Mitigating the Impact of Cellulose Particles on the Performance of Biopolyester-Based Composites by Gas-Phase Esterification


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
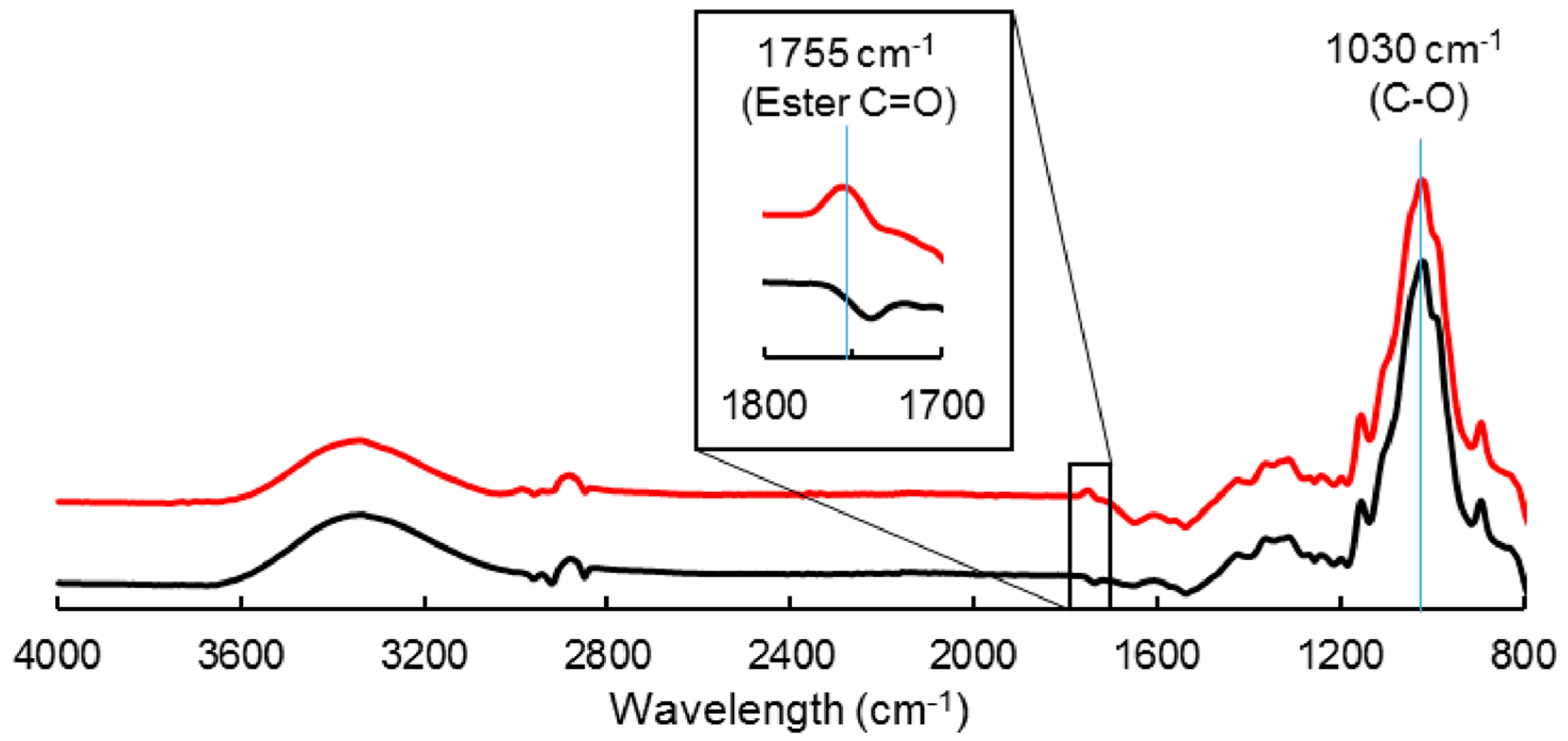
2.2.1. Grafting of Cellulose
2.2.2. Preparation of Composite Materials
2.2.3. Characterization of Films
2.2.4. Modeling
3. Results and Discussion
3.1. Impact of Gas-Phase Esterification on Some Macromolecular Parameters of PHBV
3.1.1. Molecular Weight
3.1.2. Differential Scanning Calorimetry
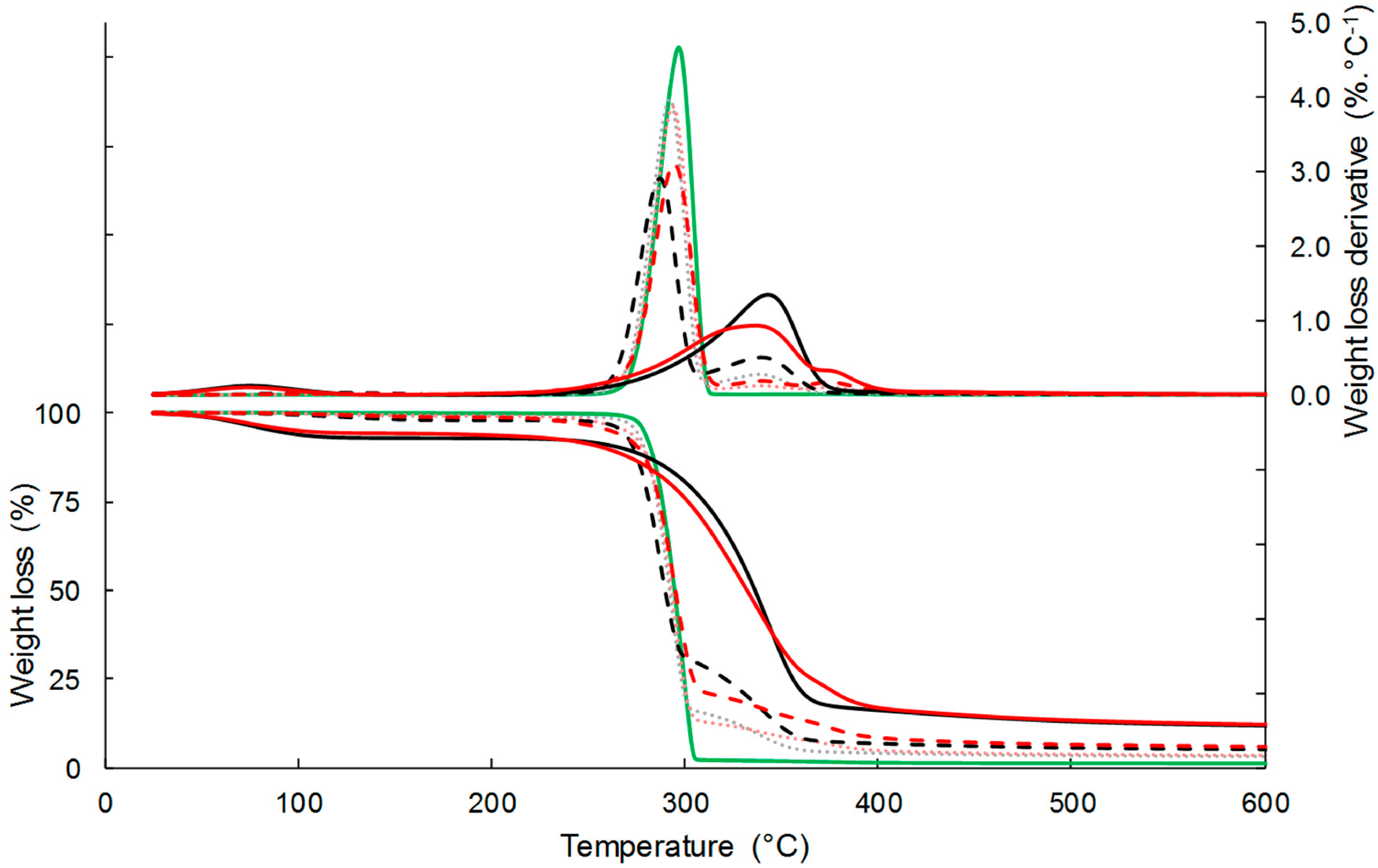
3.1.3. Thermal Stability
3.2. Impact of Gas-Phase Esterification on Interfacial Adhesion: Qualitative Evaluation
3.3. Impact on Water Transfer Properties in Resulting Composites
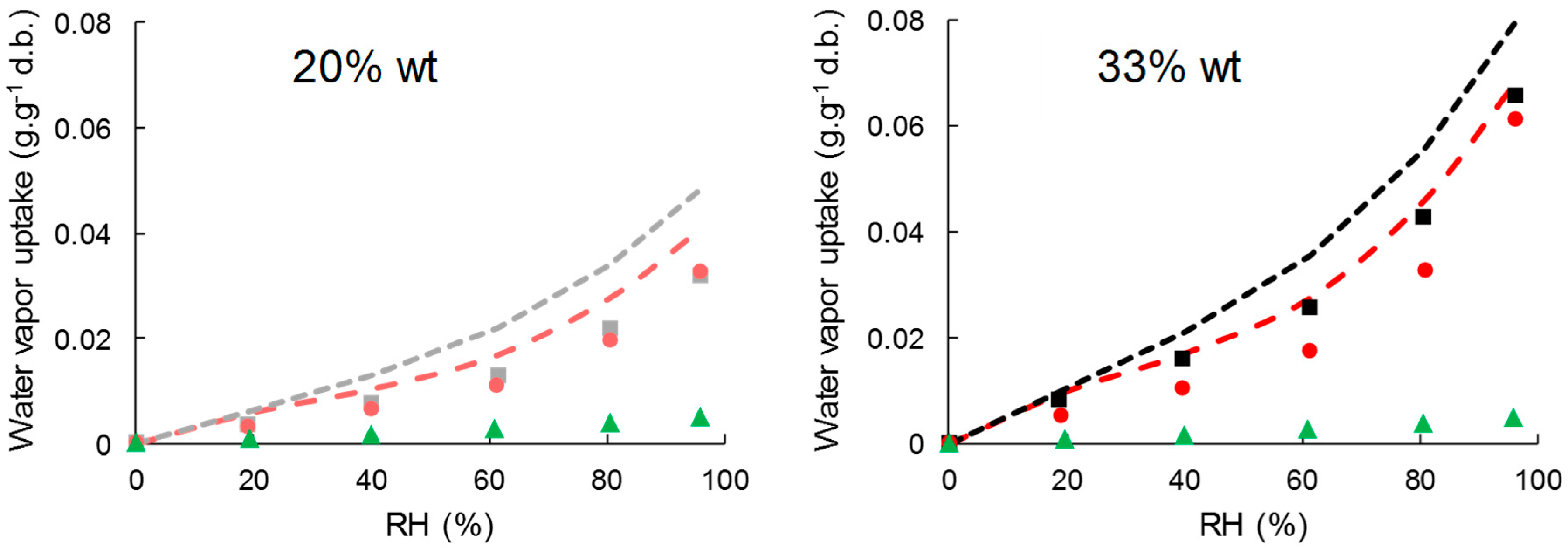
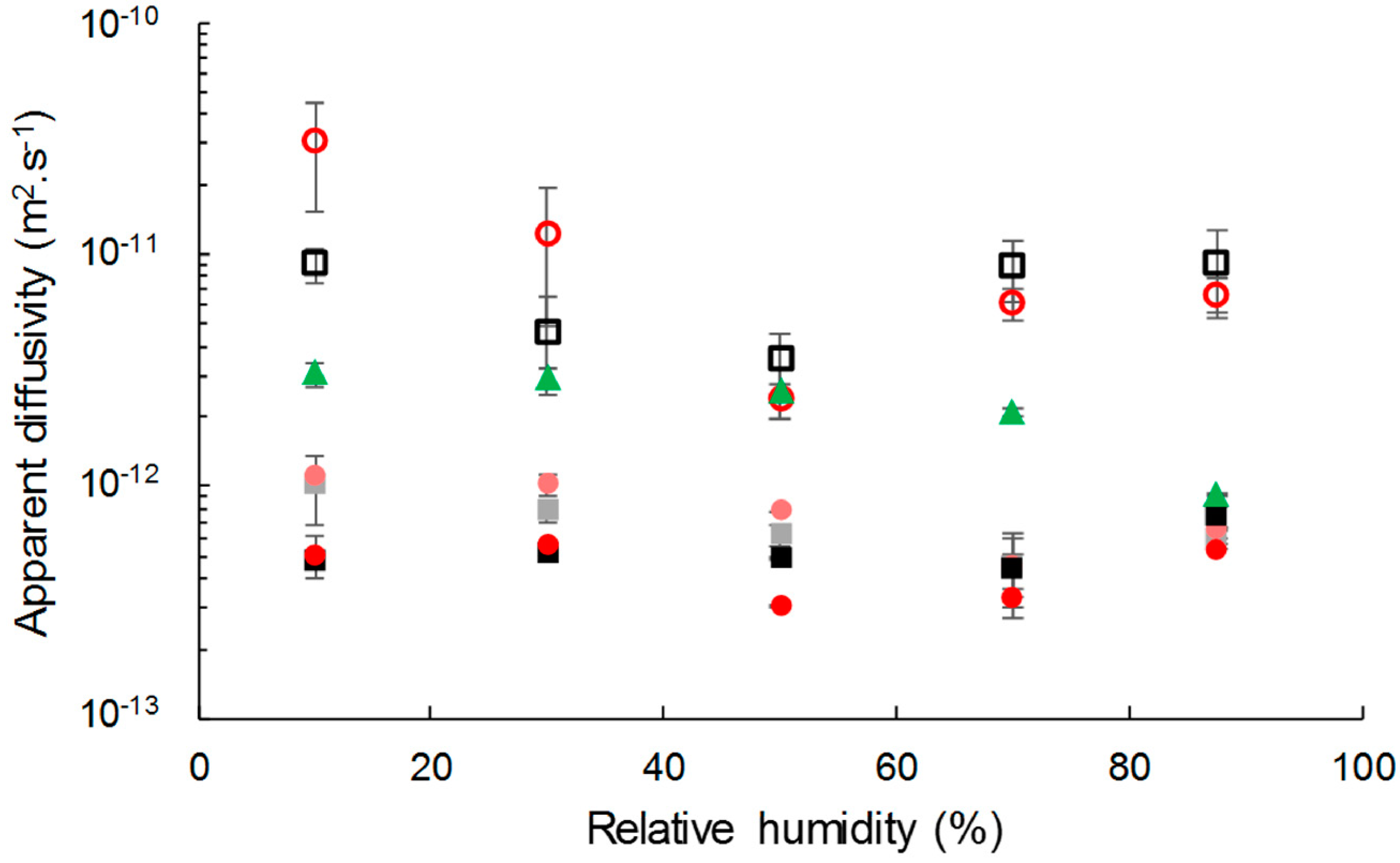
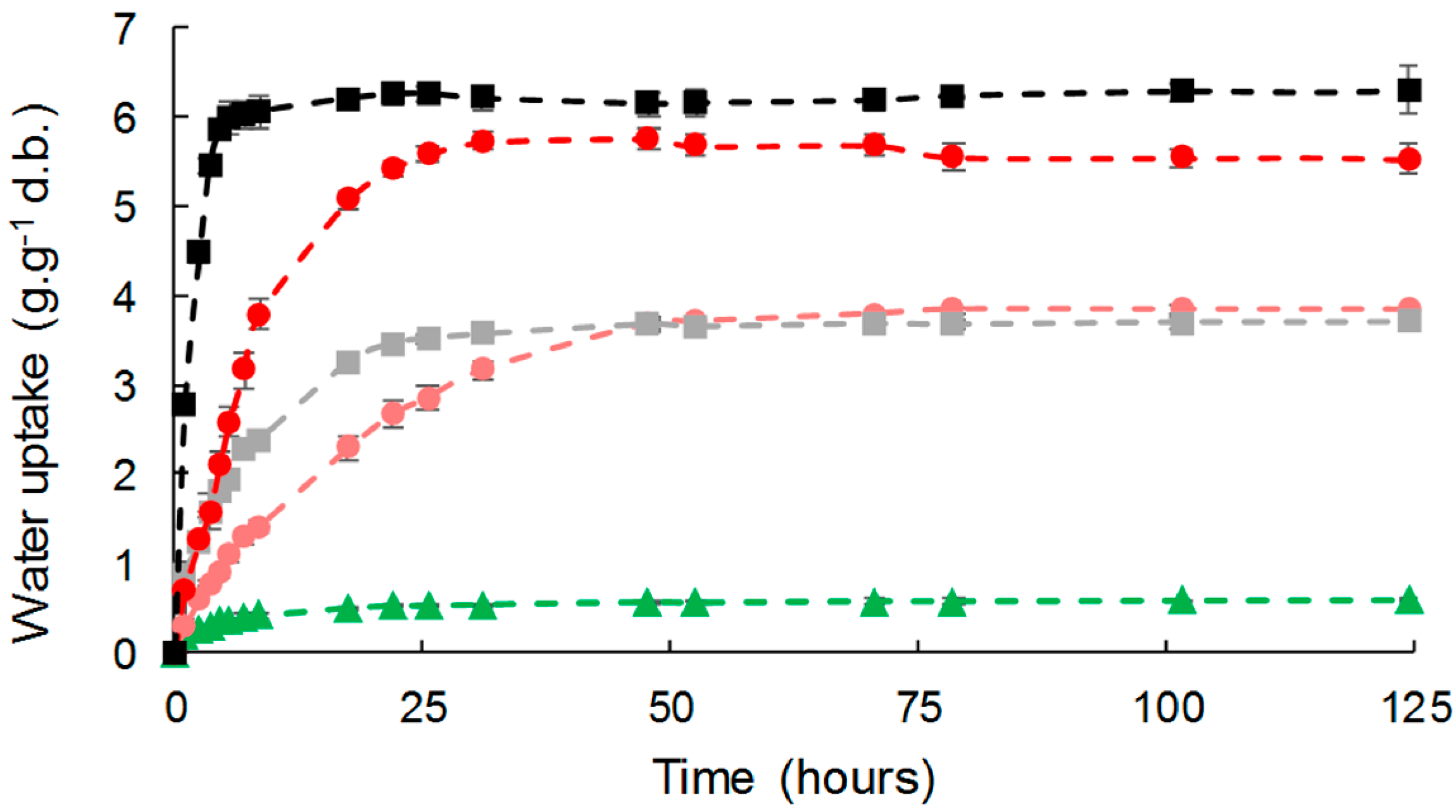
3.3.1. Water Vapor Sorption Kinetics
3.3.2. Water Vapor Permeability (WVP)
3.3.3. Liquid Water Absorption
3.4. Impact on Gas-Phase Esterification on Mechanical Properties of the Resulting Composites
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hon, D.N.S. Cellulose: A random walk along its historical path. Cellulose 1994, 1, 1–25. [Google Scholar] [CrossRef]
- Bledzki, A.K.; Gassan, J. Composites reinforced with cellulose based fibers. Prog. Polym. Sci. 1999, 24, 221–274. [Google Scholar] [CrossRef]
- Mohanty, A.K.; Misra, M.; Hinrichsen, G. Biofibres, biodegradable polymers and biocomposites: An overview. Macromol. Mater. Eng. 2000, 276–277, 1–24. [Google Scholar] [CrossRef]
- Eichhorn, S.J.; Baillie, C.A.; Zafeiropoulos, N.; Mwaikambo, L.Y.; Ansell, M.P.; Dufresne, A.; Entwistle, K.M.; Herrera-Franco, P.J.; Escamilla, G.C.; Groom, L.; et al. Current international research into cellulosic fibres and composites. J. Mater. Sci. 2001, 36, 2107–2131. [Google Scholar] [CrossRef]
- John, M.J.; Thomas, S. Biofibres and biocomposites. Carbohydr. Polym. 2008, 71, 343–364. [Google Scholar] [CrossRef]
- La Mantia, F.P.; Morreale, M. Green composites: A brief review. Compos. Part A Appl. Sci. Manuf. 2011, 42, 579–588. [Google Scholar] [CrossRef]
- Faruk, O.; Bledzki, A.K.; Fink, H.P.; Sain, M. Biocomposites reinforced with natural fibers: 2000–2010. Prog. Polym. Sci. 2012, 37, 1552–1596. [Google Scholar] [CrossRef]
