On the Mechanisms of the Effects of Ionizing Radiation on Diblock and Random Copolymers of Poly(Lactic Acid) and Poly(Trimethylene Carbonate)


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials and Polymers Synthesis
2.2. Preparation of Samples and Irradiation
2.3. Analytical Procedures
3. Results and Discussion
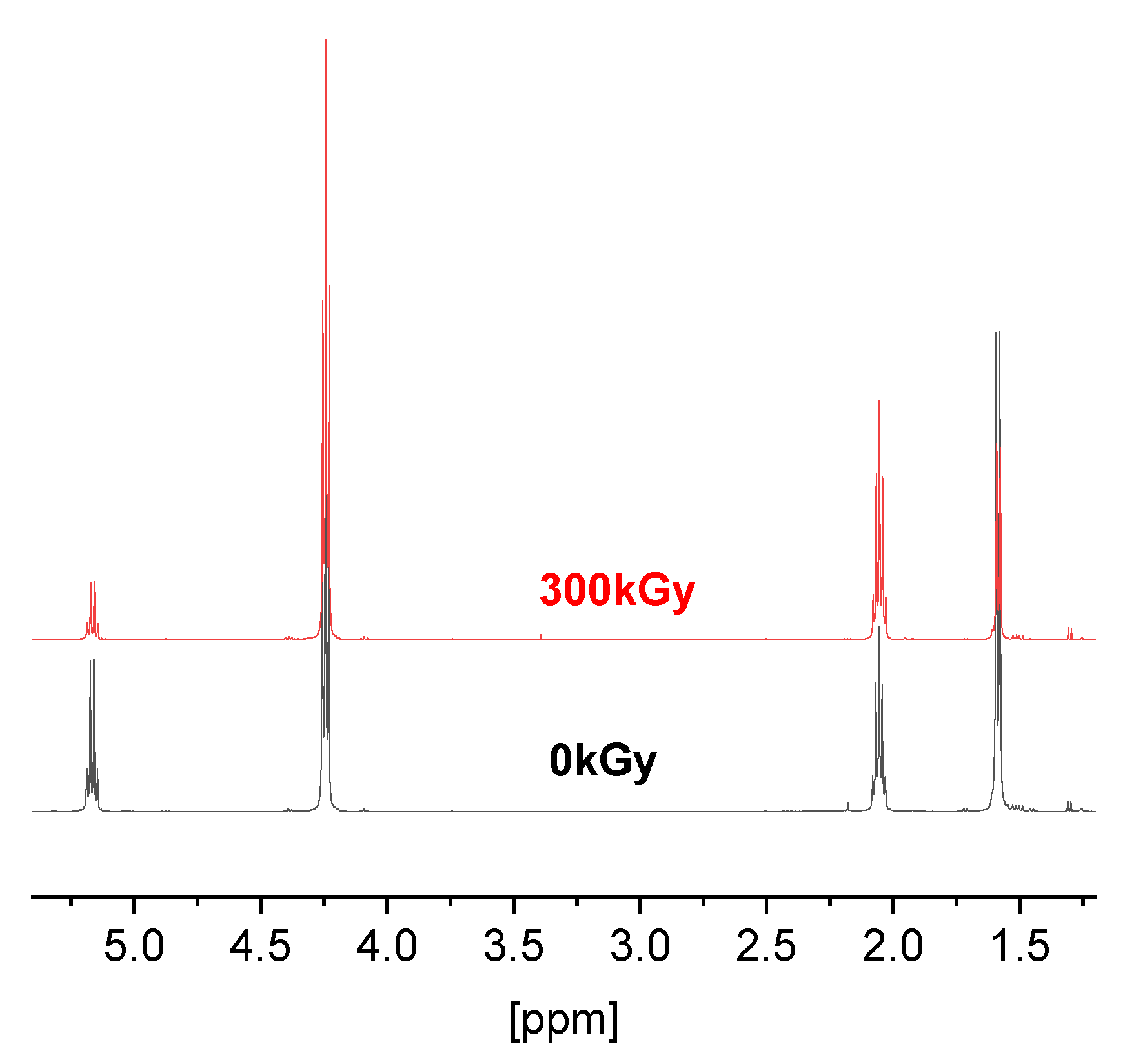
3.1. Synthesis of Copolymers
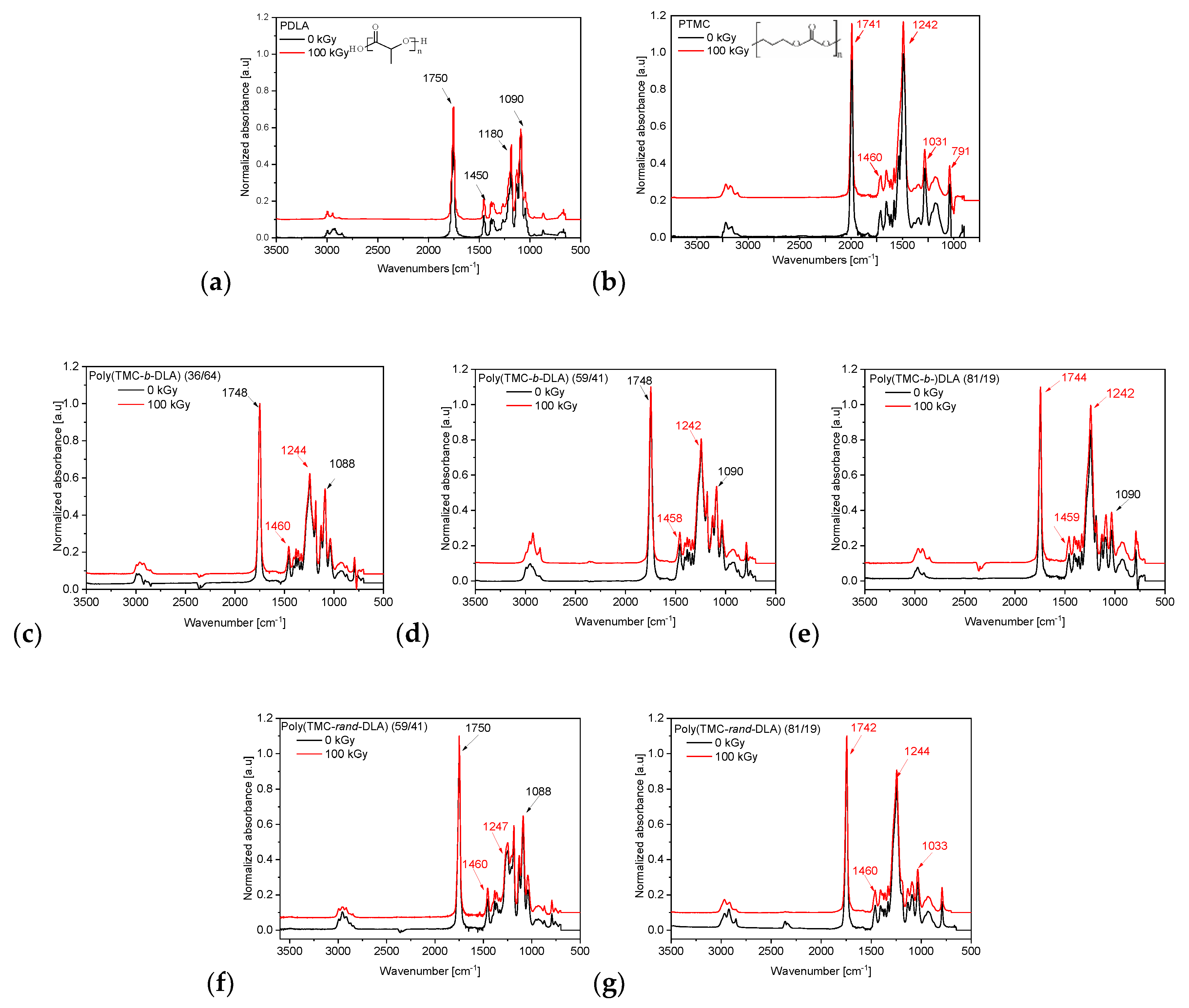
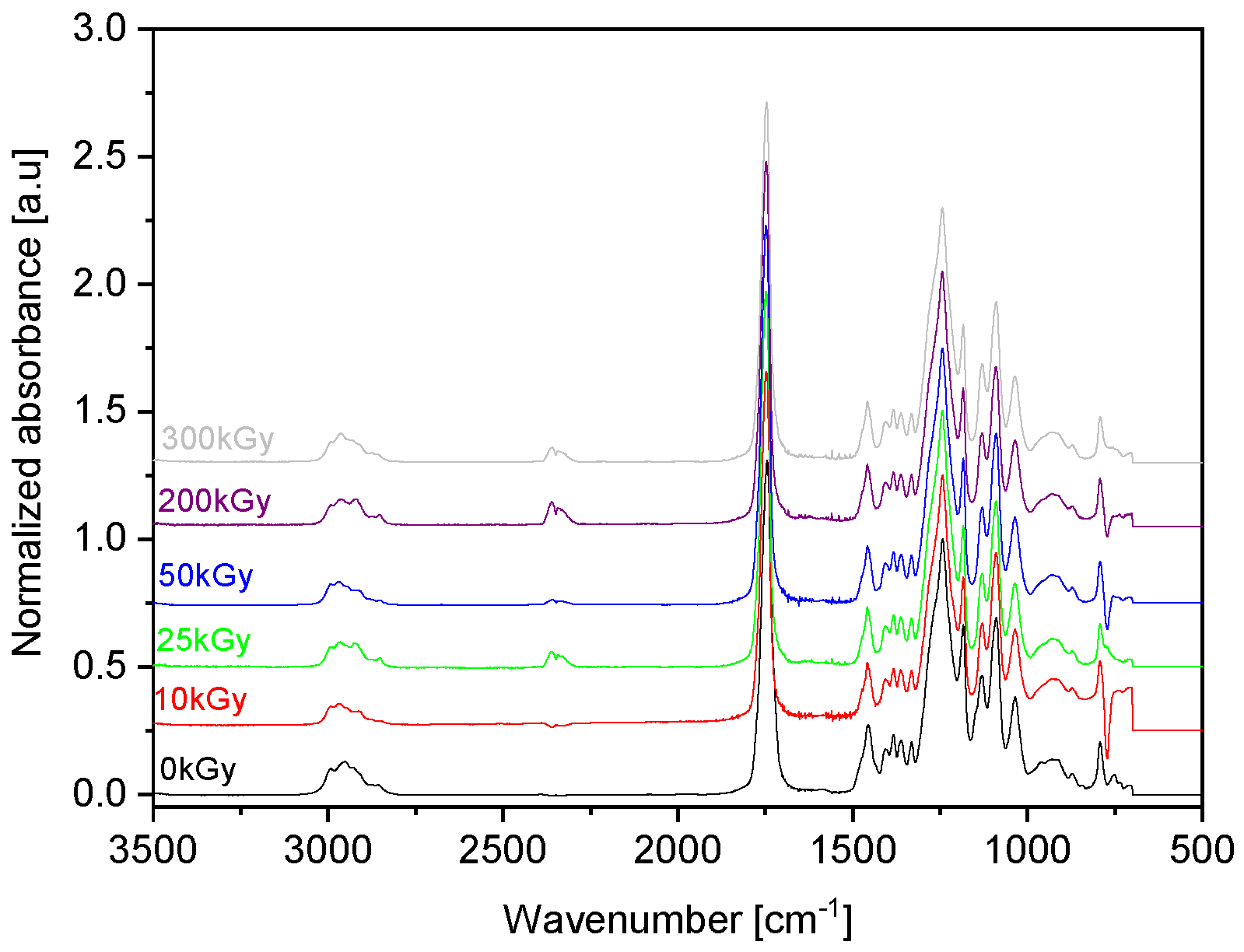
3.2. Irradiation Effects on the Structure of Copolymers
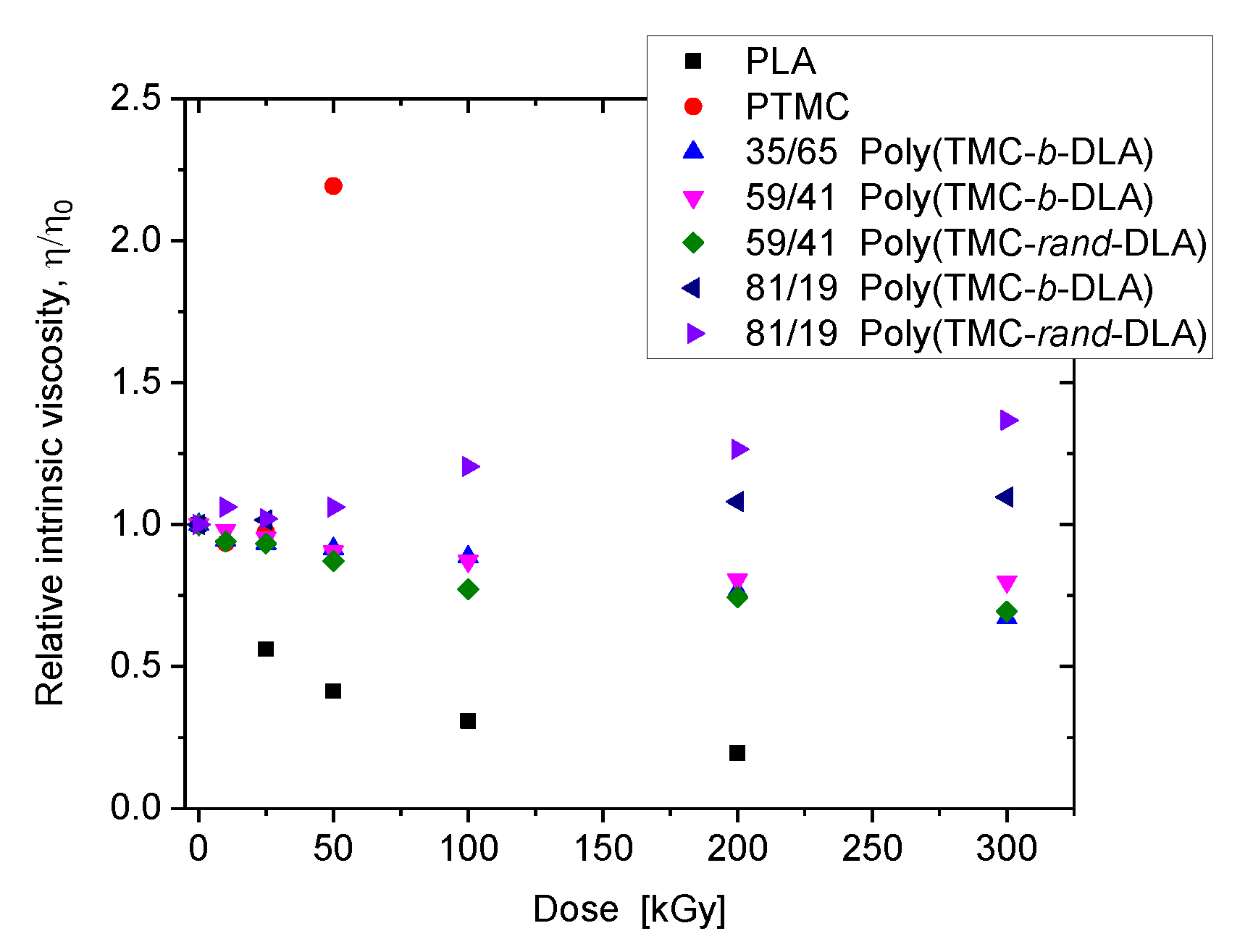
3.3. Intrinsic Viscosity
3.4. Molecular Weight
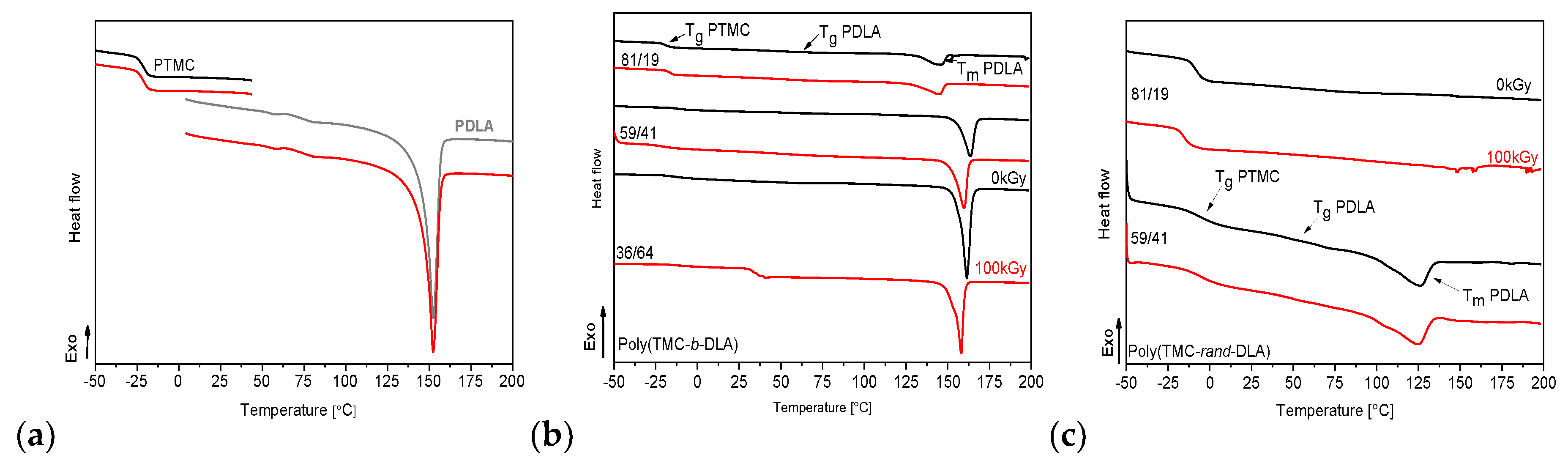
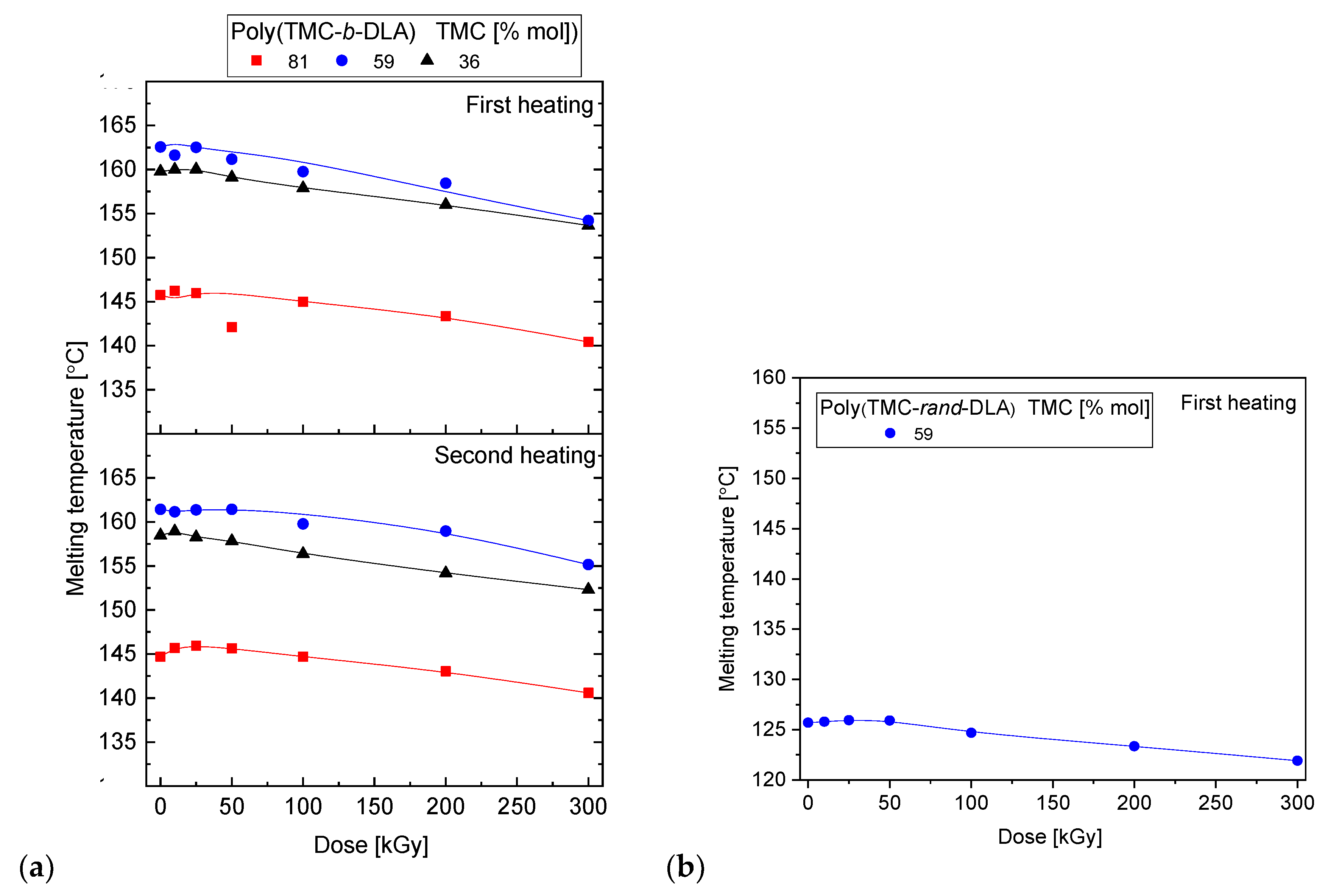
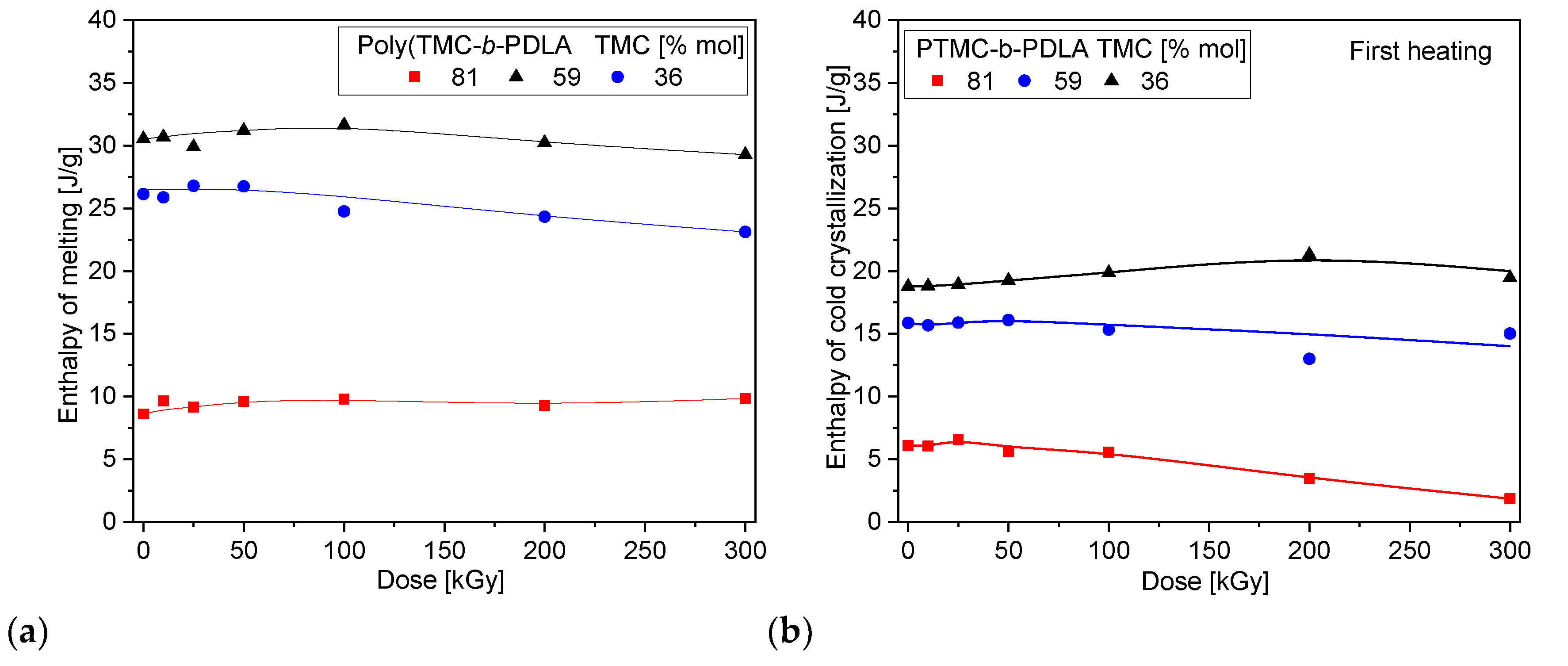
3.5. Thermal Properties
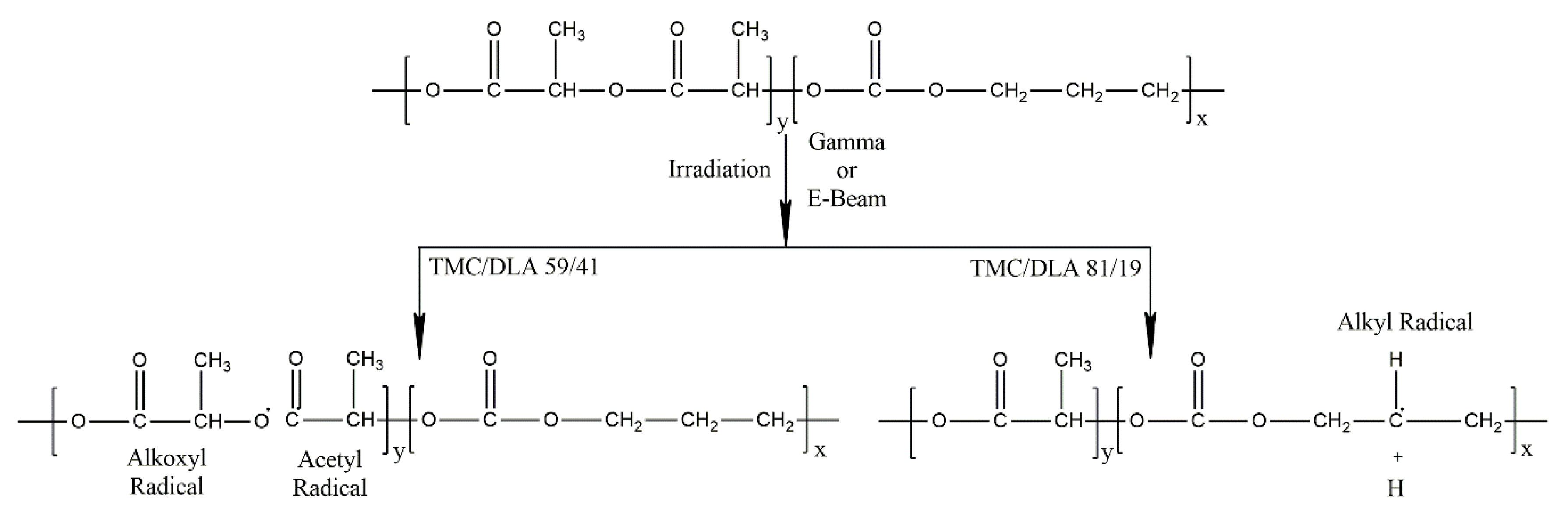
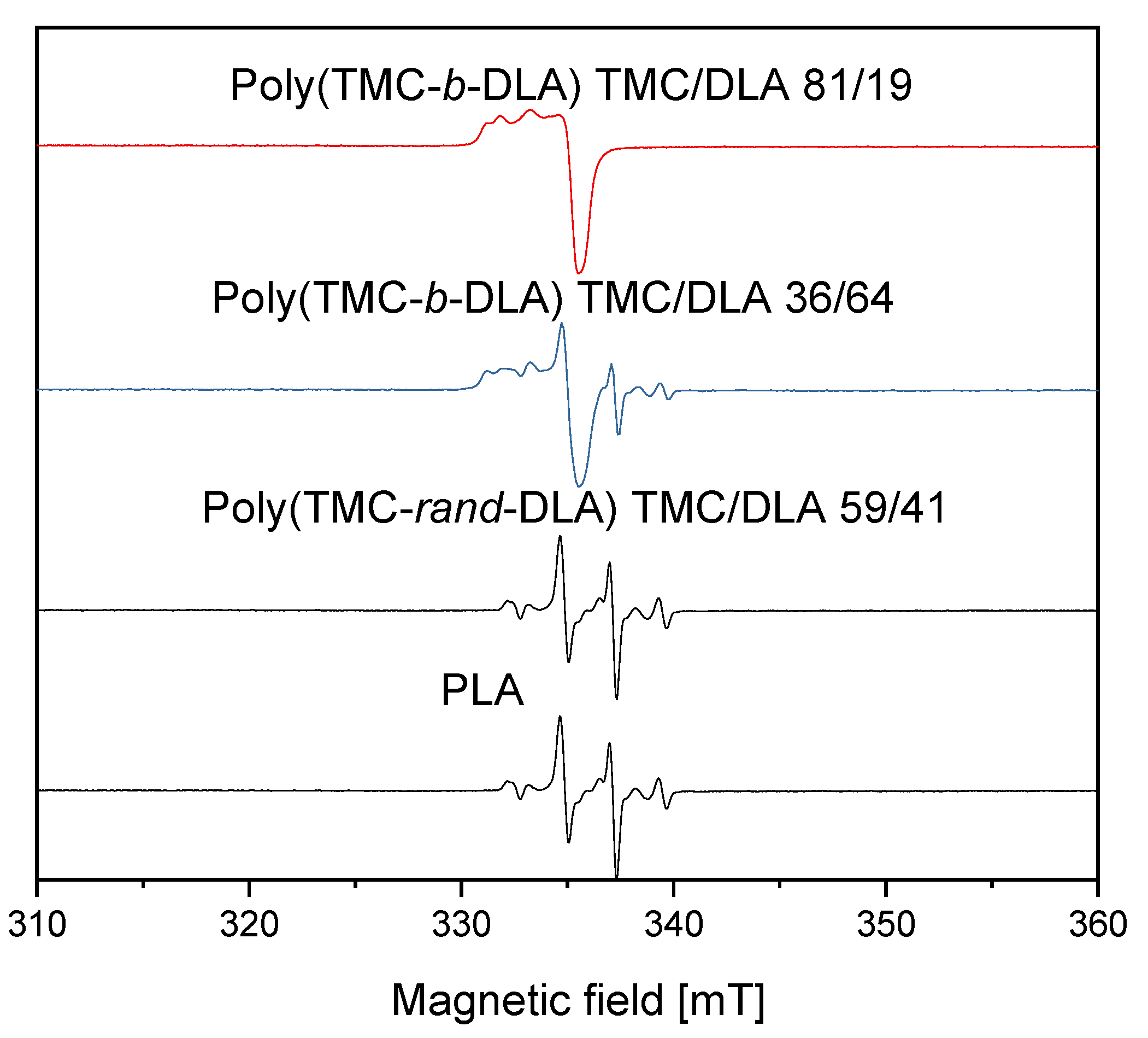
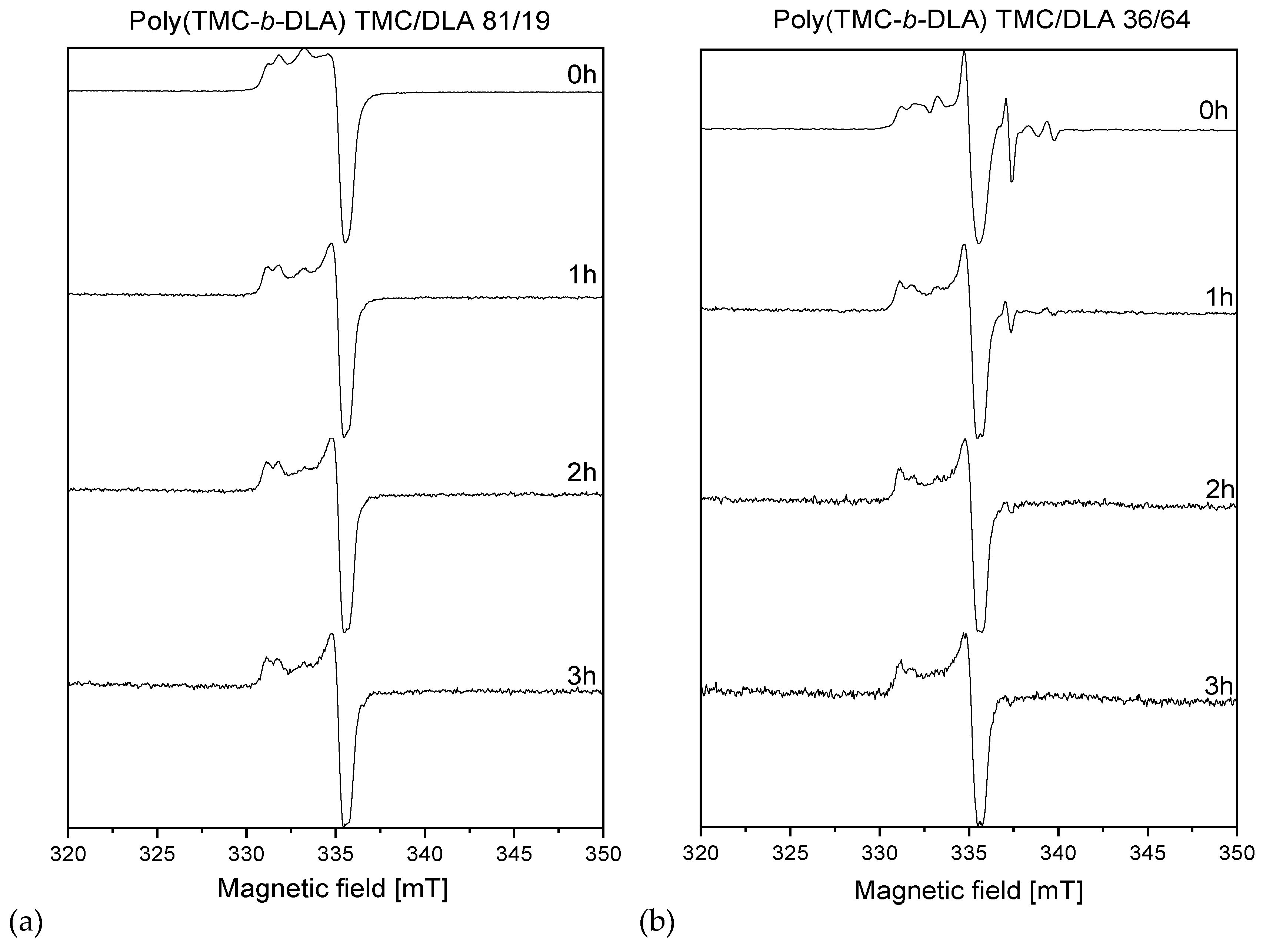
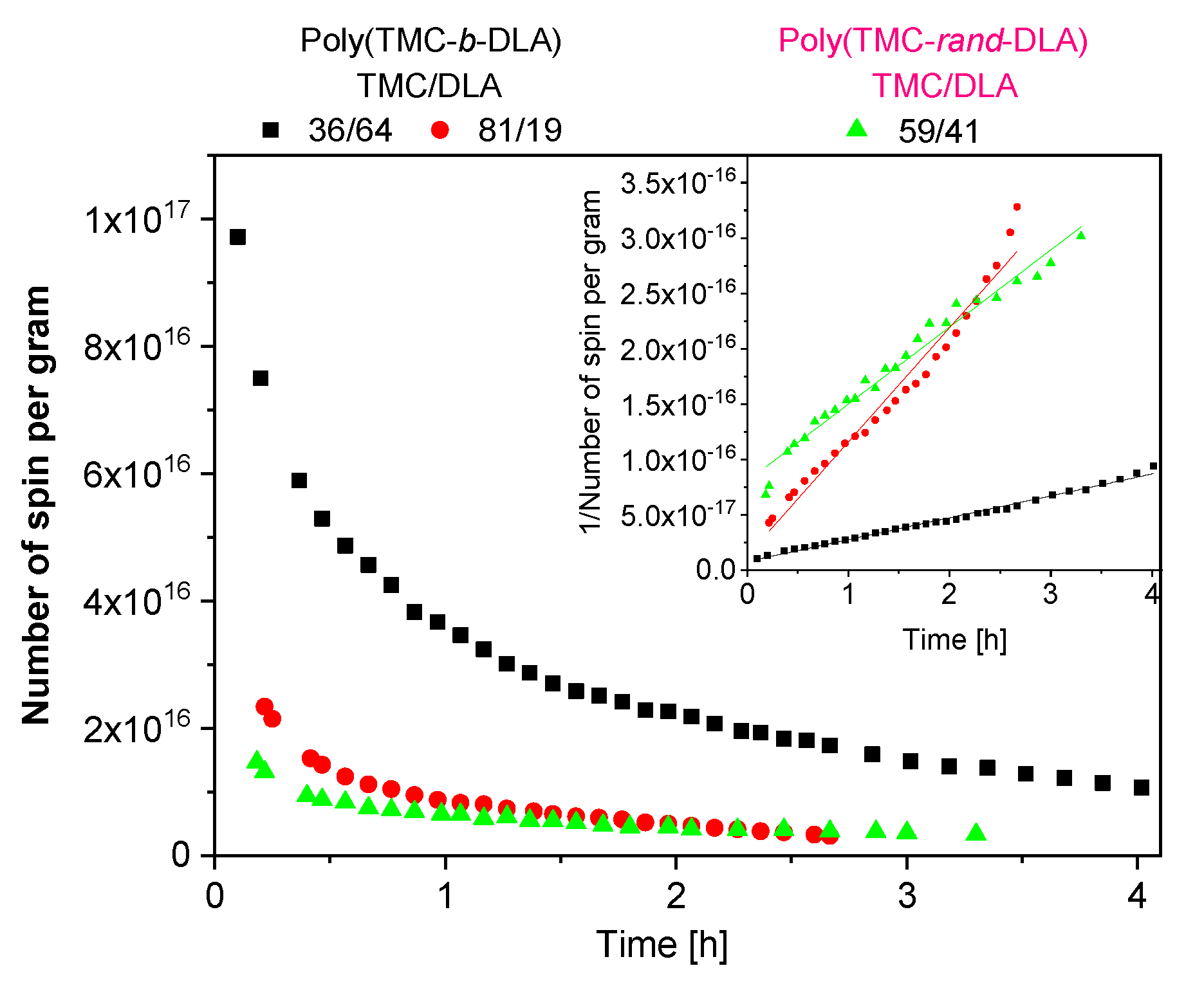
3.6. Radiolytically Produced Free Radicals
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Albertsson, A.-C.; Varma, I.K. Aliphatic Polyesters: Synthesis, Properties and Applications. Adv. Polym. Sci. 2002, 157, 1–40. [Google Scholar]
- Zhang, Z.; Grijpma, D.W.; Feijen, J. Triblock Copolymers Based on 1,3-Trimethylene Carbonate and Lactide as Biodegradable Thermoplastic Elastomers. Macromol. Chem. Phys. 2004, 205, 867–875. [Google Scholar] [CrossRef]
- Vert, M.; Li, S.M.; Speniehauer, G.; Guerir, P. Bioresorbability and biocomapatibility of aliphatic polyesters. J. Mater. Sci. Mater. M. 1992, 3, 43–46. [Google Scholar] [CrossRef]
- Södergård, A.; Stolt, M. Properties of lactic acid based polymers and their correlation with composition. Prog. Polym. Sci. 2002, 27, 1123–1163. [Google Scholar] [CrossRef]
- Drumright, B.R.E.; Gruber, P.R.; Henton, D.E. Polylactic Acid Technology. Adv. Mater. 2000, 12, 1841–1846. [Google Scholar] [CrossRef]
- Krikorian, V.; Pochan, D.J. Poly(l-Lactic Acid)/Layered Silicate Nanocomposite: Fabrication, Characterization, and Properties. Chem. Mater. 2003, 15, 4317–4324. [Google Scholar] [CrossRef]
