1. Introduction
The outstanding mechanical performance of semicrystalline polymers, including their ability for large-scale plastic deformation, can be attributed to their unique morphology, consisting of crystalline and amorphous elements in the form of thin alternating layers. Additionally very important is the specific structure of layers—more or less regularly folded chains in crystalline lamellae vs. highly entangled chains constituting the amorphous phase—and the robust phase interconnectivity, which is provided by numerous chains intersecting the amorphous-crystalline interface and ensuring the extremely strong covalent bonding of adjacent amorphous and crystalline layers. These chains allow for the load transfer between neighboring lamellae across the stack. It is commonly accepted that the performance of semi-crystalline polymers crucially depends on such molecular links, which are called stress transmitters (STs) [
1]. Therefore, detailed knowledge, not only about the individual phases but also the interplay between them, is essential for understanding the deformation habits of semicrystalline polymers.
The deformation of a semicrystalline polymer appears to be a complicated process, involving all elements of their complex morphology. In this process, different micromechanisms are activated at various stages [
1,
2,
3,
4,
5]. It is well documented that the crystalline part primarily deforms through mechanisms of a crystallographic nature [
2,
4,
5,
6], including various modes of crystallographic slip (which is principally the dominant mechanism), twinning or stress-induced crystallographic (martensitic) transformation. These mechanisms generally appear similar to those operating in low-molecular crystalline solids. A strain-induced ‘melting-recrystallization’ has also been proposed as a plausible mechanism involved in deformation [
5,
7,
8]. The plastic deformation of polymer lamellar crystals is accompanied by several mechanisms operating in the amorphous phase: interlamellar shear (slip), lamella separation, and rotations of the stacks of lamellae. These mechanisms support the deformation of crystalline lamellae. Moreover, interlamellar shear can substitute the lacking slip systems that would intersect the chain direction. The joint activity of all these mechanisms allows for a full accommodation of the strain [
4]. The deformation mechanisms mentioned above can be employed in a complex deformation sequence, with particular mechanisms activated and terminated at specific strains. Furthermore, the active mechanisms can be adjusted with advancing strain due to interactions between the adjacent amorphous and crystalline layers, which are intimately connected and therefore forced to jointly deform. These interactions can also lead to some deformation instabilities, which in turn, may effect in opening new deformation paths and/or launch alternative mechanisms, previously inactive. Therefore, such instabilities may appear very important, sometimes essential, in the entire deformation sequence. Recent studies have pointed out a very substantial role of the amorphous phase, including the ST chains in the process of plastic deformation and a deep mutual relationship between the deformation of the amorphous and crystalline components [
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24,
25,
26,
27,
28,
29]. The role of the amorphous phase and its topology-dependent properties have turned out to be much more important in the deformation process than merely the role of a compliant medium transferring the load and adapting to the deformation of crystalline lamellae, as it was commonly assumed in the past. Amorphous phase layers have proven to be an active part of the structure, capable of tuning the deformation of crystalline lamellae. They can even take control of deformation, especially in the strain-hardening range, when the network stresses in amorphous layers become higher than the shear stresses needed to deform adjacent crystallites [
11,
13,
19,
30]. Frequently, the partial damage of crystalline lamellae has been observed in the same strain range—reaching the extensibility limit by the molecular network leads to a significant increase in resistance to the deformation of the amorphous phase and, consequently, to a substantial increase in stress [
10,
11,
20,
31]. The stretched ST chains, immobilized at the entry points to the crystal, give rise to localized stress concentrations on the lamellar faces. Because at this point the lamellae became notably thinner due to a well-advanced activity of the slip [
2], these stress concentrations may trigger instabilities in that slip through its severe localization, which can quickly lead to lamellae disruption and its fragmentation into smaller blocks. Such fragmentation relieves much of the structural constraints imposed on deforming lamellae by their neighborhood and makes further strain accommodation easier.
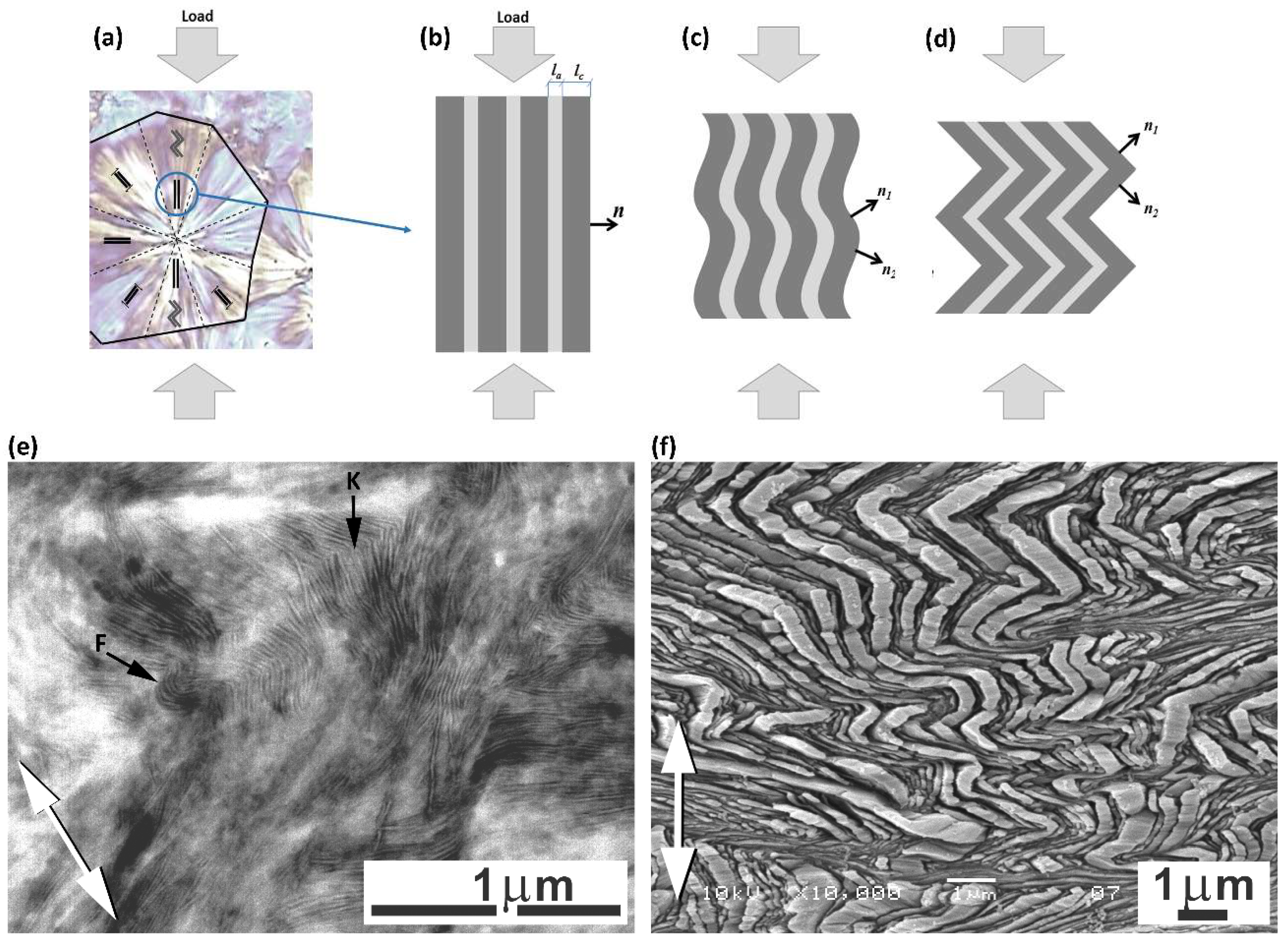
The main deformation instabilities found in the plastic deformation of semicrystalline polymers are associated primarily with cavitation, the formation of lamellar kinks, and lamellae fragmentation, which often lead to profound changes of the material morphology, as e.g., the transformation of the initial lamellar structure into microfibrillar due to lamellae fragmentation, frequently observed in tension. All of these instabilities are essentially related to the properties of the amorphous phase as well as to its stress response. For example, the lamella stacks respond to the tensile stress perpendicular to the lamella plane, initially with the dilatation of the amorphous layers, which can end up either with cavitation within this layer or with the cooperative bucking of layers, the formation of kinks, and, subsequently, the chevron-like morphology, depending on the properties of the amorphous layer, including its coupling to adjacent lamellae and additionally on the stress field, which can either promote or suppress internal cavitation [
9].
