3.1. Preparation of Host-Molecule Functionalized Microgels
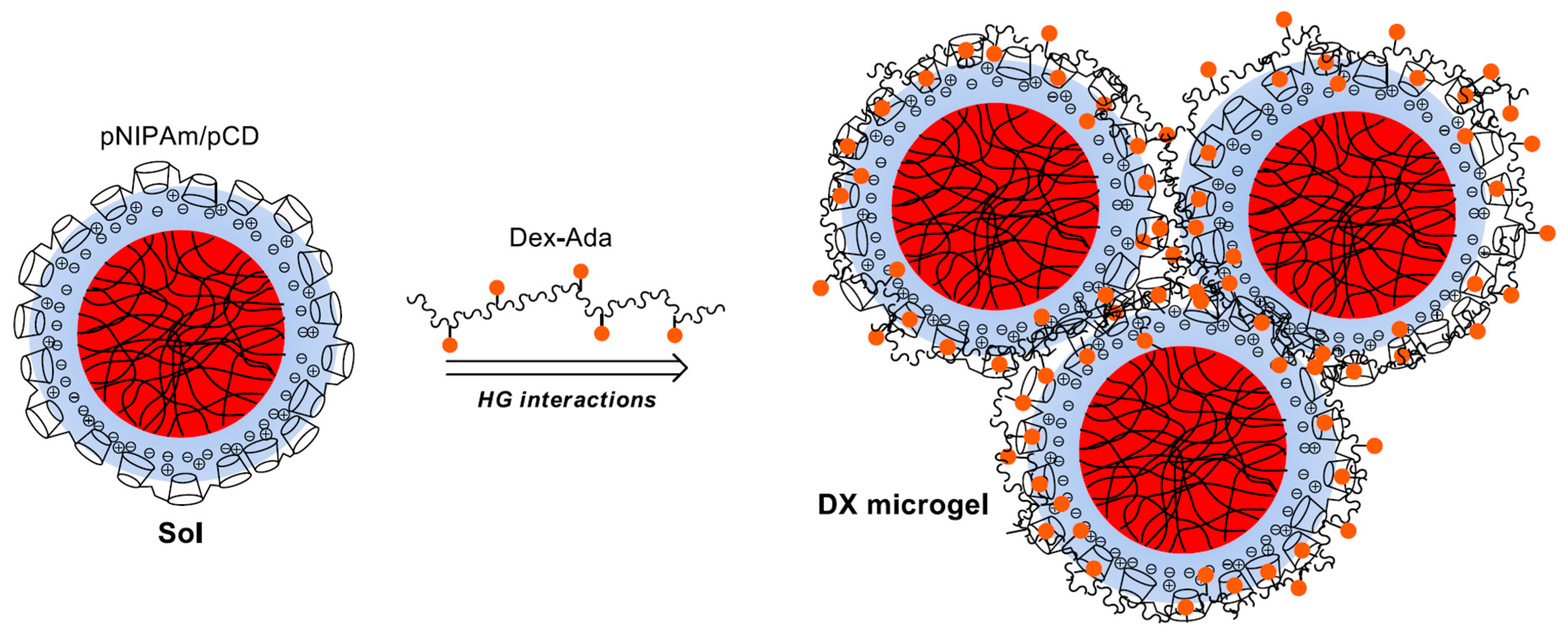
To facilitate the formation of 3D host/guest macrogels using pNIPAm microgels as the building blocks we prepared host-molecule-decorated microgels and guest-molecule-grafted polymers. We hypothesized that in the mixture of these materials, the host-molecule-decorated microgels can act as multivalent crosslinkers and enable the formation of a 3D gel matrix. However, it could be expected that efficient gel formation can take place only if the surfaces of the microgel beads are functionalized by the host molecules in high-enough density. To achieve high
βCD coverage on the pNIPAm microgel surface, we used polyelectrolyte complex formation between pNIPAm–shell–AAc microgels and cationic
β-cyclodextrin polymers (pCD,
Mw = 238 kDa, with 1.62 positive charge per
βCD). The characteristics of these compounds are summarized in
Table 1 where c
MG, c
AAc, c
CD, c
N+, c
Ada are the concentrations in microgel, AAC, CD, N+ and Ada respectively and n
N+/n
CD is the molar ratio of N+ over CD. The main advantage of this approach is that the interaction of the highly charged non-crosslinked acrylic acid shell of the microgel and the small positively charged pCD coils leads to the accumulation of the pCD on the surface of the microgel and exposes the
βCD moieties for host–guest interactions.