- Gurunathan, T.; Mohanty, S.; Nayak, S.K. A review of the recent developments in biocomposites based on natural fibres and their application perspectives. Compos. Part A Appl. Sci. Manuf. 2015, 77, 1–25. [Google Scholar] [CrossRef]
- Henrique, P.; Pereira, F.; Rosa, M.D.F.; Odila, M.; Cioffi, H.; Cristina, K.; De Carvalho, C.; Milanese, A.C.; Jacobus, H.; Voorwald, C.; et al. Vegetal fibers in polymeric composites: A review. Polimeros 2015, 25, 9–22. [Google Scholar] [CrossRef]
- Pickering, K.L.; Efendy, M.G.A.; Le, T.M. A review of recent developments in natural fibre composites and their mechanical performance. Compos. Part A Appl. Sci. Manuf. 2016, 83, 98–112. [Google Scholar] [CrossRef] [Green Version]
- Pöllänen, M.; Suvanto, M.; Pakkanen, T.T. Cellulose reinforced high density polyethylene composites—Morphology, Mechanical and thermal expansion properties. Compos. Sci. Technol. 2013, 76, 21–28. [Google Scholar] [CrossRef]
- Le Moigne, N.; Otazaghine, B.; Corn, S.; Angellier-Coussy, H.; Bergeret, A. Surfaces and Interfaces in Natural Fibre Reinforced Composites—Fundamentals, Modifications and Characterization; Navard, P., Ed.; Springer Briefs in Molecular Science: Cham, Switzerland, 2018; ISBN 978-3-319-71409-7. [Google Scholar]
- Belgacem, M.N.; Gandini, A. The surface modification of cellulose fibres for use as reinforcing elements in composite materials. Compos. Interfaces 2005, 12, 41–75. [Google Scholar] [CrossRef]
- Trejo-O’Reilly, J.-A.; Cavaille, J.-Y.; Gandini, A. The surface chemical modification of cellulosic fibres in view of their use in composite materials. Cellulose 1997, 4, 305–320. [Google Scholar] [CrossRef]
- Gauthier, R.; Joly, C.; Coupas, A.C.; Gauthier, H.; Escoubes, M. Interfaces in polyolefin/cellulosic fiber composites: Chemical coupling, morphology, correlation with adhesion and aging in moisture. Polym. Compos. 1998, 19, 287–300. [Google Scholar] [CrossRef]
- George, J.; Sreekala, M.S.; Thomas, S. A review on interface modification and characterization of natural fiber reinforced plastic composites. Polym. Eng. Sci. 2001, 41, 1471–1485. [Google Scholar] [CrossRef]
- Li, X.; Tabil, L.G.; Panigrahi, S. Chemical treatments of natural fiber for use in natural fiber-reinforced composites: A review. J. Polym. Environ. 2007, 15, 25–33. [Google Scholar] [CrossRef]
- Hubbe, M.A.; Rojas, O.J.; Lucia, L.A. Green modification of surface characteristics of cellulosic materials at the molecular or nano scale: A review. BioResources 2015, 10, 6095–6206. [Google Scholar] [CrossRef]