While the cavitation, commonly observed in tension, has been studied (see, e.g., [
32] and references therein), much less attention has been paid to other deformation instabilities, like microbuckling [
29,
33] or lamellae fragmentation [
8,
11,
20,
21,
24,
31]. This shortage motivated our recent study [
33], in which we probed these phenomena with X-ray and differential scanning calorimetry(DSC) techniques and tried to find how the deformation process and its stability are influenced by the amorphous layers and its properties, including their connection to the adjacent crystalline layers. For this purpose, polyethylene samples of diverse structure and morphology were examined in plane-strain compression. Two main deformation instabilities accompanying plastic deformation were investigated: the microbuckling instability (observed around the true strain of e = 0.3–0.4) and the fragmentation of crystalline lamellae due to the localization of the advanced crystallographic slip (e = 0.6–1.0). Microbuckling leads to the cooperative folding and kinking of lamellae that were initially parallel to the compression direction; hence, this subsequently leads to their rapid and substantial reorientation. This transformation can show up macroscopically as the so-called ‘second yield.’ For a given stiffness of layers, which is practically constant within the studied set of samples, the microbuckling appears dependent on the ratio of the amorphous and crystalline layer thicknesses. The second instability—an extensive fragmentation of crystalline lamellae—has been postulated to be prompted by local stress concentrations generated at lamella faces by taut ST chains. The critical strain related to this phenomenon has been found dependent on the surface fraction of STs at the amorphous-crystals interface—when this fraction is low, fewer but stronger stress concentrations are expected, which apparently cause an earlier localization of the crystallographic slip [
34] that cause the rapid local thinning of lamellae and quickly leads to their fragmentation. Both deformation instabilities deeply influence the further deformation path of the material and affect its final texture.
The goal of the present study was to extend the previous work [
33] and investigate how modifying the topology in the amorphous phase can affect the course and stability of the process of plastic deformation of a polymer. In this previous work, the samples demonstrated various crystallinity and layer thickness levels but a similar topology of the amorphous phase, consisting of the network of entangled chains. In contrast, the polyethylene samples prepared for this study had a nearly identical crystalline phase and layers thickness but significantly differed in the chain topology within the amorphous phase. The macromolecular network in amorphous layers of samples used here consisted of both chain entanglements and chemical crosslinks, introduced by electron beam irradiation. As a consequence of solid chemical crosslinks, the amorphous component demonstrated various stiffness levels as well as the concentration of stress transmitters.
All samples were deformed by compression in the plane-strain conditions using a channel-die, similarly to a process employed in the previous studies [
18,
19,
20,
31,
33]. The main advantage of this deformation mode is its macroscopic homogeneity (no neck formation) and a lack of cavitation, which is prevented by the compressive stress components. Thus, the plane-compression allows for the study of the course of deformation that is not concealed by any other unwanted or inessential effects, in contrast to other deformation modes, e.g., tensile deformation, where the strong cavitation is usually observed.
4. Discussion
In our recent paper [
33] the deformation instabilities accompanying the plastic deformation of polyethylene of diverse structure and morphology were investigated. The first instability, observed at the true strain e = 0.3–0.4, was the microbuckling that led to the joint, cooperative folding or kinking of lamellae and frequently macroscopically manifested as the second yield. It was found that for a given stiffness of individual phases, the initiation of microbuckling depended on the ratio of the thickness of the crystalline and amorphous layers
la/lc, the dependence of which seemed to be consistent with the general concept that the buckling of layered materials is driven by different stiffness degrees of adjacent layers. At e = 0.6–1.0, another significant instability was observed: the fragmentation of crystalline lamellae due to the significant localization of the crystallographic slip. We postulated that the fragmentation was initiated in already highly deformed lamellae by the stress concentrations generated at the amorphous-crystal interface by stress transmitter chains, stretched and taut due to the interlamellar shear in amorphous layers that accompanies the deformation of crystalline lamellae. As a consequence, the critical strain for lamellae fragmentation appeared to depend on the surface fraction of stress transmitters at the interface (
FS)—when
FS was low, the stress concentrations were fewer but grew stronger than in the system of high
FS. This, in turn, could trigger an earlier localization of the slip, quickly leading to lamella break-up. Such instability had a large impact on further deformation of the material and strongly affected its final texture—the massive fragmentation of lamellae significantly reduced structural constraints imposed on deforming material and facilitated the formation of a new ordering of fragmented crystals along the FD.