In order to utilize the pCD-covered microgels in host–guest gel formation, first we had to understand the phase behavior of the microgel/pCD mixtures. We found that upon mixing the microgels and pCD dilute solutions (60 ppm for the microgel) in stoichiometric ratio (1:1 ratio of negative charges of the pAAc monomers to the positive charges of the pCD), fast aggregation of the components took place resulting in a high-turbidity mixture. The turbidity of the samples decreased dramatically upon aging, as precipitate settled to the bottom of the container leaving a polymer-free transparent supernatant. Electrophoretic mobility measurements indicated that the fresh aggregates formed during mixing were charge–neutral, confirming the formation of colloidally unstable complexes.
However, when pCD was added to the microgel solution either in excess or in lower-than-stoichiometric positive-to-negative charge (c(+)/c(−)) ratios, the turbidity quickly decreased and transparent, stable mixtures formed. In the case of microgel excess, the c(+)/c(−) ratio had to be decreased to 0.8 or below to avoid precipitation. These samples had negative electrophoretic mobility indicating the excess of polyacrylic acid chains in the shell of the microgel/pCD complexes. In the case of pCD excess, the (c(+)/c(−)) ratio had to be increased to 1.5 to avoid precipitation. The microgel/pCD complexes had a positive electrophoretic mobility (~1 × 10
−8 m
2·V
−1·s
−1) showing that pCD is in excess of pAAc in the microgel shell. However, if the positive-to-negative ratio was further increased to 2, the electrophoretic mobility could be increased significantly further (~2 × 10
−8 m
2·V
−1·s
−1), demonstrating the formation of a more robust pCD shell on the microgel particles. To confirm that in excess pCD the formed microgel/pCD complexes are present as individual particles, cryo-transmission electron microscopy (cryo-TEM) experiments were performed. As shown in
Figure 2, in the case of excess pCD (2:1 positive-to-negative charge ratio), no sign of aggregation can be detected, but well-separated individual particles are present in the sample.
3.2. Hydrogel Formation
Having established that stable microgel/pCD complexes can be prepared, we aimed at using these complexes for the preparation of 3D host–guest gels. We aimed at using the pCD-coated microgels as multivalent crosslinkers of adamantane-grafted dextran (
Table 1). It can be expected that gels could form only if the adamantane-grafted dextran can bridge the gap between the neighboring microgels and strong (multiple) bonds can form between the microgel particles. To meet these requirements, the volume fraction of the microgel particles has to be high enough to ensure the close proximity of the microgels. According to Senff and Richtering, the rheological properties and phase behavior of pNIPAm microgel suspensions are strongly dependent on their effective volume fraction (
) [
27]. For instance, by measuring zero shear viscosity of microgels at different temperatures below lower critical solution temperature (LCST) as a function of
, they observed a sharp increase in viscosity at
0.5 [
28]. The phenomenon was ascribed to the microgel soft spheres being close to contact and interacting with each other at these concentrations. Hence, we decided to use microgel samples for the 3D gel formation with this effective volume fraction (
= 0.5).
Previously it was found by dynamic light-scattering measurements [
29] that the molar weight of microgel particles (MG) prepared with similar collapsed size (
dh = 150 nm determined by DLS at 40 °C) always exceeded the value of
MMG ~10
8 g·mol
−1. Using this molecular weight as a lower limit and the hydrodynamic diameter of the pCD-coated microgels measured in their swollen state (305 nm at 25 °C), we could calculate a lower limit for the microgel concentration in terms of dry weight per solution volume required to reach the desired effective volume fraction (
= 0.5). Using this simple estimation, we found that 6.0 g/L microgel concentration (15 mM for acrylic acid monomer concentration) should be sufficient in the final mixture to ensure the close proximity of the microgel beads required for gel formation.
To connect the neighboring microgel particles by multiple host–guest interactions, preferably large-molecular-weight Dex-Ada polymer has to be used with high-enough graft density of adamantane in high-enough concentration. To meet these requirements, we used dextran molecules with a molecular weight of 115 kDa (~700 glucose units) and 4.7% graft density (ca. 30 adamantane moieties on each polymer chain), and the concentration of the Dex-Ada polymer was chosen to ensure at least 1:1 adamantane:CD ratio in the samples.