- Wei, L.; McDonald, A.G. A review on grafting of biofibers for biocomposites. Materials 2016, 9, 303. [Google Scholar] [CrossRef]
- Jandura, P.; Riedl, B.; Kokta, B.V. Inverse gas chromatography study on partially esterified paper fiber. J. Chromatogr. A 2002, 969, 301–311. [Google Scholar] [CrossRef]
- Pasquini, D.; de Morais Teixeira, E.; da Silva Curvelo, A.A.; Belgacem, M.N.; Dufresne, A. Surface esterification of cellulose fibres: Processing and characterisation of low-density polyethylene/cellulose fibres composites. Compos. Sci. Technol. 2008, 68, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Freire, C.S.R.; Silvestre, A.J.D.; Neto, C.P.; Gandini, A.; Martin, L.; Mondragon, I. Composites based on acylated cellulose fibers and low-density polyethylene: Effect of the fiber content, degree of substitution and fatty acid chain length on final properties. Compos. Sci. Technol. 2008, 68, 3358–3364. [Google Scholar] [CrossRef] [Green Version]
- Junior De Menezes, A.; Siqueira, G.; Curvelo, A.A.S.; Dufresne, A. Extrusion and characterization of functionalized cellulose whiskers reinforced polyethylene nanocomposites. Polymer 2009, 50, 4552–4563. [Google Scholar] [CrossRef]
- Reulier, M.; Perrin, R.; Avérous, L. Biocomposites based on chemically modified cellulose fibers with renewable fatty-acid-based thermoplastic systems: Effect of different fiber treatments. J. Appl. Polym. Sci. 2016, 133, 1–13. [Google Scholar] [CrossRef]
- Yano, H.; Omura, H.; Honma, Y.; Okumura, H.; Sano, H.; Nakatsubo, F. Designing cellulose nanofiber surface for high density polyethylene reinforcement. Cellulose 2018, 25, 3351–3362. [Google Scholar] [CrossRef]
- David, G.; Gontard, N.; Guerin, D.; Heux, L.; Lecomte, J.; Angellier-Coussy, H. Gas-phase esterification of cellulose particles for the production of PHBV based biocomposites. In Proceedings of the 3rd International EPNOE Junior Scientists Meeting, Maribor, Slovenia, 14–15 May 2018. [Google Scholar]
- Berlioz, S.; Molina-Boisseau, S.; Nishiyama, Y.; Heux, L. Gas-phase surface esterification of cellulose microfibrils and whiskers. Biomacromolecules 2009, 10, 2144–2151. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.; Ouhab, D.; Boisseau, S.M.; Heux, L. Versatile gas-phase reactions for surface to bulk esterification of cellulose microfibrils aerogels. Biomacromolecules 2013, 14, 3246–3255. [Google Scholar] [CrossRef] [PubMed]