In contrast to the previous work, where the samples demonstrated various degrees of crystallinity and layer thickness but had a similar topology of the amorphous phase, consisting of the network of entangled chains (although exhibiting different effective network densities) [
33], the samples studied in this work exhibited a nearly identical crystalline phase (the same orthorhombic crystal modification, similar crystal thickness, degree of crystallinity, etc.) but significantly different topology in the amorphous phase, where the molecular network contained both chain entanglements and solid chemical crosslinks, introduced in various amounts by electron beam irradiation. As a consequence, the amorphous component in irradiated samples demonstrated larger stiffness and concentration degrees of stress transmitters, which were significantly higher than in the raw material (
cf. Table 2).
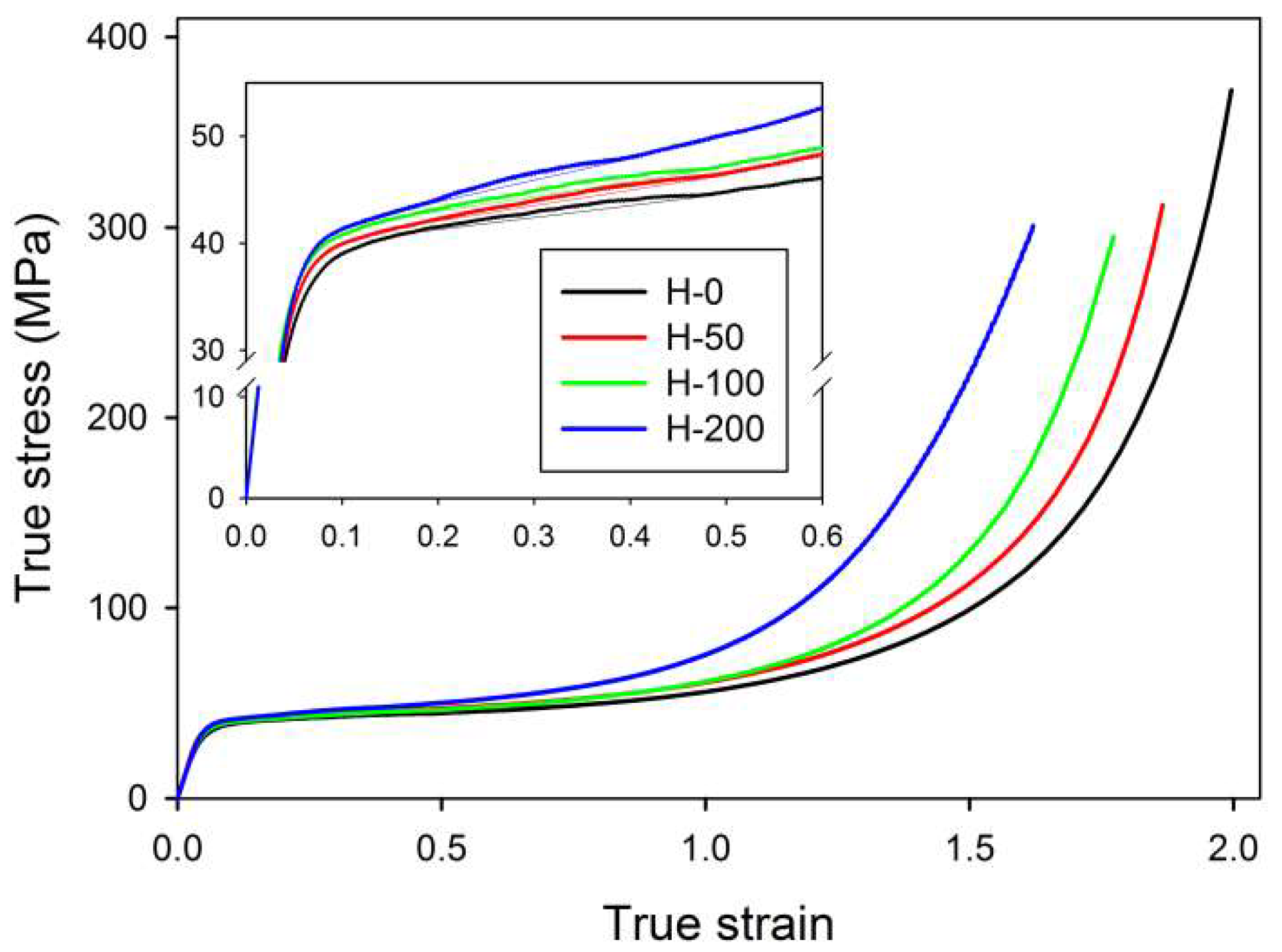
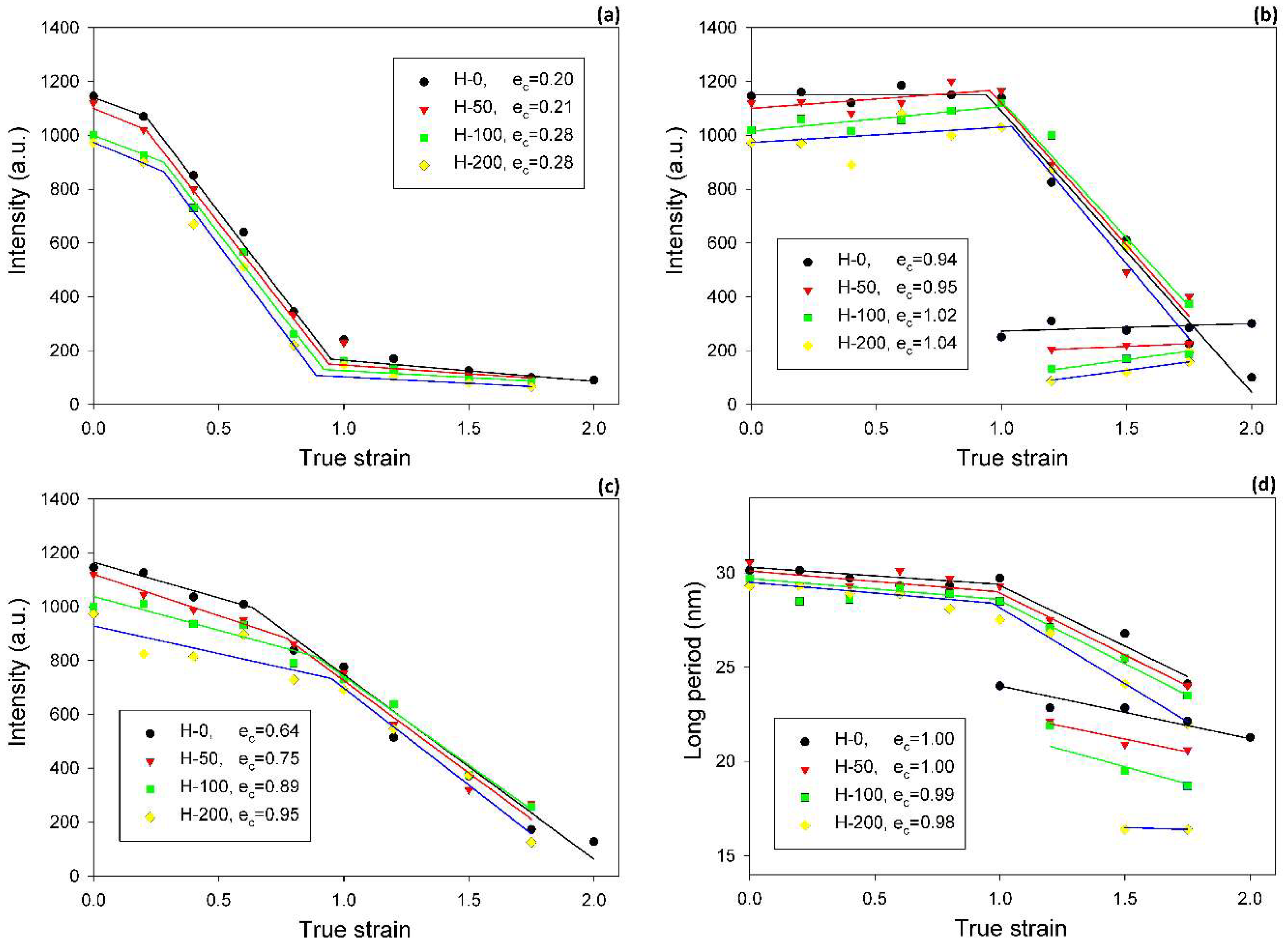
The effects of buckling instability were observed in crosslinked samples in the range of low strain, similarly to the non-crosslinked samples studied previously [
33]. This instability resulted in the development of the macroscopic second yield, which was observed in the true stress–true strain curves as a broad and low maximum, located at approximately e ≈ 0.2–0.45 (