Unfortunately, when we tried to prepare the 3D gel network by the direct mixing of the concentrated components at room temperature, the loss of colloid stability of the system resulted in precipitation. To circumvent this limitation, an indirect gelation approach was applied. In the first step, stable β-cyclodextrin polymer-coated microgel particles (MG/pCD) were prepared at lower concentrations (typically cAAc = 6 mM, c(+) = 4.8–12 mM in the final mixture) at room temperature. These coated microgels were mixed with the Dex-Ada polymer solution at close to stoichiometric Ada/CD molar ratios. Then, the resulting homogenous mixtures were concentrated in vacuum at 50 °C to reach the desired 6.0 g/L microgel concentration. After cooling down the concentrated mixtures to room temperature, the pNIPAm core of the MG/pCD complexes could reswell, leading to the formation of final samples.
First, we investigated the effect of the pCD coverage of microgel particles on the gel formation. We used the three different surface coverages providing colloidally stable MG/pCD complexes: a low pCD coverage that provides stable complexes with acrylic acid excess in the shell (c(+)/c(−) = 0.8); a pCD coverage that provides stable complexes with a slight pCD excess in the shell (c(+)/c(−) = 1.5); and a full pCD excess where electrophoretic mobility reaches the plateau for the MG/pCD complexes (c(+)/c(−) = 2). In each case we used the same final microgel concentration (c
tot ~6 g/L), while the ratio of the CD and the adamantane moieties was kept at a fixed value (n
Ada/n
CD ~ 1.3). For further details see
Table 2. These experiments indicated that the pCD coverage of the microgel particles has a profound effect on the gel formation. In the case of the lowest pCD coverage (
G1), precipitation occurred during the sample concentration. This can be explained by the loss of the colloid stability of the complex particles, whose charge could not counteract the increased van der Waals interaction among the collapsed complex particles (T > LCST). In the case of the overcharged complexes (excess pCD), no precipitation was observed and stable concentrated samples could be produced. At the same time, the lower pCD coverage (
G2: c(+)/c(−) = 1.5) resulted in a viscous liquid sample, while the sample with the largest pCD coverage (
G3) behaved as a physical gel (see insets in
Figure 3). This indicates that although the MG/pCD complexes preserved their colloid stability in both cases during sample concentration, connectivity among the microgel particles could reach a percolation threshold only in the latter case due to the larger number of links performed by the adamantyl units, increasing the amount of Dex-Ada polymer being needed to ensure the constant adamantane/cyclodextrin ratio in the samples.
To confirm that the gel formation is indeed related to the host–guest complex formation, we prepared two control samples without the addition of the guest polymer (
Table 2). Since it has been reported in the literature that uniform pNIPAm microgel dispersions with concentrations corresponding to close-packing (~5 wt %) undergo sol–gel transition below the LCST [
30], we also concentrated the uncoated pNIPAm–shell–pAAc microgels to test if they can give a sol/gel transition in the investigated concentration range. This experiment indicated that the core/shell microgel particles did not form a gel even at concentrations as high as 10.1 wt % (
R1), where the acrylic acid shells of the microgel particles strongly interpenetrate each other (semidilute systems). This can be explained by the strong electrostatic repulsion of the highly charged acrylic acid chains in the shell of the microgel particles (pH = 7).
In addition, the bare MG/pCD complexes (c(+)/c(−) = 2) were also concentrated to 10.5 wt % (R2), which resulted in the precipitation of the samples. The observed precipitation is a consequence of the loss of the colloid stability of the collapsed MG/pCD complexes. This clearly indicates that the colloid stability of the microgel complexes observed during the concentration of G2 and G3 samples is provided by the adamantane-grafted dextran chains and not exclusively by the charge of the MG/pCD complexes. Thus we can conclude that the host–guest complex formation indeed took place, and guest-polymer-wrapped hierarchical complexes formed upon mixing.
To get a better insight into the mechanical characteristics of the prepared samples, the viscoelastic properties of
G2 and
G3 were studied by oscillatory rheological measurements. Frequency sweep measurements of storage G′ and loss G″ moduli are shown in the two upper panels of
Figure 3. The G′ and G″ values prove to be frequency dependent in both cases, which was previously reported as a typical behavior for associating systems [
31,
32,
33]. At higher frequencies and shorter observation times, materials show gel character with the elastic component G′ dominating the viscous component G″, while at lower frequencies the system starts to behave as a viscous liquid with G″ > G′. In a number of earlier studies on host–guest polymeric hydrogels, such behavior is explained by the weak and reversible character of the host–guest links, which are being broken and reformed under applied stress [
32,
34,
35].