- Ku, H.; Wang, H.; Pattarachaiyakoop, N.; Trada, M. A review on the tensile properties of natural fiber reinforced polymer composites. Compos. Part B Eng. 2011, 42, 856–873. [Google Scholar] [CrossRef] [Green Version]
- Espert, A.; Vilaplana, F.; Karlsson, S. Comparison of water absorption in natural cellulosic fibres from wood and one-year crops in polypropylene composites and its influence on their mechanical properties. Compos. Part A Appl. Sci. Manuf. 2004, 35, 1267–1276. [Google Scholar] [CrossRef]
- Tănase, E.E.; Popa, M.E.; Râpă, M.; Popa, O. PHB/Cellulose Fibers Based Materials: Physical, Mechanical and Barrier Properties. Agric. Agric. Sci. Procedia 2015, 6, 608–615. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Sanz, M.; Vicente, A.; Gontard, N.; Lopez-Rubio, A.; Lagaron, J.M. On the extraction of cellulose nanowhiskers from food by-products and their comparative reinforcing effect on a polyhydroxybutyrate-co-valerate polymer. Cellulose 2015, 22, 535–551. [Google Scholar] [CrossRef]
- Ambrosio-Martin, J.; Fabra, M.J.; Lopez-Rubio, A.; Gorrasi, G.; Sorrentino, A.; Lagaron, J.M. Assessment of Ball Milling as a Compounding Technique to Develop Nanocomposites of Poly(3-Hydroxybutyrate-co-3-Hydroxyvalerate) and Bacterial Cellulose Nanowhiskers. J. Polym. Environ. 2016, 24, 241–254. [Google Scholar] [CrossRef]
- Wolf, C.; Guillard, V.; Angellier-Coussy, H.; Silva, G.G.D.; Gontard, N. Water vapor sorption and diffusion in wheat straw particles and their impact on the mass transfer properties of biocomposites. J. Appl. Polym. Sci. 2016, 133, 1–10. [Google Scholar] [CrossRef]
- Bhardwaj, R.; Mohanty, A.K.; Drzal, L.T.; Pourboghrat, F.; Misra, M. Renewable Resource-Based Green Composites from Recycled Cellulose Fiber and Poly (3-hydroxybutyrate-co-3-hydroxyvalerate) Bioplastic. Biomacromolecules 2006, 7, 2044–2051. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Huang, J.; Qian, J.; Chen, F.; Zhang, J.; Wolcott, M.P.; Zhu, Y. Study of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV)/bamboo pulp fiber composites: Effects of nucleation agent and compatibilizer. J. Polym. Environ. 2008, 16, 83–93. [Google Scholar] [CrossRef]