cf. Figure 2 and
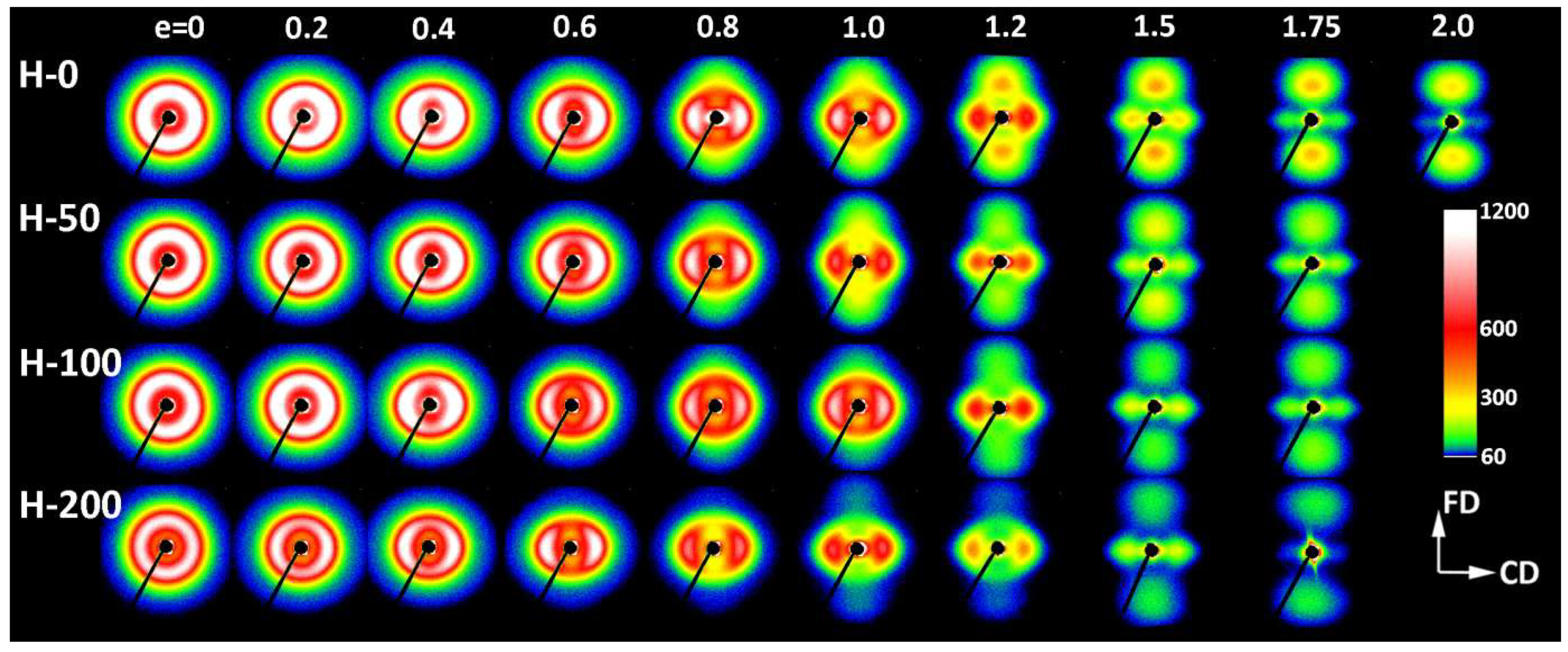
Table 3). In the same strain range, a steep decrease in intensity of scattering along the FD was detected in 2-D SAXS images (
cf.
Figure 4,
Figure 5 and
Figure 8a). This fast reduction of the scattering intensity along the FD indicates that the progressing deformation led to the rapid elimination of these lamellae that were oriented initially along the LD. Moreover, the four-point pattern developed in the CD view SAXS images at slightly higher strains could be easily observed at e ≥ 0.4 (
Figure 4). These changes in SAXS images suggest a swift rotation of lamella initially oriented along the LD to the new preferred orientation, seen approximately 45–50° away from the FD. All observations reported above probably evidence the same mechanism, which, in our opinion, was the buckling instability, activated around e = 0.3 and quickly leading to the development of joint kinks in lamellae. The drop of SAXS intensity along the FD, which can be considered as the signature of initiation of such microbuckling and subsequent kinking, progressively shifted towards higher strain with increasing irradiation dose, from e
c(
IFD) = 0.2 (in H-0) to 0.28 (in H-200)—
cf.
Table 3.