- Hamad, K.; Kaseem, M.; Yang, H.W.; Deri, F.; Ko, Y.G. Properties and medical applications of polylactic acid: A review. eXPRESS Polym. Lett. 2015, 9, 435–455. [Google Scholar] [CrossRef]
- Nampoothiri, K.M.; Nair, N.R.; John, R.P. An overview of the recent developments in polylactide (PLA) research. Bioresour. Technol. 2010, 101, 8493–8501. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, T.; Chow, L.C.; Yang, M.; Mitchell, J.W. Effects of Inorganic Fillers on the Thermal and Mechanical Properties of Poly(lactic acid). Int. J. Polym. Sci. 2014, 827028. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yanga, Q.; Wang, Y.; Yu, H.; Chena, X.; Jing, X. Biodegradable electrospun poly(l-lactide) fibers containing antibacterial silver nanoparticles. Eur. Polym. J. 2006, 42, 2081–2087. [Google Scholar] [CrossRef]
- Yang, T.-C.; Hung, K.-C.; Wu, T.-L.; Wu, T.-M.; Wu, J.-H. A comparison of annealing process and nucleating agent (zinc phenylphosphonate) on the crystallization, viscoelasticity, and creep behavior of compression-molded poly(lactic acid) blends. Polym. Degrad. Stab. 2015, 121, 230–237. [Google Scholar] [CrossRef]
- Mathew, A.P.; Oksman, K.; Sain, M. Mechanical Properties of Biodegradable Composites from Poly Lactic Acid (PLA) and Microcrystalline Cellulose (MCC). J. Appl. Polym. Sci. 2005, 97, 2014–2025. [Google Scholar] [CrossRef]
- Pluta, M.; Paul, M.-A.; Alexandre, M.; Dubois, P. Plasticized Polylactide/Clay Nanocomposites. II. The Effect of Aging on Structure and Properties in Relation to the Filler Content and the Nature of its Organo-Modification. J. Polym. Sci. Part B 2006, 44, 312–325. [Google Scholar] [CrossRef]
- Yang, S.; Wu, Z.-H.; Yang, W.; Yang, M.-B. Thermal and mechanical properties of chemical crosslinked polylactide (PLA). Polym. Test. 2008, 27, 957–963. [Google Scholar] [CrossRef]
- Quynh, T.M.; Mitomo, H.; Nagasawa, N.; Wada, Y.; Yoshii, F.; Tamada, M. Properties of crosslinked polylactides (PLLA & PDLA) by radiation and its biodegradability. Eur. Polym. J. 2007, 43, 1779–1785. [Google Scholar] [CrossRef]
