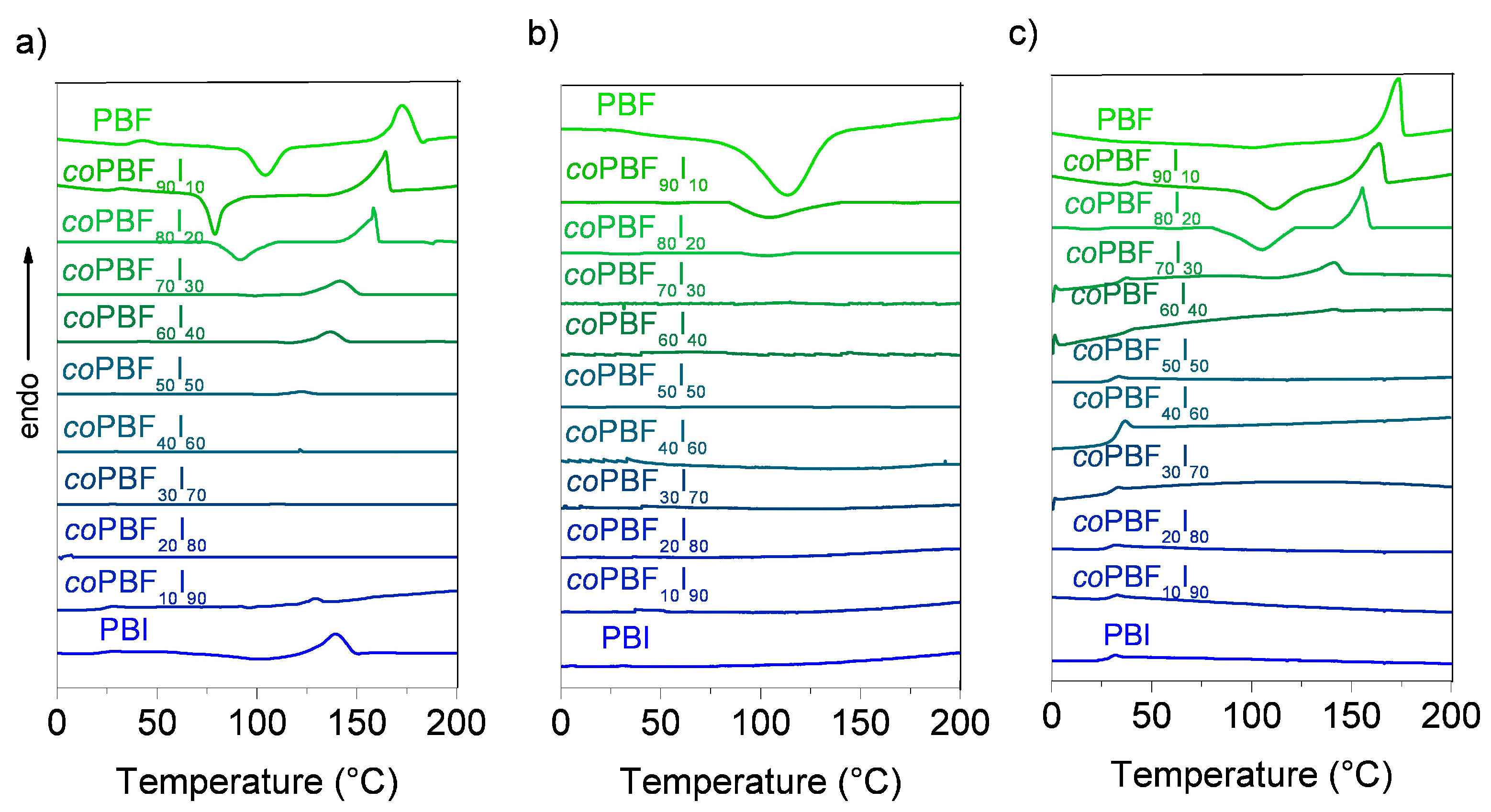
Thermal properties of the copolyesters, such as melting and glass transition temperatures, have been evaluated by DSC. The DSC traces obtained at heating from samples coming directly from synthesis are depicted in
Figure 6a–c and data taken from these thermograms are collected in
Table 3.