Buckling instability, resulting in the cooperative folding or kinking of layers, was observed in various layered materials in response to the compressive load along the direction of layers or to the tensile load, perpendicular to them. It seems to be a general phenomenon occurring in various materials on remarkably different length scales that can range from the molecular scale (e.g., liquid crystals) up to the macroscopic scale (like in geological formations) [
9]. The precondition of buckling is layered morphology, which must consist of stacks of alternating soft and hard relatively thin layers. They must be also strongly coupled, especially when loaded in tension. In such systems, buckling originates from very different stiffness degrees of the soft and hard layers [
9]. Buckling instability, ending up with the formation of lamellar kinks, has also been observed in semicrystalline polymers, e.g., in PE [
47,
48,
49], iPP [
29,
50], POM [
51] and iPS [
52]. Angular kinks initiated by layer buckling cause only limited damage to the crystalline lamellae, mainly at the kink tip [
33] (see also
Figure 3f). Nevertheless, kinking appeared a vitally important phenomenon for the subsequent deformation behavior of the material—this induced a swift reorientation of the lamellae, which were not able to plastically deform earlier, and it additionally caused the skeleton of the stiff lamellae constituting initial structure to rapidly collapse. Prior to kink formation, the lamellae parallel to the compression direction could not deform by crystallographic slip due to very low resolved shear stress in the plane and the direction of any potential slip system. The reorientation of lamellae in kinks led to a significant increase in the resolved shear stress in the slip plane, which could initiate the crystallographic slip process in such lamellae, now becoming relatively easy, just like the crystallographic slip that was already been active in other lamellae that oriented obliquely to the LD, These phenomena opened up new paths for the plastic deformation of the whole material. Microbuckling and subsequent kinking, triggering new mechanisms of plastic deformation, often macroscopically manifest as the second yield point.
In a recent work [
33], we found that for a given stiffness of the crystalline and amorphous phases, the initiation of lamellae microbuckling depended on the ratio of the amorphous and crystalline layer thickness
la/lc, and kinks started to develop at a lower strain when
la/lc decreased. In this study, the samples did not markedly differ in thickness of the amorphous and crystalline layers, as well as in degree of crystallinity, i.e.,
la/lc and the stiffness of crystalline layers were roughly constant. Instead, they differed significantly in the properties of the amorphous phase, which exhibited increasing stiffness with increasing crosslink density. As reported above, initiation of microbuckling, indicated by the SAXS data, shifted toward higher strains with an increasing irradiation dose (increasing crosslink density). It can be concluded then that lamellae buckling was controlled in this case (
la/lc ≈ const, E
cr = const) by the stiffness of the amorphous phase, which increased with the dose—see
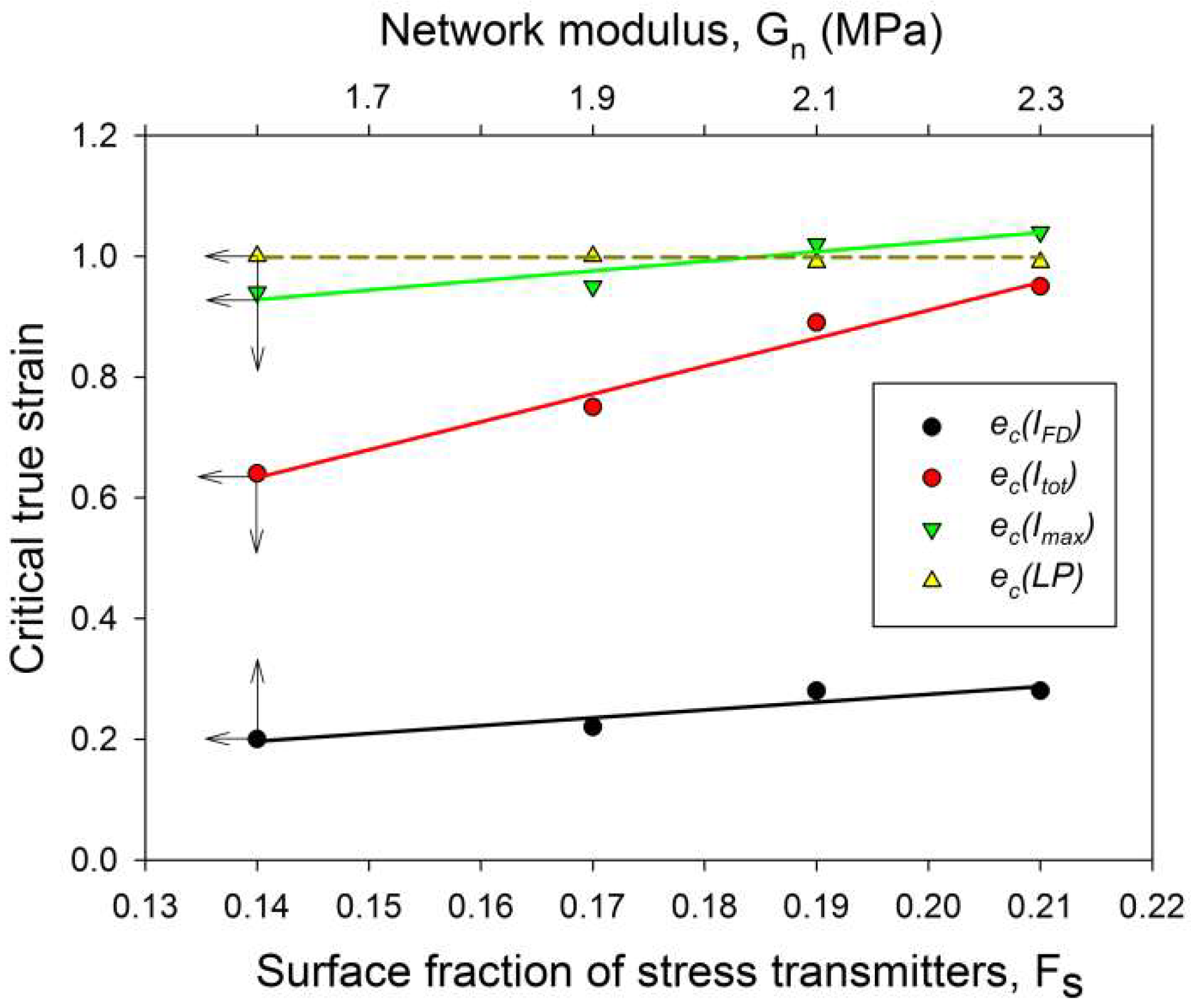
Table 2, which reports the network modulus of raw and irradiated samples. The relationship between buckling instability and the stiffness of amorphous layers is illustrated in
Figure 11, which presents the dependence of the critical strain e
c(
IFD) on the strain hardening (network) modulus of the amorphous component. The conclusion drawn here agrees with the general prediction that buckling is driven by different stiffness degrees of the layers (which are determined by the material stiffness of the respective phase and the layer thickness) [
9]. If the stiffness of the crystalline and amorphous phase remains unchanged, then other morphological parameters, like the thickness ratio of hard and soft layers, can also play a role in the activation of buckling instability; this was indeed observed in the previous study [
33].