- Yu, H.; Qin, Z. Effect of Cellulose nanocrystal on Crystallization Behavior of Poly (3-hydroxybutyrate-co-3-hydroxyvalerate). Adv. Mater. Res. 2012, 432, 20–23. [Google Scholar] [CrossRef]
- Ten, E.; Jiang, L.; Wolcott, M.P. Crystallization kinetics of poly (3-hydroxybutyrate-co-3-hydroxyvalerate)/cellulose nanowhiskers composites. Carbohydr. Polym. 2012, 90, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Srithep, Y.; Ellingham, T.; Peng, J.; Sabo, R.; Clemons, C.; Turng, L.; Pilla, S. Melt compounding of poly (3-hydroxybutyrate-co-3-hydroxyvalerate)/nano fibrillated cellulose nanocomposites. Polym. Degrad. Stab. 2013, 98, 1439–1449. [Google Scholar] [CrossRef]
- Yu, H.; Yan, C.; Yao, J. Fully biodegradable food packaging materials based on functionalized cellulose nanocrystals/poly(3-hydroxybutyrate-co-3-hydroxyvalerate) nanocomposites. RSC Adv. 2014. [Google Scholar] [CrossRef]
- Sánchez-Safont, E.L.; González-Ausejo, J.; Gámez-Pérez, J.; Lagarón, J.M.; Cabedo, L. Poly(3-Hydroxybutyrate-co-3-Hydroxyvalerate)/Purified Cellulose Fiber Composites by Melt Blending: Characterization and Degradation in Composting Conditions. J. Renew. Mater. 2016, 4, 123–132. [Google Scholar] [CrossRef]
- Malmir, S.; Montero, B.; Rico, M.; Barral, L.; Bouza, R. Morphology, thermal and barrier properties of biodegradable films of poly (3-hydroxybutyrate-co-3-hydroxyvalerate) containing cellulose nanocrystals. Compos. Part A 2017, 93, 41–48. [Google Scholar] [CrossRef]
- Thoury-Monbrun, V.; Angellier-Coussy, H.; Guillard, V.; Legland, D.; Gaucel, S. Impact of two-dimensional particle size distribution on estimation of water vapor diffusivity in micrometric size cellulose particles. Materials 2018, 11, 1712. [Google Scholar] [CrossRef] [PubMed]
- Barham, P.J.; Keller, A.; Otun, E.L.; Holmes, P.A. Crystallization and morphology of a bacterial thermoplastic: Poly-3-hydroxybutyrate. J. Mater. Sci. 1984, 19, 2781–2794. [Google Scholar] [CrossRef]
- Thoury-Monbrun, V. Formalisation des relations structure/propriétés de transfert de matière dans un biocomposite modèle. Ph.D. Thesis, Université de Montpellier, Montpellier, France, 2018. [Google Scholar] [CrossRef]
- Crank, J. The Mathematics of Diffusion, 2nd ed.; Oxford University Press: London, UK, 1975; ISBN 0198533446. [Google Scholar]
- Spitalsky, Z.; Lacik, I.; Lathova, E.; Janigova, I.; Chodak, I. Controlled degradation of polyhydroxybutyrate via alcoholysis with ethylene glycol or glycerol. Polym. Degrad. Stab. 2006, 91, 856–861. [Google Scholar] [CrossRef]