- Zhang, Z.; Kuijer, R.; Bulstra, S.K.; Grijpma, D.W.; Feijen, J. The in vivo and in vitro degradation behavior of poly(trimethylene carbonate). Biomaterials 2006, 27, 1741–1748. [Google Scholar] [CrossRef] [PubMed]
- Habraken, W.J.; Zhang, Z.; Wolke, J.G.; Grijpma, D.W.; Mikos, A.G.; Feijen, J.; Jansen, J.A. Introduction of enzymatically degradable poly(trimethylene carbonate) microspheres into an injectable calcium phosphate cement. Biomaterials 2008, 29, 2464–2476. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhuo, R.-X.; Zhang, X.-Z. Construction of functional aliphatic polycarbonates for biomedical applications. Prog. Polym. Sci. 2012, 37, 211–236. [Google Scholar] [CrossRef]
- Ruckenstein, E.; Yuan, Y. Molten Ring-Open Copolymerization of l-Lactideand Cyclic Trimethylene Carbonate. J. Appl. Polym. Sci. 1998, 69, 1429–1434. [Google Scholar] [CrossRef]
- Pêgo, A.P.; Poot, A.A.; Grijpma, D.W.; Feijen, J. Copolymers of trimethylene carbonate and epsilon-caprolactone for porous nerve guides: Synthesis and properties. J. Biomater. Sci. Polym. Ed. 2001, 12, 35–53. [Google Scholar] [CrossRef] [PubMed]
- Jie, C.; Zhu, K.J.; Shilin, Y. Preparation, characterization and biodegradable characteristics of poly(1,3-trimethylene carbonate-co-glycolide). Polym. Int. 1996, 41, 369–375. [Google Scholar] [CrossRef]
- Han, Y.; Fan, Z.; Lu, Z.; Zhang, Y.; Li, S. In vitro degradation of poly[(l-lactide)-co(trimethylene carbonate)] copolymers and a composite with poly[(l-lactide)-co-glycolide] fibers as cardiovascular stent material. Macromol. Mater. Eng. 2012, 297, 128–135. [Google Scholar] [CrossRef]
- Zhu, K.J.; Zhang, J.X.; Wang, C.; Yasuda, H.; Ichimaru, A.; Yamamoto, K. Preparation and in vitro release behaviour of 5-fluorouracil-loaded microspheres based on poly(l-lactide) and its carbonate copolymers. J. Microencapsul. 2003, 20, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, F.; Tu, S.; Chen, Y.; Luo, X.; Lu, Z.; Wei, J.; Li, S. Haemo- and cytocompatibility of bioresorbable homo- and copolymers prepared from 1,3-trimethylene carbonate, lactides, and e-caprolactone. J. Biomed. Mater. Res. A 2010, 94, 396–407. [Google Scholar] [CrossRef]