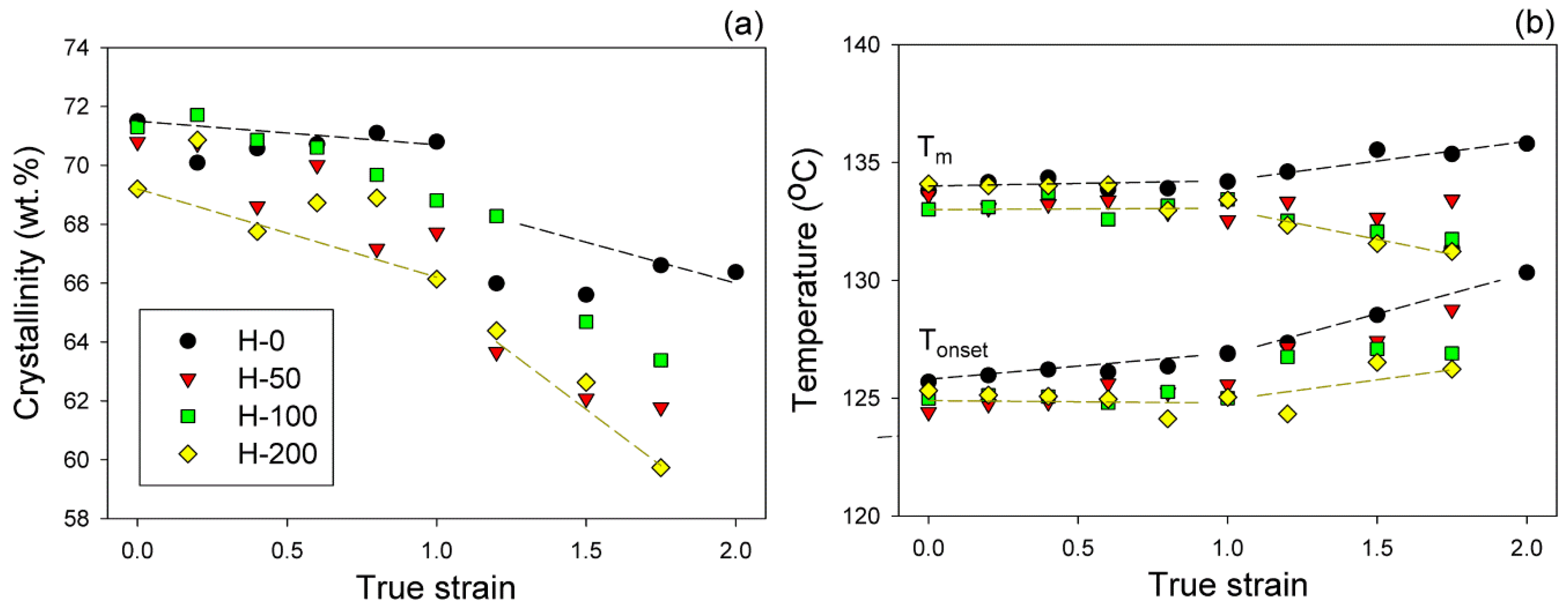
Another deformation instability that was detected in crosslinked samples at higher strain, around e = 1.0, manifested as curve break points on the dependencies of the SAXS intensity (maximum or average), long period, average crystal size, crystallinity, and melting temperature on strain (see
Figure 8,
Figure 9 and
Figure 10, respectively). All of these specific changes observed on the respective plots could be related to the same lamellae fragmentation phenomena, which were induced in this strain range by an advancing deformation [
33]. Fragmentation results in partial damage to crystal ordering. Moreover, it can lead to the formation of new stacked lamellae that were thinner than the parent and oriented normally along the FD.
One of the clear marks of lamellae fragmentation is a sudden decrease of crystallinity, here observed around e
c(
Xc) ≈ 1.0 (
Figure 10a). Crystallinity significantly fell in a narrow strain range, even by 4 wt.% in the neat sample H-0 and slightly less in irradiated samples. This means that the damage of a part of the lamellar crystals, most likely due to their fragmentation into smaller species, was accompanied by local, yet notable, destruction of the crystalline ordering. Such structure damage phenomena were reported earlier to occur in PE in this range of strain [
11,
20,
31,
33]. The increase of the onset temperature of melting, seen in the same range of strain (
Figure 10b), suggests that fragmentation involved primarily the thinnest crystallites, which were apparently destroyed first.
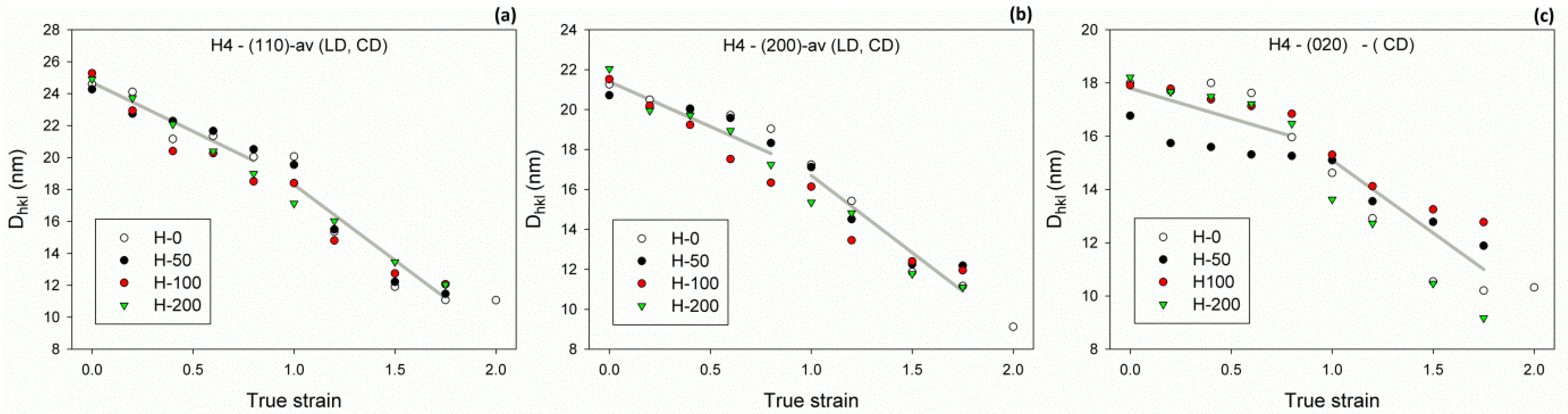
Another indication of lamellae damage by extensive fragmentation can be found in the relationship between the average crystal size and strain. The average X-ray coherent crystal sizes
Dhkl (estimated in various directions in crystal) tended to decrease with strain but with a varying rate, which appeared notably higher in the high strain range than in the low strain range (see
Figure 9). The cross-over point could be located approximately at the strain of e
c(
Dhkl) = 0.9–1.0, irrespective of the crystallographic direction. The faster reduction of average crystal size above this critical strain could be considered as another signature of lamellae fragmentation, which appeared to markedly intensify above e
c(
Dhkl). This coincided well with the conclusion derived from DSC data, discussed earlier. Regrettably, any deeper analysis of the presented dependencies is problematic due to the noticeable scatter of data points. In particular, it is hard to assess whether the critical strain associated with a reduction of the average crystal size e
c(
Dhkl) showed any dependence on the irradiation dose (hence, the density of crosslinks) or did not.
