The Chain Distribution Tensor: Linking Nonlinear Rheology and Chain Anisotropy in Transient Polymers



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Transient Network Theory for Polymer Networks with Reversible Bonds
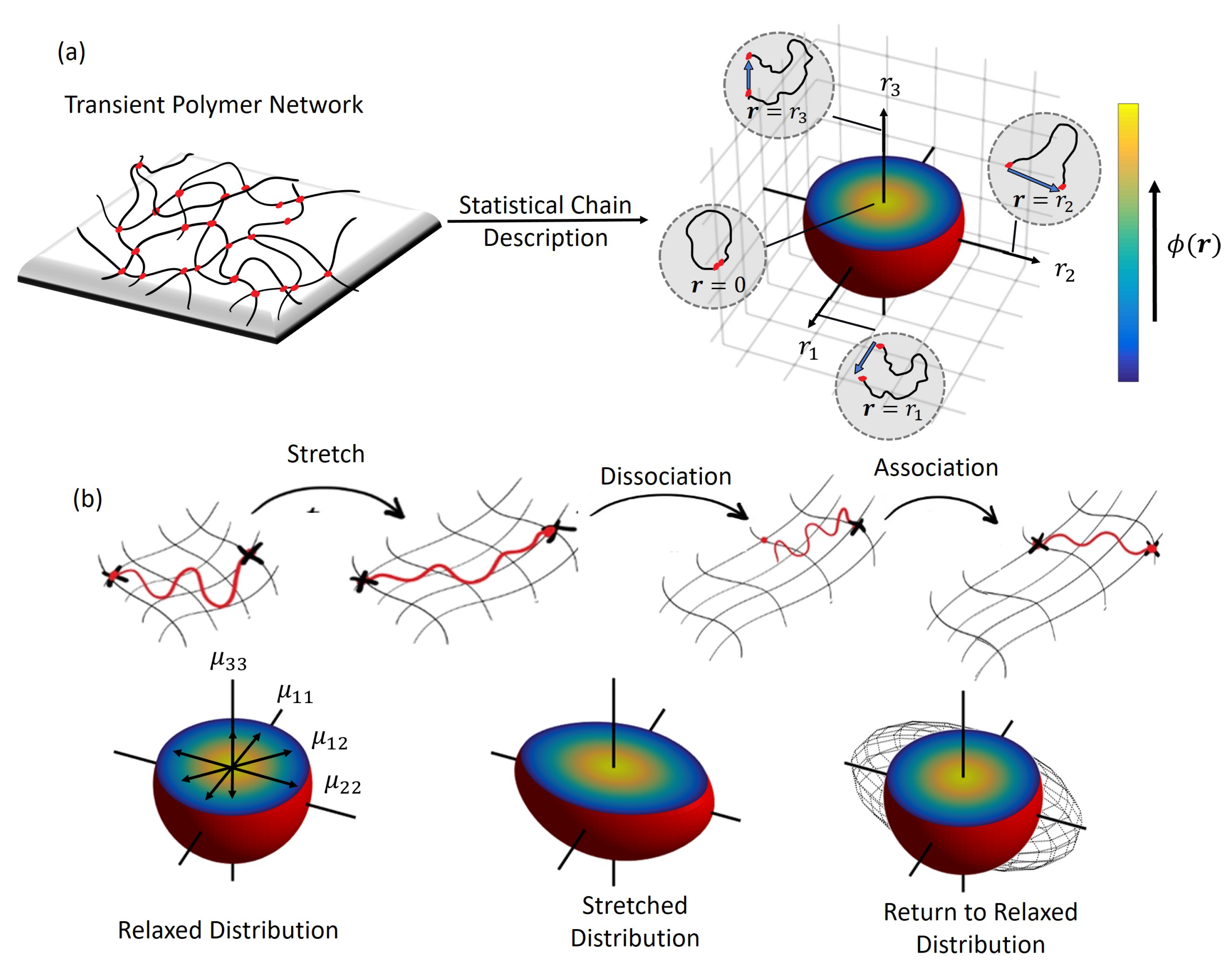
2.1. Statistical Description of Polymer Chains.
2.2. Evolution equations.
- (a)
- The rate of change of chain stretch, , due to network deformation that is governed by the macroscopic velocity gradient where: is the velocity field in the material and ∇ is the differential operator. If we assume that the chains undergo affine deformation (i.e., they follow the macroscopic deformation ), the stretch rate is given by .