- Berthet, M.A.; Angellier-Coussy, H.; Machado, D.; Hilliou, L.; Staebler, A.; Vicente, A.; Gontard, N. Exploring the potentialities of using lignocellulosic fibres derived from three food by-products as constituents of biocomposites for food packaging. Ind. Crops Prod. 2015, 69, 110–122. [Google Scholar] [CrossRef]
- Berthet, M.A.; Mayer-Laigle, C.; Rouau, X.; Gontard, N.; Angellier-Coussy, H. Sorting natural fibres: A way to better understand the role of fibre size polydispersity on the mechanical properties of biocomposites. Compos. Part A Appl. Sci. Manuf. 2017, 95, 12–21. [Google Scholar] [CrossRef]
- Lammi, S.; Le Moigne, N.; Djenane, D.; Gontard, N.; Angellier-Coussy, H. Dry fractionation of olive pomace for the development of food packaging biocomposites. Ind. Crops Prod. 2018, 120, 250–261. [Google Scholar] [CrossRef]
- Banik, G.; Brückle, I. Principles of Water Absorption and Desorption in Cellulosic Materials. Restaur. Int. J. Preserv. Libr. Arch. Mater. 2010, 31, 164–177. [Google Scholar] [CrossRef]
- Bugnicourt, E.; Cinelli, P.; Lazzeri, A.; Alvarez, V. Polyhydroxyalkanoate (PHA): Review of synthesis, characteristics, processing and potential applications in packaging. Express Polym. Lett. 2014, 8, 791–808. [Google Scholar] [CrossRef] [Green Version]
- Marais, S.; Nguyen, Q.T.; Devallencourt, C.; Metayer, M.; Nguyen, T.U.; Schaetzel, P. Permeation of water through polar and nonpolar polymers and copolymers: Determination of the concentration-dependent diffusion coefficient. J. Polym. Sci. Part B Polym. Phys. 2000, 38, 1998–2008. [Google Scholar] [CrossRef]
- Shogren, R. Water vapor permeability of biodegradable polymers. J. Environ. Polym. Degrad. 1997, 5, 91–95. [Google Scholar] [CrossRef]
- Tomé, L.C.; Pinto, R.J.B.; Trovatti, E.; Freire, C.S.R.; Silvestre, A.J.D.; Neto, C.P.; Gandini, A. Transparent bionanocomposites with improved properties prepared from acetylated bacterial cellulose and poly(lactic acid) through a simple approach. Green Chem. 2011, 13, 419–427. [Google Scholar] [CrossRef]
- Avella, M.; La Rota, G.; Martuscelli, E.; Raimo, M.; Sadocco, P.; Elegir, G.; Riva, R. Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) and wheat straw fibre composites: Thermal, mechanical properties and biodegradation behaviour. J. Mater. Sci. 2000, 35, 829–836. [Google Scholar] [CrossRef]