The finite lifetimes of the interpolymer crosslinks are due to the fluctuations in the relative kinetic energy of the interacting adamantane and
βCD-groups. These lifetimes (
), also known as relaxation times, may be estimated from the frequency (
fco) of the crossover point of G′ and G″ in the frequency sweeps:
fco should decrease with the number of host–guest links between individual microgels. Indeed, the
G2 sample, which behaves as a viscous liquid (
Figure 2a), has the higher
value of 12.6 Hz (
= 0.013 s), whereas in the case of
G3, which behaves as a gel, the
is shifted down to 1.7 Hz (
= 0.094 s) (
Figure 2b). The strength of the hydrogel structures was also studied by strain-amplitude sweep performed at
= 10 Hz (
Figure 2c). The plateau G′ moduli vary from 232 Pa for
G2, behaving as a viscous liquid, to 3257 Pa for the
G3 hydrogel.
G2 and
G3 exhibit quite high strain resistance and maintain their structure up to
γ ~100% (
Figure 2c). Similar values of elastic storage modulus were found for recently described cyclodextrin-based hierarchical hydrogels such as
βCD-coated quantum dots crosslinked with azobenzene-modified thermoresponsive copolymers (G′ = 200–400 Pa, c
tot = 12.5 wt % and T > 40 °C) [
36] or
βCD vesicles interconnected with Ada-modified cellulose polymer (G′ = 200–400 Pa, c
tot = 2–3 wt %) [
18].
We also attempted to fit the frequency sweep data of the samples by the Maxwell model. This basic viscoelasticity model was widely used for describing the behavior of entangled physically bonded polymer networks [
37,
38] and polymeric host–guest hydrogels [
35]. The Maxwell model implies that storage G and loss G″ moduli of a viscoelastic body vary as a function of the strain angular frequency
according to the following equations:
where
is the angular frequency [rad·s
−1],
is the relaxation time and
is the limiting value of G′ at high frequencies where it typically reaches a plateau. From the equations above, it also follows that
. In our case,
was obtained from the G′ and G″ crossover frequency value and was set as a fixed parameter during the fitting. Since G′ did not reach the plateau in the available range of angular frequencies,
was set as a free optimized parameter. Despite the viscoelastic character of the samples evidenced by the frequency sweep data (
Figure 3), rather poor fits to the Maxwell model were obtained. An example of such a fit for the
G3 sample is illustrated in
Figure S3. Only the low-frequency part (0.1–1.6 rad·s
−1) of the loss modulus G″ curve showed a reasonable agreement with the experimental data. The failure of the fitting might be related to the oversimplifications of the Maxwell model, which is suitable only for the description of the viscoelastic materials with a single and well-defined relaxation time,
. For instance, van de Manakker et al. obtained relatively good fits for host–guest hydrogel networks composed of linear and four-arm star PEG polymers end-functionalized with
βCD and cholesterol groups [
35]. However, they found that the fitting to the Maxwell model was no longer possible for structurally more complex hydrogels composed of eight-arm PEGs with the same end-functionalities. They ascribed this discrepancy to the presence of a broader range of relaxation mechanisms with different relaxation times involved in the stress relaxation in the latter case. We assume that the same arguments hold for the structurally rather complex pNIPAm/pCD/DexAda doubly crosslinked microgels. Indeed, a good fit of the frequency sweep data has been obtained using a generalized Maxwell model using a distribution of relaxation times (
Figure S3).