- (b)
- The association of new chains into the network with rate, , at a near stretch-free configuration that follows the well-known Gaussian probability density function for polymer chains, .
- (c)
- The dissociation of attached chains that may be in a stretched state at the rate .
2.3. Macroscopic energy and stress
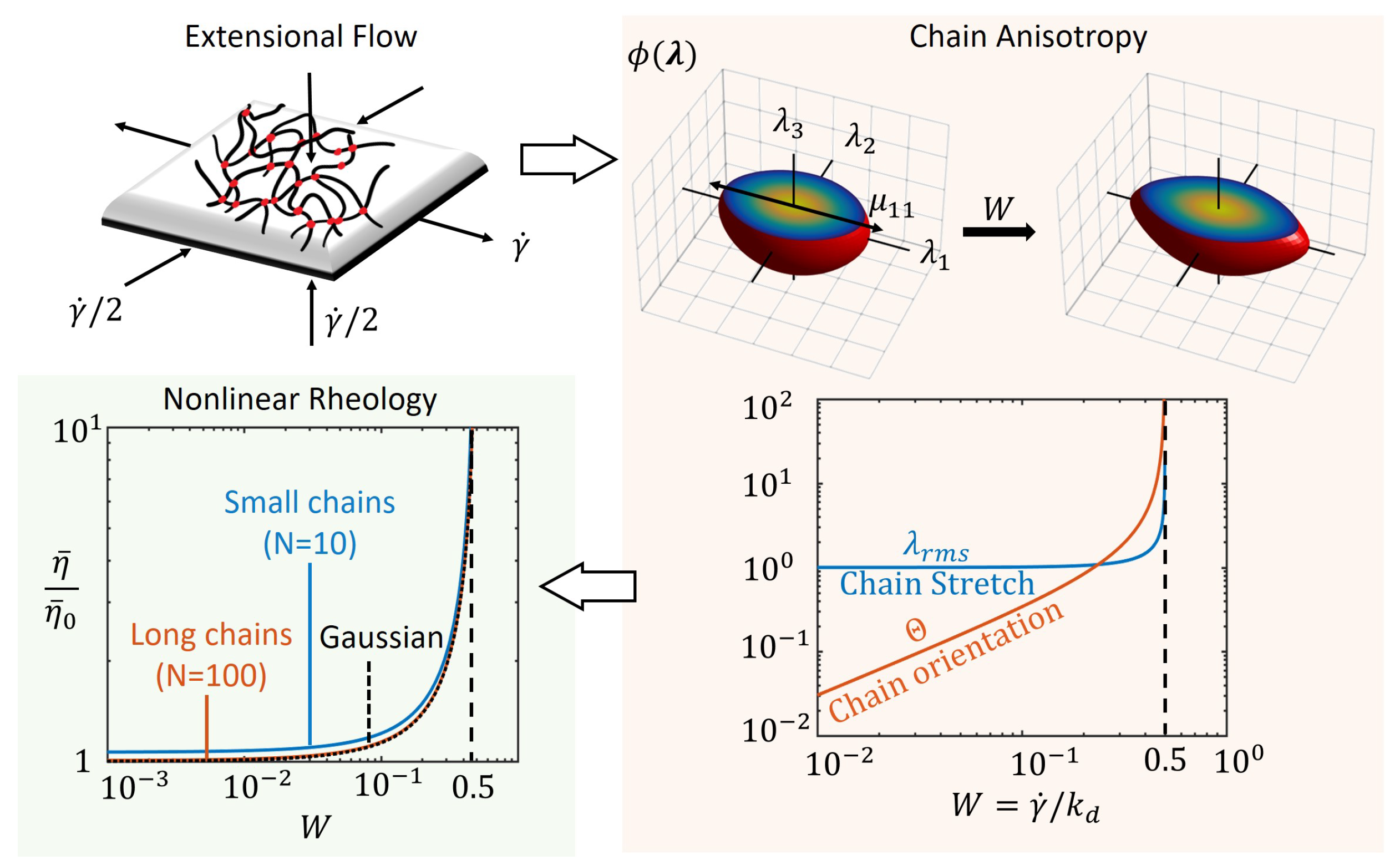
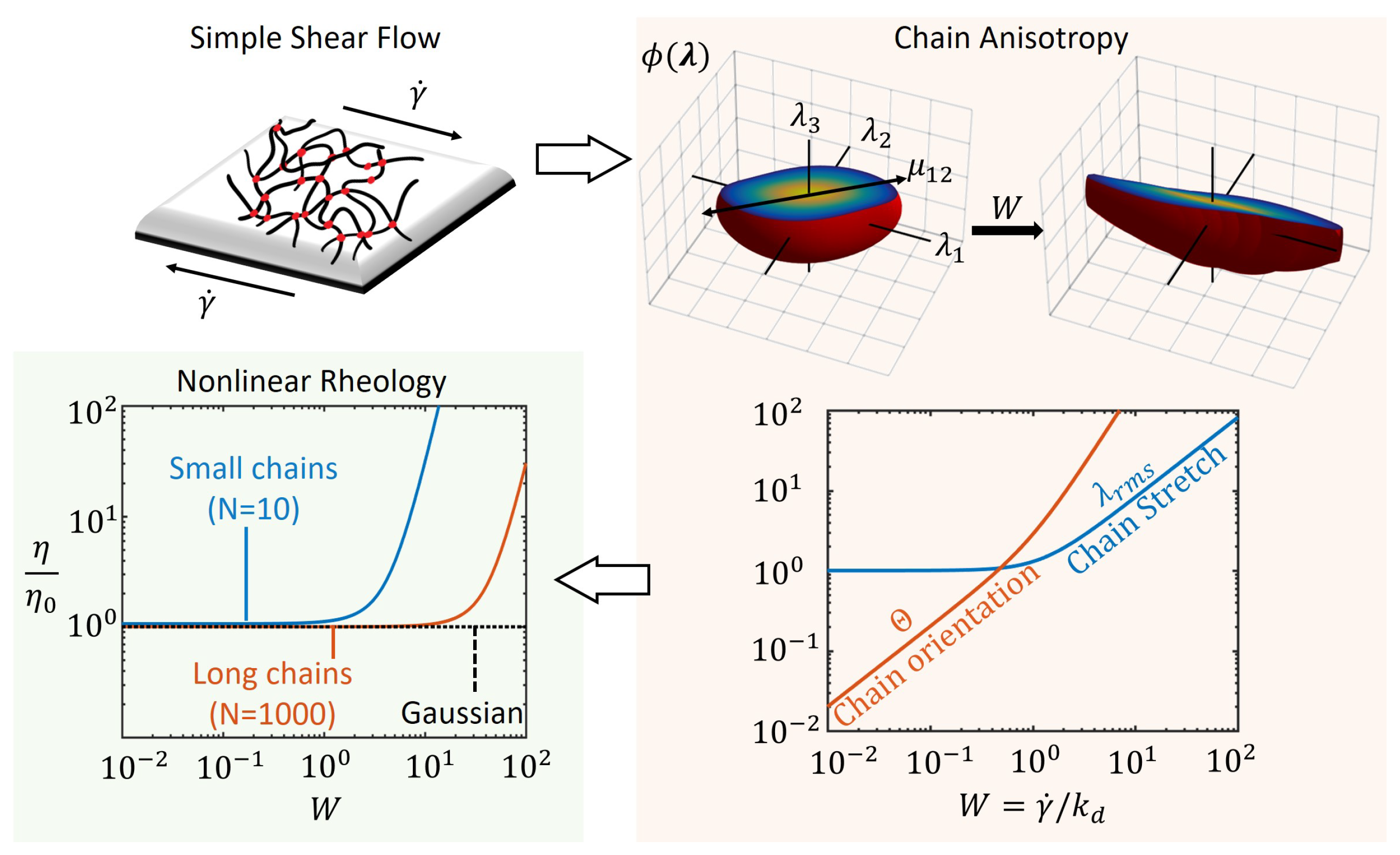
3. Flow Regimes under Steady Deformation Rates
3.1. Extensional Viscosity
3.2. Steady Shear Viscosity
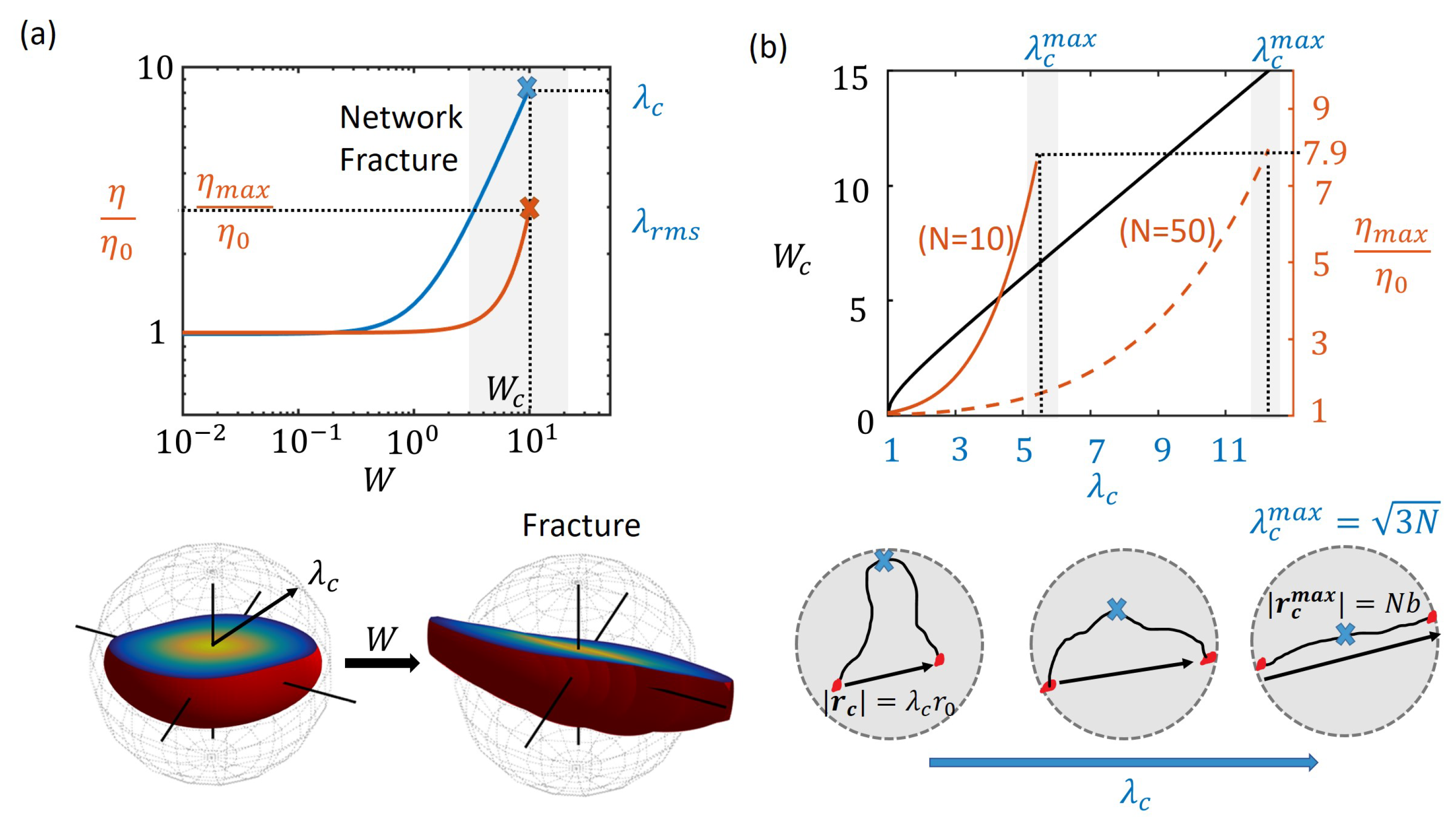
3.3. Network Fracture at Critical Strain Rates
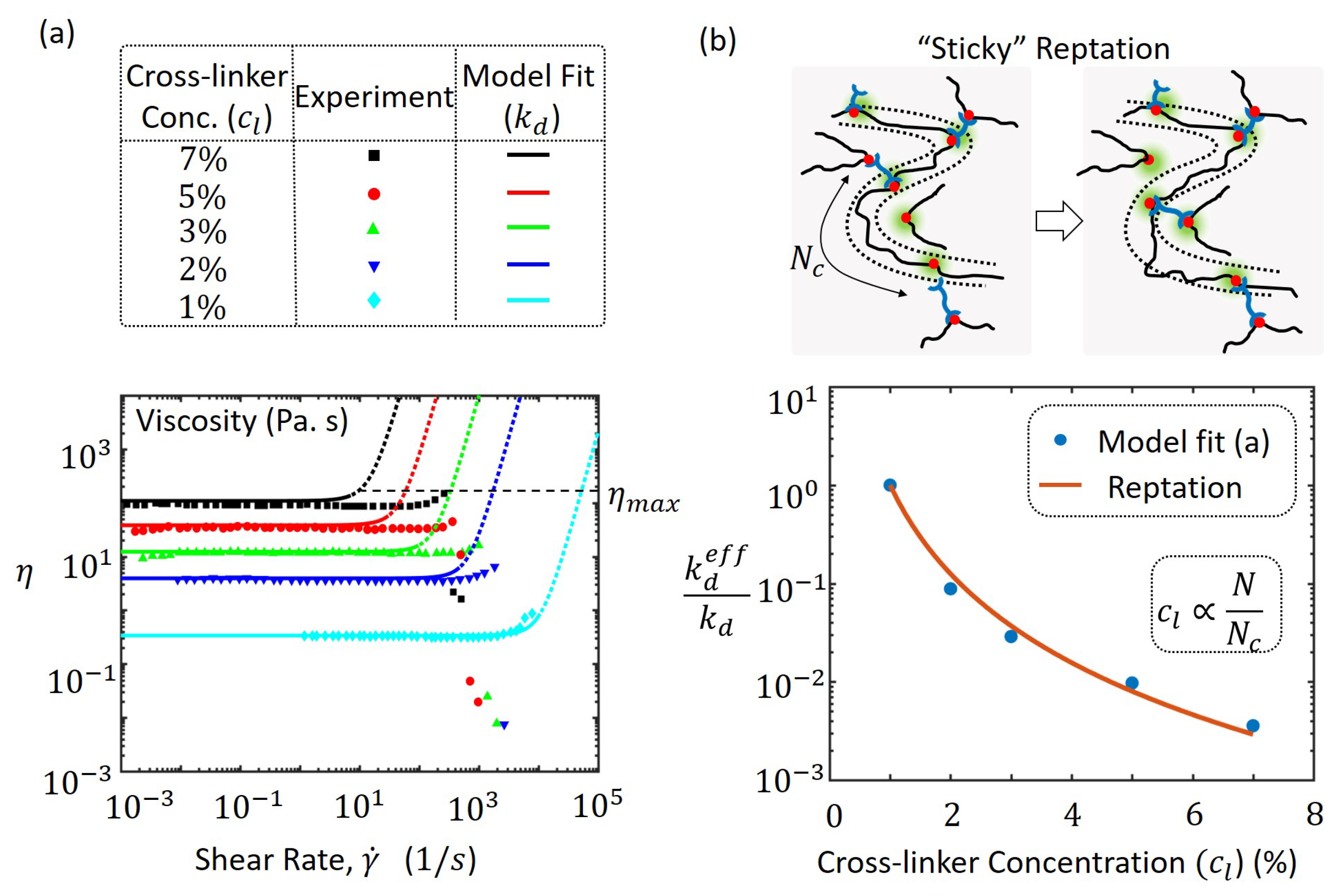
3.4. Viscosity at High Cross-Linker Densities
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| TNT | Transient Network Theory |
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of Open Access Journals |
Appendix A. Derivation of Solution for Shear Thickening Equations
Appendix B. Derivation of Friction for “Sticky” Reptation Model
References
- Green, M.S.; Tobolsky, A.V. A New Approach to the Theory of Relaxing Polymeric Media. J. Chem. Phys. 1946, 14, 80–92. [Google Scholar] [CrossRef]
- Tanaka, F.; Edwards, S.F. Viscoelastic properties of physically crosslinked networks. 1. Transient network theory. Macromolecules 1992, 25, 1516–1523. [Google Scholar] [CrossRef]
- Kramer, O. Biological and Synthetic Polymer Networks; Springer Science & Business Media: Berlin, Germany, 2012. [Google Scholar]
- Terech, P.; Schaffhauser, V.; Maldivi, P.; Guenet, J.M. Living polymers in organic solvents. Langmuir 1992, 8, 2104–2106. [Google Scholar] [CrossRef]