- Dufresne, A.; Dupeyre, D.; Paillet, M. Lignocellulosic Flour-Reinforced Poly(hydroxybutyrate-co-valerate) Composites. J. Appl. Polym. Sci. 2003, 87, 1302–1315. [Google Scholar] [CrossRef]
- Singh, S.; Mohanty, A.K. Wood fiber reinforced bacterial bioplastic composites: Fabrication and performance evaluation. Compos. Sci. Technol. 2007, 67, 1753–1763. [Google Scholar] [CrossRef]
- Ludueña, L.; Vázquez, A.; Alvarez, V. Effect of lignocellulosic filler type and content on the behavior of polycaprolactone based eco-composites for packaging applications. Carbohydr. Polym. 2012, 87, 411–421. [Google Scholar] [CrossRef]
- Pukánszky, B. Influence of interface interaction on the ultimate tensile properties of polymer composites. Composites 1990, 21, 255–262. [Google Scholar] [CrossRef]
- Dányádi, L.; Janecska, T.; Szabó, Z.; Nagy, G.; Móczó, J.; Pukánszky, B. Wood flour filled PP composites: Compatibilization and adhesion. Compos. Sci. Technol. 2007, 67, 2838–2846. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | Mw 1 (kDa) | Ip | Tm 2 (°C) | Tm 3 (°C) | Tc (°C) | Xc 2 (%) | Xc 3 (%) |
|---|---|---|---|---|---|---|---|
| PHBV | 241 | 3.2 | 177 ± 1 | 172 ± 1 | 124 ± 1 | 63 ± 1 | 73 ± 1 |
| PHBV-20VC | 245 | 2.7 | 173 ± 1 | 171 ± 1 | 124 ± 1 | 64 ± 1 | 74 ± 1 |
| PHBV-20GC | 231 | 3.2 | 171 ± 3 | 169 ± 1 | 111 ± 1 | 59 ± 1 | 66 ± 1 |
| PHBV-33VC | 230 | 3.6 | 170 ± 1 | 169 ± 1 | 120 ± 1 | 62 ± 9 | 67 ± 1 |
| PHBV-33GC | 230 | 3.2 | 172 ± 4 | 171 ± 1 | 108 ± 1 | 69 ± 2 | 66 ± 1 |
| Materials | Tdeg(1) (°C) | Tdeg(2) (°C) | Tonset (°C) | Toffset (°C) |
|---|---|---|---|---|
| PHBV | 297 ± 1 | - | 267 ± 1 | 312 ± 1 |
| C-virgin | - | 343 ± 1 | 259 ± 1 | 375 ± 0 |
| C-grafted | - | 336 ± 1 | 247 ± 1 | 398 ± 0 |
| PHBV-10CV | 295 ± 5 | 335 ± 1 | 248 ± 4 | 380 ± 4 |
| PHBV-10CG | 298 ± 1 | 339 ± 1 | 237 ± 1 | 413 ± 2 |
| PHBV-20CV | 292 ± 1 | 336 ± 4 | 264 ± 2 | 357 ± 4 |
| PHBV-20CG | 294 ± 1 | 340 ± 1 | 254 ± 1 | 377 ± 0 |
| PHBV-33CV | 287 ± 1 | 339 ± 1 | 256 ± 1 | 365 ± 1 |
| PHBV-33CG | 294 ± 1 | 339 ± 2 | 248 ± 1 | 389 ± 0 |
| Water Vapor | Liquid Water | ||||||
|---|---|---|---|---|---|---|---|
| Materials | WVP (×1013 mol·m/(m2·s·Pa) | Apparent Diffusivity at 50% of RH (×10−13·m2·s−1) | Equilibrium Uptake at 50% RH (%) | Apparent Diffusivity at 95% of RH (×10−13·m2·s−1) | Equilibrium Uptake at 95% RH (%) | Apparent Diffusivity (×10−13·m2·s−1) | Equilibrium Uptake (%) |