3.3. Influence of Host and Guest Polymer Characteristics on the Gel Formation
To develop a better understanding of the key parameters controlling the supramolecular gel formation, we also tested the effect of the host polymer (pCD) charge density on the gel formation. We used a host polymer to prepare the MG/pCD complexes, which had twice-as-large charge density (pCD3.2N+) as the host polymer used in the previous experiments (pCD1.6N+). This meant that while we could prepare the MG/pCD complexes with the same overall charge and colloid stability, the number of the CD groups present in the shell could be halved. The prepared complexes indeed preserved their colloid stability, however, when the system was concentrated in the presence of stoichiometric amount of guest polymer (n
Ada/n
CD = 1), gel formation could not be observed. To facilitate the gel formation, we repeated the same experiment with doubling the MG concentration (
G4 in
Table 2), thus ensuring that the same amount of adamantane moieties were present in the sample as in the case of the successfully gelled
G3 sample, while the increased microgel concentration resulted in tighter contact of the microgel particles. Surprisingly, the mixture remained a viscous liquid even at as high a total polymer content as c
tot = 14.8 wt %. Taking into account that doubling the charge density of the pCD polymer while keeping the charge ratio constant (c(+)/c(−) = 2) decreased the surface concentration of the pCD coverage by half, we hypothesized that the lack of gel formation was related to the decreased number of host/guest interactions of the guest polymers bridging the microgel particles.
In an attempt to overcome this problem, we used a guest polymer with increased number of grafted adamantane groups. This was achieved by using a larger-molecular-weight and graft density guest polymer (see Dex
500Ada
6, in
Table 1) as a crosslinker. This polymer had ~200 grafted adamantane groups compared to the 32 groups of the previously used polymer (Dex
110Ada
5). This approach proved successful: A stable transparent gel was formed at c
tot = 11.0 wt % (
G5 in
Table 2), confirming that the number of host–guest interactions established by the bridging polymer chains by the connected microgel particles is a key parameter in gel formation.
We also made frequency sweep rheological measurements on the prepared gel sample (
G5). The measured curves (
Figure S4) had the same qualitative features as measured previously for the
G3 sample (
Figure 3), but we found a lower crossover frequency (
= 1.1 Hz compared to the 1.7 Hz observed for
G3), which represents a slower relaxation time (
= 0.145 s vs.
= 0.094 s for
G3). This observation is in good agreement with the expected increased cooperativity of the interactions.
3.4. Thermoresponsive Behavior of the Supramolecular Gels
Since the obtained supramolecular hydrogels are composed of crosslinked thermoresponsive pNIPAm beads, their swelling and mechanical properties are expected to be temperature dependent. Previously, it has been found that hydrogels filled either noncovalently or covalently with pNIPAm microgels [
12,
13] became mechanically more robust at high temperatures (
t > volume phase-transition temperature) due to changing the nature of the microgel particles from a soft to a hard filler. It has also been shown that the nanostructured gels formed by chemically crosslinked pNIPAm microgels or by metal–ligand interactions [
39] shrunk with increasing temperature, and their volume deswelling closely mimicked the volume deswelling of the microgel building blocks.
Interestingly, the supramolecular gel samples we prepared did not shrink as a uniform body on temperature increase. Instead they remained a single-phase system with practically constant volume, and the gel sample turned into a liquid system at high temperatures. In order to follow the gel–sol transition of the supramolecular hydrogels, the storage and loss moduli were recorded as a function of temperature between 25 and 60 °C for both
G3 and
G5 (
Figure 4). The measurements provided very similar results for the two gel samples. Both G′ and G″ decreased monotonously with increasing temperature. Furthermore, in the temperature range where the volume phase transition of the microgel particles occurs (~30 °C <
t < 35 °C), the decrease of the storage and loss moduli accelerated then leveled off again, giving rise to a step-like decrease of the moduli. However, it is interesting to note this step-like decrease was significantly larger for G′ than for G″, decreasing the gap between the two curves. Since G′ decreased faster than G″, at some point G″ became larger (crossover temperature—
Tco), and the gel-to-sol phase transition occurred. It should also be noted that in the case of the
G3 sample, the crossover temperature was in a good agreement with the end of the temperature range where the pNIPAm/pCD microgels collapsed (VPTT: 37 °C), however, in the case of the
G5 sample, the crossover temperature was shifted to a higher value (41 °C) and it seemed independent of the microgel collapse. Finally, when the samples were gradually cooled back from 60 to 25 °C, the moduli showed full reversibility; hysteresis could not be observed neither for G′ nor for G″.
Contrary to previous investigations [
12,
13,
39], our measurements clearly showed that the prepared supramolecular gels became softer with increasing temperature, and finally turned into a liquid at the crossover temperature. Since the main difference between the previously investigated samples and our supramolecular gels was that we used host–guest interaction for the crosslinking of the microgel particles, we assumed that the observed temperature dependence was related to the characteristics of the host–guest complex formation. Indeed, the group of B. Ravoo has also reported decreasing mechanical strength in the case of host–guest supramolecular hydrogels connected by vesicles [
18].