- Geitmann, A.; Ortega, J.K.E. Mechanics and modeling of plant cell growth. Trends Plant Sci. 2009, 14, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Vernerey, F.J.; Akalp, U. Role of catch bonds in actomyosin mechanics and cell mechanosensitivity. Phys. Rev. E 2016, 94, 012403. [Google Scholar] [CrossRef] [PubMed]
- Akalp, U.; Schnatwinkel, C.; Stoykovich, M.P.; Bryant, S.J.; Vernerey, F.J. Structural Modeling of Mechanosensitivity in Non-Muscle Cells: Multiscale Approach to Understand Cell Sensing. ACS Biomater. Sci. Eng. 2017, 3, 2934–2942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtecki, R.J.; Meador, M.A.; Rowan, S.J. Using the dynamic bond to access macroscopically responsive structurally dynamic polymers. Nat. Mater. 2011, 10, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.; Bruchmann, B.; Lehn, J.M. DYNAMERS: Dynamic polymers as self-healing materials. Chem. Soc. Rev. 2015, 44, 3786–3807. [Google Scholar] [CrossRef] [PubMed]
- Cordier, P.; Tournilhac, F.; Soulié-Ziakovic, C.; Leibler, L. Self-healing and thermoreversible rubber from supramolecular assembly. Nature 2008, 451, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Akalp, U.; Bryant, J.S.; Vernerey, J.F. Tuning tissue growth with scaffold degradation in enzyme-sensitive hydrogels: A mathematical model. Soft Matter 2016, 12, 7505–7520. [Google Scholar] [CrossRef] [PubMed]