- Pego, A.P.; Van Luyn, M.J.A.; Brouwer, L.A.; Van Wachem, P.B.; Poot, A.A.; Grijpma, D.W.; Feijen, J. In vivo behavior of poly(1,3-trimethylene carbonate) and copolymers of 1,3-trimethylene carbonate with D,L-lactide or ∊-caprolactone: Degradation and tissue response. J. Biomed. Mater. Res. 2003, 67A, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Savaris, M.; dos Santos, V.; Brandalise, R.N. Influence of different sterilization processes on the properties of commercial poly(lactic acid). Mater. Sci. Eng. C 2016, 69, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, D.J.; Thirucote, R.R. Sterilization of medical devices: A review. J. Biomater. Appl. 1989, 3, 454–523. [Google Scholar] [CrossRef] [PubMed]
- Weir, N.A.; Buchanan, F.J.; Orr, J.F.; Farrar, D.F.; Boyd, A. Processing, annealing and sterilisation of poly-l-lactide. Biomaterials 2004, 25, 3939–3949. [Google Scholar] [CrossRef] [PubMed]
- Melski, K.; Kubera, H.; Głuszewski, W.; Zimek, Z. Effect of ionizing radiation on the properties of PLA packaging materials. Nukleonika 2011, 56, 65–69. [Google Scholar]
- Mansouri, M.; Berrayah, A.; Beyens, C.; Rosenauer, C.; Jama, C.; Maschke, U. Effects of electron beam irradiation on thermal and mechanical properties of poly(lactic acid) films. Polym. Degrad. Stab. 2016, 133, 293–302. [Google Scholar] [CrossRef]
- Leonard, D.J.; Pick, L.T.; Farrar, D.F.; Dickson, G.R.; Orr, J.F.; Buchanan, F.J. The modification of PLA and PLGA using electron-beam radiation. J. Biomed. Mater. Res. Part A 2009, 89A, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Chakoli, A.N.; He, J.; Chayjan, M.A.; Huang, Y.; Zhang, B. Irradiation of poly(l-lactide) biopolymer reinforced with functionalized MWCNTs. RSC Adv. 2015, 5, 55544–55549. [Google Scholar] [CrossRef]
- Chapiro, A.; Tabata, Y.; Charlesby, A.; Stannett, V.; Dole, M.; Dobo, J. Future of radiation processing of polymers. Radiat. Phys. Chem. 1990, 35, 15–17. [Google Scholar]