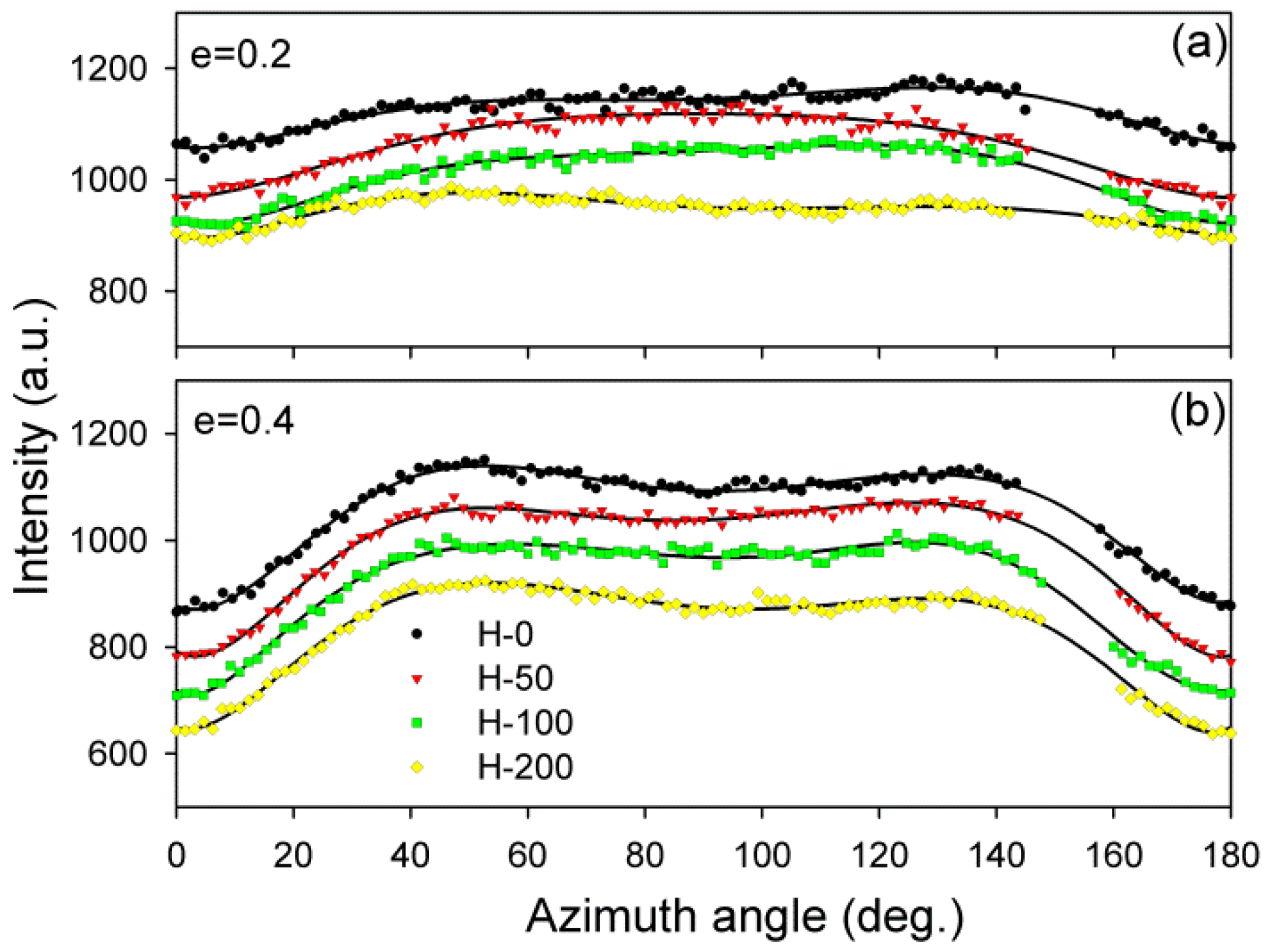
Some characteristic changes indicating lamellae fragmentation could be, furthermore, recognized in the lamellar structure at the strain around e = 1, when probed with 2-D SAXS. The CD view SAXS images of the deformed samples (
Figure 4) demonstrated that the intensity in the maxima of the four-point pattern, which initially increased with increasing strain up to e ≈ 1, started to quickly decrease at higher strains, e > 1 (
cf.
Figure 8b). As reported in
Table 3, the crossover point, determined from the plots presented in
Figure 8b gradually moved to higher strain with increasing irradiation dose, from e
c(
Imax) = 0.94 to e
c(
Imax) = 1.04, for H-0 and H-200, respectively. The quickly decreasing intensity in the maxima of the pattern indicated a fast reduction of the population of lamellae stacks that were normally oriented in their current preferred orientation direction. This notion is supported by the results of the average intensity obtained by integration along the azimuth, which also showed a quick decrease of
Itot at high strain, although for every dose, the critical strain e
c(
Itot) = 0.65–0.95 (
Figure 8c) was smaller than the respective critical strain e
c(
Imax) (
cf. Figure 8b,c and
Table 3). This was probably a result of the contribution of the scattering in directions around the FD, which already rapidly decreased at low strains because of microbuckling and kinking, as discussed earlier.
Another cross-over point was found in the curve of the primary long period vs. applied true strain, approximately at ec(LP) ≈ 1.0—LP decreases notably faster at e > ec(LP) than below ec. The decrease of LP is associated with advancing plastic deformation by chain slip, which results in an increasing chain tilt in lamella and consequently decreasing lamella thickness, hence decreasing LP. The steeper decrease of LP above ec(LP) could have been a result of an extensive lamellae fragmentation, which seriously relieved the structural constraints that were imposed earlier on deforming lamellae by the initial structure. This makes the further deformation by slip mechanism in survived lamellae fragments easier and faster than in the initial, more constrained structure. Consequently, the thickness of these lamellae, and thus the LP, could decrease at a rate higher than prior to fragmentation.