- Bryant, S.J.; Vernerey, F.J. Programmable Hydrogels for Cell Encapsulation and Neo-Tissue Growth to Enable Personalized Tissue Engineering. Adv. Healthc. Mater. 2017, 7, 1700605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridhar, S.L.; Schneider, C.M.; Chu, S.; Roucy, G.D.; Bryant, J.S.; Vernerey, J.F. Heterogeneity is key to hydrogel-based cartilage tissue regeneration. Soft Matter 2017, 13, 4841–4855. [Google Scholar] [CrossRef] [PubMed]
- De Greef, T.F.A.; Meijer, E.W. Materials science: Supramolecular polymers. Nature 2008, 453, 171–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serpe, M.J.; Craig, S.L. Physical Organic Chemistry of Supramolecular Polymers. Langmuir 2007, 23, 1626–1634. [Google Scholar] [CrossRef] [PubMed]
- Seiffert, S.; Sprakel, J. Physical chemistry of supramolecular polymer networks. Chem. Soc. Rev. 2012, 41, 909–930. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H. Viscoelasticity and dynamics of entangled polymers. Prog. Polym. Sci. 1999, 24, 1253–1403. [Google Scholar] [CrossRef]
- Leibler, L.; Rubinstein, M.; Colby, R.H. Dynamics of reversible networks. Macromolecules 1991, 24, 4701–4707. [Google Scholar] [CrossRef] [Green Version]
- Loveless, D.M.; Jeon, S.L.; Craig, S.L. Rational Control of Viscoelastic Properties in Multicomponent Associative Polymer Networks. Macromolecules 2005, 38, 10171–10177. [Google Scholar] [CrossRef]
- Meng, F.; Pritchard, R.H.; Terentjev, E.M. Stress Relaxation, Dynamics, and Plasticity of Transient Polymer Networks. Macromolecules 2016, 49, 2843–2852. [Google Scholar] [CrossRef]