- O’Donnell, J.H. Radiation chemistry of polymers. In The Effects of Radiation on High-Technology Polymers; Reichmanis, E., O’Donnell, J.H., Eds.; ACS Symposium Series 381; American Chemical Society: Washington, DC, USA, 1989. [Google Scholar]
- Gupta, M.C.; Deshmukh, V.G. Radiation effects on poly(lactic acid). Polymer 1983, 24, 827–830. [Google Scholar] [CrossRef]
- Rytlewski, P.; Malinowski, R.; Moraczewski, K.; Zenkiewicz, M. Influence of some crosslinking agents on thermal and mechanical properties of electron beam irradiated polylactide. Radiat. Phys. Chem. 2010, 79, 1052–1057. [Google Scholar] [CrossRef]
- Jozwiakowska, J.; Wach, R.A.; Rokita, B.; Ulanski, P.; Nalawade, S.P.; Grijpma, D.W.; Feijen, J.; Rosiak, J.M. Influence of electron beam irradiation on physicochemical properties of poly(trimethylene carbonate). Polym. Degrad. Stab. 2011, 96, 1430–1437. [Google Scholar] [CrossRef]
- Miao, P.; Zhao, C.; Xu, G.; Fu, Q.; Tang, W.; Zeng, K.; Wang, Y.; Zhou, H.; Yang, G. Degradation of Poly(d,l-lactic acid)-b-poly(ethyleneglycol)-b-poly(d,l-lactic acid) Copolymer by Electron Beam Radiation. J. Appl. Polym. Sci. 2009, 112, 2981–2987. [Google Scholar] [CrossRef]
- Calıs, S.; Bozdag, S.; Kas, H.S.; Tuncay, M.; Hıncal, A.A. Influence of irradiation sterilization on poly(lactide-co-glycolide) microspheres containing anti-inflammatory drugs. II Farmaco 2002, 57, 55–62. [Google Scholar] [CrossRef]
- Loo, S.C.J.; Ooi, C.P.; Boey, F.Y.C. Radiation effects on poly(lactide-co-glycolide) (PLGA) and poly(l-lactide) (PLLA). Polym. Degrad. Stab. 2004, 83, 259–265. [Google Scholar] [CrossRef]
- Kowalski, A.; Libiszowski, J.; Duda, A.; Penczek, S. Polymerization of l,l-Dilactide Initiated by Tin(II) Butoxide. Macromolecules 2000, 33, 1964–1971. [Google Scholar] [CrossRef]
- Duda, A.; Penczek, S. On the difference of reactivities of various aggregated forms of aluminum isopropoxide in initiating ring-opening polymerizations. Macromol. Rapid Commun. 1995, 16, 67–76. [Google Scholar] [CrossRef]