To confirm this interpretation, we prepared a reference sample that had the same concentrations of the host and guest polymers as
G5 but did not contain microgel particles (
R3). The storage and loss moduli recorded for this sample as a function of temperature are plotted in panel (c) of
Figure 4. Similarly to the microgel-containing samples, G′ and G″ decreased monotonously with increasing temperature. However, the step-like steep decrease of the moduli in the temperature range of the microgel collapse was missing in this case, and as a consequence, the crossover temperature shifted to a much higher value (
Tco = 51 °C).
Based on these experimental results, it can be concluded that two independent processes take place in the supramolecular gel samples on temperature increase. On the one hand, as the volume phase-transition temperature is approached, the microgel building blocks shrink and become fully collapsed when the VPTT is reached. This should lead to the uniform shrinking of the gel samples and give rise to mechanically more-robust gels. However, this is counteracted by the fact that the host–guest association is an equilibrium process that has a negative enthalpy change (H). Thus, as the temperature is increased, the host–guest complex formation is shifted towards dissociation. This decreases the number of host–guest complexes in the system, and as a consequence, the average strength of the bonds bridging the microgel particles (the average number of host–guest complexes formed by a guest polymer) also decreases. This is evidenced by the continuously decreasing moduli and the gel–sol transition taking place even in the pure mixture of the host and guest polymers. At the same time, in the presence of the microgels, an osmotic effect attributed to the Dex-Ada chains opposes the deswelling of the gel sample. When the microgels shrink, the interstitial volume between the microgels is increased and the Dex-Ada chains accommodate this space to oppose the deswelling. This is the reason why there is no phase separation for these gels, contrary to the covalently crosslinked DX hydrogels [
12,
13,
39]. As a consequence, the local Dex-Ada concentration decreases, inducing a decrease of the number of host–guest complexes. In a previous study of host–guest hydrogels, it has been shown that moduli were decreasing with the concentrations of host and/or guest moieties [
29]. As a result, the average number of host/guest complexes formed by a guest polymer connecting the neighboring microgel beads decreases, and the supramolecular gel becomes softer, giving rise to the step-like decrease of moduli as the VPTT is approached.
The gel-to-sol transition temperature (TCO) corresponds to a temperature at which a percolation threshold in the connected microgel beads is attained. Increasing the number of adamantyl groups on the guest polymer should result in a larger number of host–guest complexes at any temperature, and thus to a shift of TCO to larger temperature. This explains why the TCO of G3, made with Dex110-Ada5 (32 adamantyl groups per chain), is lower by 4 °C than TCO of G5 made with Dex500-Ada6 (200 adamantyl groups per chain). An important message of this conclusion is that the gel–sol transition could be tuned within a wide temperature range by varying the size and graft density of the guest molecules, as well as by tuning the VPTT of the microgel beads.
To test the response dynamics of the supramolecular gels, the sample was subjected to several heating–cooling cycles, where its temperature was alternated between 25 and 50 °C (
Figure 5). As indicated by the figure, the temperature jumps required only a few seconds. G′ and G″ were recorded at a constant frequency (10 Hz). Both moduli changed promptly when the temperature jump was triggered, significant delay in their response to the temperature change could not be observed on the applied timescale. G″ was larger than G′ at 50 °C, whereas the inverse situation was obtained at 25 °C, meaning that a gel-to-sol transition occurred. The cycle was repeated several times, and the initial G′ and G″ values were fully restored in each case, proving the reversibility of the temperature-induced phase transition. One should note that the sample in
Figure 5 has the same nominal concentration as G3, but the final concentrated sample was a softer gel at the end due to the uncertainties of the sample preparation. We are working on a more robust and simple sample-preparation method.
The observed fast response of the supramolecular hydrogel to the temperature change is in contrast with the observations made for the chemically crosslinked pNIPAM hydrogels, which have characteristic relaxation times on the order of 100 s under the same geometry conditions [
40]. This can be explained by the fact that in this case, the overall volume of the gel does not change, thus the transport of the liquid out of and into the macroscopic gel structure is not required. Instead, the liquid transport is localized to the close neighborhood of the microgel beads and confined to a few-hundred-nanometer length scale. This renders the response time of the system to the subsecond timescale.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}