- Gold, B.J.; Hövelmann, C.H.; Lühmann, N.; Pyckhout-Hintzen, W.; Wischnewski, A.; Richter, D. The microscopic origin of the rheology in supramolecular entangled polymer networks. J. Rheol. 2017, 61, 1211–1226. [Google Scholar] [CrossRef]
- White, Z.W.; Vernerey, F.J. Armours for soft bodies: How far can bioinspiration take us? Bioinspir. Biomim. 2018, 13, 041004. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.G. The Structure and Rheology of Complex Fluids; Oxford University Press: Oxford, UK, 1999; Volume 150. [Google Scholar]
- Tripathi, A.; Tam, K.C.; McKinley, G.H. Rheology and Dynamics of Associative Polymers in Shear and Extension: Theory and Experiments. Macromolecules 2006, 39, 1981–1999. [Google Scholar] [CrossRef] [Green Version]
- Yount, W.C.; Juwarker, H.; Craig, S.L. Orthogonal Control of Dissociation Dynamics Relative to Thermodynamics in a Main-Chain Reversible Polymer. J. Am. Chem. Soc. 2003, 125, 15302–15303. [Google Scholar] [CrossRef] [PubMed]
- Yount, W.C.; Loveless, D.M.; Craig, S.L. Small-Molecule Dynamics and Mechanisms Underlying the Macroscopic Mechanical Properties of Coordinatively Cross-Linked Polymer Networks. J. Am. Chem. Soc. 2005, 127, 14488–14496. [Google Scholar] [CrossRef] [PubMed]
- Berret, J.F.; Séréro, Y.; Winkelman, B.; Calvet, D.; Collet, A.; Viguier, M. Nonlinear rheology of telechelic polymer networks. J. Rheol. 2001, 45, 477–492. [Google Scholar] [CrossRef]
- Amin, D.; Likhtman, A.E.; Wang, Z. Dynamics in Supramolecular Polymer Networks Formed by Associating Telechelic Chains. Macromolecules 2016, 49, 7510–7524. [Google Scholar] [CrossRef]