- Bernardo, K.D.S.; Robert, A.; Dahan, F.; Meunier, B. Preparation of new chiral Schiff base ligands containing a binaphthyl moiety. X-ray structure of the H2Cl4 Salbinapht ligand. New J. Chem. 1995, 19, 129–131. [Google Scholar]
- Socka, M.; Duda, A.; Adamus, A.; Wach, R.A.; Ulanski, P. Lactide/trimethylene carbonate triblock copolymers: Controlled sequential polymerization and properties. Polymer 2016, 87, 50–63. [Google Scholar] [CrossRef]
- Fischer, E.W.; Sterzel, H.J.; Wegner, G. Investigation of the structure of solution grown crystals of lactide copolymers by means of chemical reactions. Colloid. Polym. Sci. 1973, 251, 980–990. [Google Scholar] [CrossRef]
- Eaton, G.R.; Eaton, S.S.; Barr, D.P.; Weber, R.T. Quantitative EPR; Springer: New York, NY, USA, 2010; ISBN 978-3-211-92947. [Google Scholar]
- Pamuła, E.; Błażewicz, M.; Paluszkiewicz, C.; Dobrzyński, P. FTIR study of degradation products of aliphatic polyesters–carbon fibres composites. J. Mol. Struct. 2001, 596, 69–75. [Google Scholar] [CrossRef]
- Jia, Y.T.; Kim, H.Y.; Gong, J.; Lee, D.R.; Ding, B.; Bhattarai, N. Synthesis and degradation of PLA–PCL–PLA triblock copolymer prepared by successive polymerization of ε-caprolactone and dl-lactide. Polym. Int. 2004, 53, 312–319. [Google Scholar] [CrossRef]
- Charlesby, A. Atomic Radiation and Polymers; Pergamon Press: Oxford, UK, 1960. [Google Scholar]
- Moad, C.L.; Winzor, D.J. Quantitative characterization of radiation degradation in polymers by evaluation of scission and cross-linking yields. Prog. Polym. Sci. 1998, 23, 759–813. [Google Scholar] [CrossRef]
- Solarski, S.; Ferreira, M.; Devaux, E. Characterization of the thermal properties of PLA fibers by modulated differential scanning calorimetry. Polymer 2005, 46, 11187–11192. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, J.; Bian, X.; Feng, L.; Xiang, S.; Sun, B.; Chen, Z.; Li, G.; Chen, X. Melt stereocomplexation from poly(l-lactic acid) and poly(d-lactic acid) with different optical purity. Polym. Degrad. Stab. 2013, 98, 844–852. [Google Scholar] [CrossRef]