DSC traces recorded at heating from
coPBF
xI
y samples quenched from the melt showed a single
Tg intermediate between the two homopolyesters, with a value that decreased continuously from 41 to 21 °C with the content of isophthalate units in the copolyester. This feature is usually taken as an indication of the presence of random copolymers or miscible polymers blends. On the other hand, the replacement of furanoate by terephthalate units showed the opposite effect in the copolyesters, with a small increase of
Tg with the content of terephthalate units [
21].
The DSC analysis showed that, according to expectations, both PBF and PBI are semicrystalline polyesters with melting temperatures of 173 and 141 °C, and melting enthalpies of 45 and 31 kJ·mol
−1, respectively. The insertion of butylene isophthalate units in PBF restricted its crystallinity, reducing gradually both the melting temperature and enthalpy as their content increased. In fact, copolyesters with contents between 60 mol % and 80 mol % of isophthalate units showed very low melting enthalpy values at the first heating (≤3 kJ·mol
−1), and were unable to crystallize upon cooling from the melt. This last effect was not observed for PBF copolyester series containing terephthalate units in the copolyester, and all copolyesters were observed to be semicrystalline [
21].
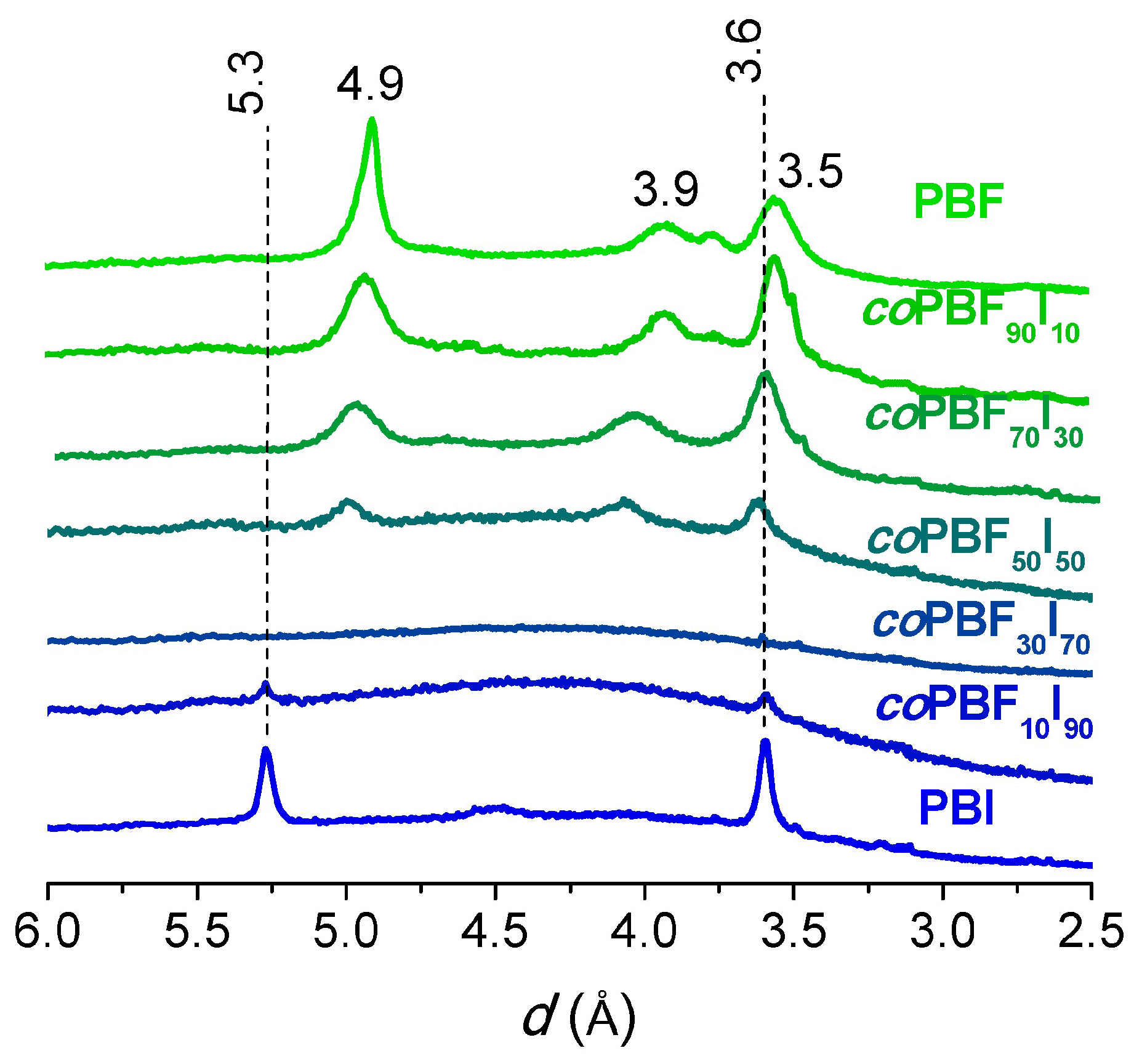
The intensity profiles of the X-rays scattered by powder pristine samples of representative
coPBF
xI
y copolyesters and the homopolyesters examined in the present work are compared in
Figure 7. In agreement with DSC results, discrete scattering was observed for copolyesters containing up to 50 mol % of isophthalate units, with spacings at around 4.9, 3.9 and 3.5 Å. Peaks broadening with the content of isophthalate units in the copolyester agree with should be expected for the impoverishment of the polymer crystallites, and their slight displacement upwards is probably due to the crystal lattice strain caused by the isophthalate units placed at the crystalline-amorphous interphase. From a comparison of these profiles with those produced by PBF, it can be inferred that semicrystalline copolyesters share the triclinic structure of the homopolyester, a fact that implies the exclusion of the isophthalate units from the crystal lattice [