- Xu, D.; Hawk, J.L.; Loveless, D.M.; Jeon, S.L.; Craig, S.L. Mechanism of Shear Thickening in Reversibly Cross-Linked Supramolecular Polymer Networks. Macromolecules 2010, 43, 3556–3565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, K.C.; Jenkins, R.D.; Winnik, M.A.; Bassett, D.R. A Structural Model of Hydrophobically Modified Urethane Ethoxylate (HEUR) Associative Polymers in Shear Flows. Macromolecules 1998, 31, 4149–4159. [Google Scholar] [CrossRef]
- Regalado, E.J.; Selb, J.; Candau, F. Viscoelastic Behavior of Semidilute Solutions of Multisticker Polymer Chains. Macromolecules 1999, 32, 8580–8588. [Google Scholar] [CrossRef]
- Wang, S.Q. Transient network theory for shear-thickening fluids and physically crosslinked networks. Macromolecules 1992, 25, 7003–7010. [Google Scholar] [CrossRef]
- Witten, T.A.; Cohen, M.H. Crosslinking in shear-thickening ionomers. Macromolecules 1985, 18, 1915–1918. [Google Scholar] [CrossRef]
- Marrucci, G.; Bhargava, S.; Cooper, S.L. Models of shear-thickening behavior in physically crosslinked networks. Macromolecules 1993, 26, 6483–6488. [Google Scholar] [CrossRef]
- Séréro, Y.; Jacobsen, V.; Berret, J.F.; May, R. Evidence of Nonlinear Chain Stretching in the Rheology of Transient Networks. Macromolecules 2000, 33, 1841–1847. [Google Scholar] [CrossRef]
- Ma, S.X.; Cooper, S.L. Shear Thickening in Aqueous Solutions of Hydrocarbon End-Capped Poly (ethylene oxide). Macromolecules 2001, 34, 3294–3301. [Google Scholar] [CrossRef]