- Samskog, P.-O.; Lund, A. The alkoxy radical RCHȮ formed in irradiated single crystals of rhamnose. Chem. Phys. Lett. 1980, 75, 525–527. [Google Scholar] [CrossRef]
- Rieger, R.H. Electron Spin Resonance-Analysis and Interpretation; Royal Society of Chemistry: Cambridge, UK, 2007; ISBN 978-0-85404-355-3. [Google Scholar]
- Barbanalbandi, A.; Hill, D.J.T.; O’Donnel, J.; Pomery, P.; Whittaker, A. An electron spin resonance study on g irradiated poly(l-lactic acid) and poly(dl-lactic acid). Polym. Degrad. Stab. 1995, 50, 297–304. [Google Scholar] [CrossRef]
- Kasser, M.J.; Silverman, J.; Al-Sheikhly, M. EPR Simulation of Polyenyl Radicals in Ultrahigh Molecular Weight Polyethylene. Macromolecules 2010, 43, 8862–8867. [Google Scholar] [CrossRef]
- Kasser, M.J.; Silverman, J.; Al-Sheikhly, M. On the Mechanism of Polyenyl Photoconversion in Irradiated Ultrahigh Molecular Weight Polyethylene. Macromolecules 2010, 43, 8868–8873. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Copolymer | TMC Fraction [mol %] | Conv. [%] | LDLA a | LTMC a | Mn | Mw | Mw/Mn | |
|---|---|---|---|---|---|---|---|---|
| Feed | Actual | |||||||
| Poly(TMC-b-DLA) | 36 | 36 | 98 | - | - | 19 | 31 | 1.63 |
| Poly(TMC-b-DLA) | 59 | 59 | 99 | - | - | 24 | 47 | 1.96 |
| Poly(TMC-b-DLA) | 81 | 81 | 99 | - | - | 18 | 35 | 1.94 |
| Poly(TMC-rand-DLA) | 59 | 58 | 100 | 13.0 | 6.6 | 18 | 20 | 1.11 |
| Poly(TMC-rand-DLA) | 81 | 81 | 100 | 3.8 | 14.0 | 17 | 19 | 1.12 |
| Dose [kGy] | PTMC | PLA | ||||
|---|---|---|---|---|---|---|
| Mn | Mw | Mw/Mn | Mn | Mw | Mw/Mn | |
| 0 | 108.9 | 270 | 2.48 | 215 | 260 | 1.21 |
| 10 | 124.4 | 260 | 2.09 | - | - | - |
| 25 | 136.5 | 277 | 2.03 | 90 | 105 | 1.17 |
| 50 | 142.6 | 290 | 2.03 | 62 | 96 | 1.55 |
| 100 | 162.3 | 308 | 1.90 | 45 | 70 | 1.56 |
| 200 | 202.5 | 336 | 1.66 | 30 | 68 | 2.27 |
| Dose [kGy] | Poly(TMC-b-DLA) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 81/19 | 59/41 | 36/64 | |||||||
| Mn | Mw | Mw/Mn | Mn | Mw | Mw/Mn | Mn | Mw | Mw/Mn | |
| 0 | 19 | 31 | 1.63 | 24 | 47 | 1.96 | 18 | 35 | 1.94 |
| 10 | - | - | - | 23 | 45 | 1.96 | 17 | 32 | 1.88 |