9]. Over 50 mol % of isophthalate units, the copolyesters produced amorphous scattering up to 90 mol %, where the profiles showed weak reflections at 5.3 and 3.6 Å, characteristic of the crystal structure of PBI [
26].
In order to better understand the effect of the copolymerization on the crystallization behavior, a preliminary isothermal crystallization study has been carried out on PBF homopolymer and a copolymer containing 10 mol % of isophthalate units. Samples that were melted and quenched to 146 °C were isothermally crystallized for one hour at this temperature, and the crystallization enthalpy values generated over time were registered by DSC. It was found that the relative crystallinity (
Xt) increased following a sigmoidal trend in both cases (
Figure 8), but that the crystallization rate decreased substantially in the
coPBF
90I
10 copolyester.
The crystallization kinetics was analyzed by means of the Avrami approach. Taking the logarithm of both sides of the Avrami equation gives following equation:
where
Xt is the fraction of crystallized material,
K is the temperature-dependent rate constant,
t and
to are the elapsed and the onset times respectively, and
n is the Avrami exponent, indicative of the type of nucleation and dimensionality of crystal growth. Both,
n and log(
K) were determined from the slope and the intercept of the linear plot of log(−ln(1 −
Xt)) against log(
t − to), respectively, and the resulting values are compared in
Table 3 (
Figure S6). These results led us to conclude that a similar nucleation/growing mechanism was operating in the crystallization of the two samples, since close values were obtained for
n in both cases. In contrast, the crystallization half time increased by a factor of around four in the copolyester, revealing that crystallizability of PBF becomes severely hindered by copolymerization, even for small comonomer contents.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}