- Ballard, M.J.; Buscall, R.; Waite, F.A. The theory of shear-thickening polymer solutions. Polymer 1988, 29, 1287–1293. [Google Scholar] [CrossRef]
- Yamamoto, M. The Visco-elastic Properties of Network Structure II. Structural Viscosity. J. Phys. Soc. Jpn. 1957, 12, 1148–1158. [Google Scholar] [CrossRef]
- Indei, T. Necessary conditions for shear thickening in associating polymer networks. J. Non-Newton. Fluid Mech. 2007, 141, 18–42. [Google Scholar] [CrossRef]
- Yamamoto, M. The Visco-elastic Properties of Network Structure I. General Formalism. J. Phys. Soc. Jpn. 1956, 11, 413–421. [Google Scholar] [CrossRef]
- Vernerey, F.J.; Long, R.; Brighenti, R. A statistically-based continuum theory for polymers with transient networks. J. Mech. Phys. Solids 2017, 107, 1–20. [Google Scholar] [CrossRef]
- Vernerey, F.J. Transient response of nonlinear polymer networks: A kinetic theory. J. Mech. Phys. Solids 2018, 115, 230–247. [Google Scholar] [CrossRef]
- Treloar, L.R.G. The Physics of Rubber Elasticity; Oxford University Press: Oxford, UK, 1975. [Google Scholar]
- Trouton, F.T. On the coefficient of viscous traction and its relation to that of viscosity. Proc. R. Soc. Lond. A 1906, 77, 426–440. [Google Scholar] [CrossRef]
- Lodge, A.S. Elastic Liquids: An Introductory Vector Treatment of Finite-Strain Polymer Rheology; Academic Press: New York, NY, USA, 1964. [Google Scholar]
- Vaccaro, A.; Marrucci, G. A model for the nonlinear rheology of associating polymers. J. Non-Newton. Fluid Mech. 2000, 92, 261–273. [Google Scholar] [CrossRef]