| C-virgin | - | 42 ± 16 | 8.1 ± 0.0 | 70.2 ± 10.1 | 22.7 ± 0.0 | - | - |
| C-grafted | - | 63 ± 36 | 6.2 ± 0.0 | 115 ± 62 | 19.0 ± 0.2 | - | - |
| PHBV | 3.7 ± 1.0 | 25.9 ± 6.2 | 0.2 ± 0.0 | 23.3 ± 8.7 | 0.5 ± 0.0 | 4.1 ± 0.2 | 0.6 ± 0.0 |
| PHBV/20VC | 5.6 ± 1.0 | 6.1 ± 0.6 | 1.0 ± 0.1 | 6.9 ± 2.2 | 3.2 ± 0.1 | 1.7 ± 0.1 | 3.7 ± 0.1 |
| PHBV/20GC | 5.0 ± 0.8 | 7.8 ± 0.0 | 0.9 ± 0.0 | 8.0 ± 2.7 | 3.3 ± 0.0 | 1.2 ± 0.0 | 3.8 ± 0.1 |
| PHBV/33VC | 36 ± 3.2 | 4.9 ± 0.1 | 2.1 ± 0.1 | 5.3 ± 1.2 | 6.6 ± 0.0 | 8.7 ± 0.6 | 6.3 ± 0.1 |
| PHBV/33GC | 15 ± 2.0 | 3.0 ± 0.0 | 1.4 ± 0.1 | 4.5 ± 1.2 | 6.1 ± 0.0 | 1.9 ± 0.1 | 5.5 ± 0.1 |
| Materials | Young’s Modulus (GPa) | Stress at Break (MPa) | Strain at Break (%) | Energy at Break (mJ·cm−3) |
|---|---|---|---|---|
| PHBV | 2.9 ± 0.2 | 39.7 ± 0.1 | 2.40 ± 0.15 | 626 ± 68 |
| PHBV-10CV | 3.2 ± 0.2 | 25.1 ± 1.3 | 0.90 ± 0.07 | 124 ± 15 |
| PHBV-10CG | 3.3 ± 0.1 | 31.0 ± 1.5 | 1.23 ± 0.21 | 229 ± 56 |
| PHBV-20CV | 3.1 ± 0.1 | 23.6 ± 1.3 | 0.91 ± 0.05 | 122 ± 12 |
| PHBV-20CG | 2.6 ± 0.1 | 25.1 ± 1.1 | 1.15 ± 0.05 | 164 ± 13 |
| PHBV-33CV | 2.6 ± 0.2 | 15.8 ± 2.4 | 0.70 ± 0.10 | 62 ± 17 |
| PHBV-33CG | 2.5 ± 0.2 | 19.0 ± 0.8 | 0.93 ± 0.06 | 101 ± 8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
David, G.; Gontard, N.; Angellier-Coussy, H. Mitigating the Impact of Cellulose Particles on the Performance of Biopolyester-Based Composites by Gas-Phase Esterification. Polymers 2019, 11, 200. https://doi.org/10.3390/polym11020200
David G, Gontard N, Angellier-Coussy H. Mitigating the Impact of Cellulose Particles on the Performance of Biopolyester-Based Composites by Gas-Phase Esterification. Polymers. 2019; 11(2):200. https://doi.org/10.3390/polym11020200
Chicago/Turabian StyleDavid, Grégoire, Nathalie Gontard, and Hélène Angellier-Coussy. 2019. "Mitigating the Impact of Cellulose Particles on the Performance of Biopolyester-Based Composites by Gas-Phase Esterification" Polymers 11, no. 2: 200. https://doi.org/10.3390/polym11020200
APA StyleDavid, G., Gontard, N., & Angellier-Coussy, H. (2019). Mitigating the Impact of Cellulose Particles on the Performance of Biopolyester-Based Composites by Gas-Phase Esterification. Polymers, 11(2), 200. https://doi.org/10.3390/polym11020200