The next consequence of lamellae fragmentation is the development of a completely new population of thin and short lamellae, preferentially oriented roughly perpendicular to the FD. They were formed by the restructuration of small crystalline blocks that survived the fragmentation of highly deformed initial lamellae [
20]. The stacked new lamellae accounted for a new feature emerging in SAXS patterns right after lamella fragmentation, at e > 1.0. The scattering by these new elements of the crystalline structure had a look similar to the scattering produced by the fibrillar structure that usually completely replaced the initial lamellar structure upon drawing. However, the new lamellae formed in compression at room temperature did not completely substitute the initial structure and rather seemed to coexist with the remains of deformed and fragmented lamellae of the original structure, even at very high strains. The long period associated with the new lamellae was shorter than that associated with the initial structure, although it still seemed to depend on the original LP [
33]. The scattering intensity of the new component of the deformed structure, once it appeared, increased only slightly with advancing deformation. This indicated that the short lamellae that contribute to this new structure component, probably formed in a single step rather than in a longer process, extended to a range of strains. As mentioned in the previous section, our observations demonstrate that the new lamellae arrangement arose from small blocks that survived the extensive lamellae fragmentation and from highly oriented amorphous chains in a process of some restructuring [
20]. However, this process cannot be identified with often postulated ‘melting-recrystallization’ mechanism [
7], because the thickness of the new lamellae seemed to depend on the thickness of the parent lamellae in initial structure rather than being related to the temperature of deformation as required in the strain-induced ‘melting-recrystallization’ mechanism [
33].
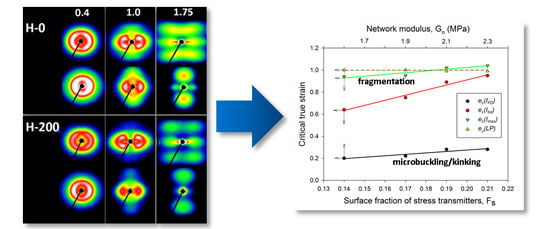
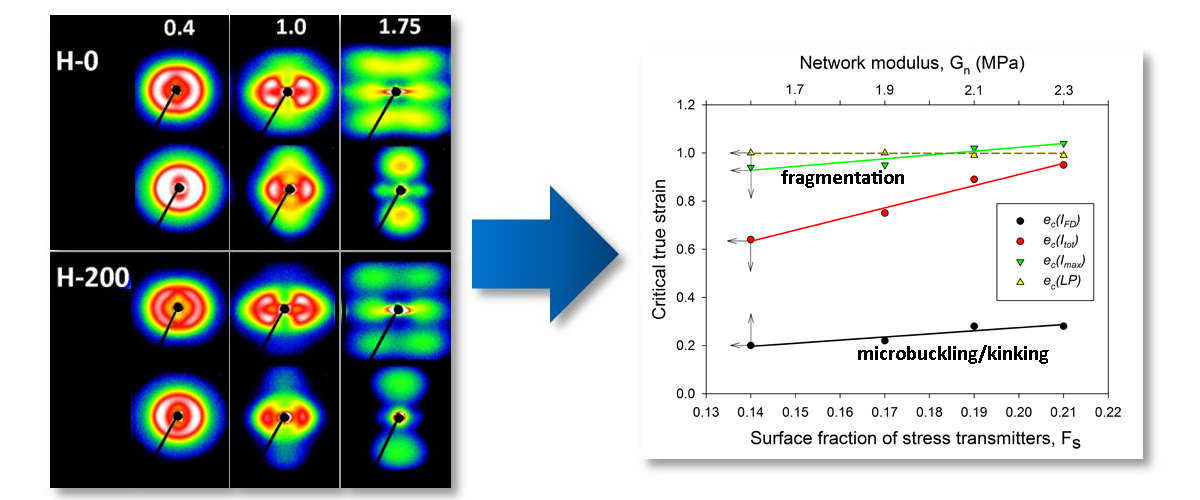
The critical strains e
c(
Imax) and e
c(
Itot) visibly increased with increasing irradiation. Regrettably, other critical strains related to lamellae fragmentation, derived from the SAXS, WAXS and DSC results, though located in the same strain range as e
c(
Itot), did not show such a clear dependence, perhaps due to the small variability and/or insufficient accuracy of estimates. Nevertheless, it seems reasonable to conclude just on the basis of the variation of e
c(
Imax) and e
c(
Itot) that the instability of deformation, which brought extensive lamellae fragmentation, was correlated with the irradiation dose. On the other hand, that radiation dose determined the number of crosslinks, hence the properties of the amorphous phase, including the molecular network density
Neff (consisting now of both chain entanglements and crosslinks) and the surface fraction of stress transmitters at the amorphous-crystal interface
Fs. In this way, a relationship was established between the deformation instability leading to lamellar fragmentation and the parameters describing the amorphous phase, either
Neff,
Fs or
Gn (the network modulus). These parameters were discussed in the previous section and their values are reported in
Table 2. The relation between
Neff and
Fs is given by Equation (5). Since the thickness of the amorphous layer was practically constant in all samples studied here, the parameters
Neff and
Fs became equivalent:
Fs = const·
Neff. In addition, there was a linear dependence of
Gn on
Neff:
Gn = kT·
Neff. In
Figure 11, which presents several critical strains derived from SAXS data and plots them in function of
Fs, a clear linear relationship between e
c(
Itot) or e
c(
Imax) and
Fs can be observed. In the previous paper [
33], we also concluded that lamellae fragmentation was controlled by the topology in adjacent layer, primarily through
Fs. The following explanation was proposed: stress transmitter chains, when stretched out due to the shear in amorphous layer, which accompanies crystallographic slip, generate stress concentrations at the amorphous-crystal interface. Then, low
Fs result in fewer but stronger stress concentrations on the lamella face, which bring the localization of the crystallographic slip and a non-uniform ‘coarse’ deformation of the lamellae [
34]. Consequently, localized slip quickly leads to lamella disruption and an earlier fragmentation of lamellae into small species. On the contrary, large
Fs result in smaller stress concentrations and thus a more homogeneous distribution of the stress on the lamella face, which promotes a more uniform, ‘homogeneous’ slip in crystalline lamellae and thus delays their fragmentation (due to other effects, e.g., non-uniform thickness) to a larger strain. The data presented in
Figure 11 fully support the above findings.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}