- Thomas Hu, Y. Mechanisms of shear thickening in transient guar network. J. Rheol. 2014, 58, 1789–1807. [Google Scholar] [CrossRef]
- Indei, T.; Koga, T.; Tanaka, F. Theory of Shear-Thickening in Transient Networks of Associating Polymers. Macromol. Rapid Commun. 2005, 26, 701–706. [Google Scholar] [CrossRef]
- Kersey, F.R.; Yount, W.C.; Craig, S.L. Single-molecule force spectroscopy of bimolecular reactions: System homology in the mechanical activation of ligand substitution reactions. J. Am. Chem. Soc. 2006, 128, 3886–3887. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.; Colby, R.H. Polymer Physics; Oxford University Press: Oxford, UK, 2003; Volume 23. [Google Scholar]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lalitha Sridhar, S.; Vernerey, F.J. The Chain Distribution Tensor: Linking Nonlinear Rheology and Chain Anisotropy in Transient Polymers. Polymers 2018, 10, 848. https://doi.org/10.3390/polym10080848
Lalitha Sridhar S, Vernerey FJ. The Chain Distribution Tensor: Linking Nonlinear Rheology and Chain Anisotropy in Transient Polymers. Polymers. 2018; 10(8):848. https://doi.org/10.3390/polym10080848
Chicago/Turabian StyleLalitha Sridhar, Shankar, and Franck J. Vernerey. 2018. "The Chain Distribution Tensor: Linking Nonlinear Rheology and Chain Anisotropy in Transient Polymers" Polymers 10, no. 8: 848. https://doi.org/10.3390/polym10080848
APA StyleLalitha Sridhar, S., & Vernerey, F. J. (2018). The Chain Distribution Tensor: Linking Nonlinear Rheology and Chain Anisotropy in Transient Polymers. Polymers, 10(8), 848. https://doi.org/10.3390/polym10080848