| 25 | 30 | 56 | 1.87 | 21 | 44 | 2.10 | 15 | 31 | 2.07 |
| 50 | 28 | 43 | 1.54 | 18 | 37 | 2.06 | 14 | 30 | 2.14 |
| 100 | 29 | 41 | 1.41 | 16 | 39 | 2.44 | 12 | 27 | 2.25 |
| 200 | 50 | 78 | 1.56 | 13 | 29 | 2.23 | 9 | 19 | 2.11 |
| 300 | 65 | 82 | 1.26 | 10 | 23 | 2.30 | 7 | 19 | 2.71 |
| Dose [kGy] | Poly(TMC-rand-DLA) | |||||
|---|---|---|---|---|---|---|
| 81/19 | 59/41 | |||||
| Mn | Mw | Mw/Mn | Mn | Mw | Mw/Mn | |
| 0 | 18 | 20 | 1.11 | 17 | 19 | 1.12 |
| 10 | 19 | 21 | 1.11 | 14 | 16 | 1.14 |
| 25 | - | - | - | 12 | 15 | 1.25 |
| 50 | 28 | 30 | 1.07 | 12 | 15 | 1.25 |
| 100 | 31 | 35 | 1.13 | 10 | 13 | 1.30 |
| 200 | 39 | 46 | 1.18 | 7 | 10 | 1.43 |
| 300 | 43 | 49 | 1.14 | 6 | 9 | 1.50 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adamus-Wlodarczyk, A.; Wach, R.A.; Ulanski, P.; Rosiak, J.M.; Socka, M.; Tsinas, Z.; Al-Sheikhly, M. On the Mechanisms of the Effects of Ionizing Radiation on Diblock and Random Copolymers of Poly(Lactic Acid) and Poly(Trimethylene Carbonate). Polymers 2018, 10, 672. https://doi.org/10.3390/polym10060672
Adamus-Wlodarczyk A, Wach RA, Ulanski P, Rosiak JM, Socka M, Tsinas Z, Al-Sheikhly M. On the Mechanisms of the Effects of Ionizing Radiation on Diblock and Random Copolymers of Poly(Lactic Acid) and Poly(Trimethylene Carbonate). Polymers. 2018; 10(6):672. https://doi.org/10.3390/polym10060672
Chicago/Turabian StyleAdamus-Wlodarczyk, Agnieszka, Radoslaw A. Wach, Piotr Ulanski, Janusz M. Rosiak, Marta Socka, Zois Tsinas, and Mohamad Al-Sheikhly. 2018. "On the Mechanisms of the Effects of Ionizing Radiation on Diblock and Random Copolymers of Poly(Lactic Acid) and Poly(Trimethylene Carbonate)" Polymers 10, no. 6: 672. https://doi.org/10.3390/polym10060672
APA StyleAdamus-Wlodarczyk, A., Wach, R. A., Ulanski, P., Rosiak, J. M., Socka, M., Tsinas, Z., & Al-Sheikhly, M. (2018). On the Mechanisms of the Effects of Ionizing Radiation on Diblock and Random Copolymers of Poly(Lactic Acid) and Poly(Trimethylene Carbonate). Polymers, 10(6), 672. https://doi.org/10.3390/polym10060672