1. Introduction
The applications of silanes are extremely diverse, including corrosion protection films [
1,
2], coupling agents and cross linkers [
1,
2,
3,
4], and corner capping agents for nanoparticle molecules such as polyhedral oligomeric silsesquioxanes (POSS) [
5]. In particular, applications of silane films on carbon substrates have been reported recently, including the formation of enzyme-based biosensors [
6], stabilising agents for water-soluble graphene dispersions [
7], modification of graphene properties [
8], incorporation of graphene [
9,
10] or carbon nanotubes into a polymer matrix [
3,
4,
11,
12,
13], polymer-electrolyte-membrane fuel cells [
9], and the formation of silicon-based nanostructures [
14].
Silane coupling agents (general formula R’Si(OR)
3) are commonly used for providing adhesion between two dissimilar materials and have many properties that make them ideal for providing the chemical attachment in the applications listed above. These properties vary depending on the R’ functional group within the silane, which imparts a chemical functionality to the modified surface. Of particular interest is the formation of the silane film on the substrate surface, and it is for this reason that propyltrimethoxysilane (PTMS) and propyldimethylmethoxysilane (PDMMS) were chosen for this study on the formation of silane films on HOPG. While there have been a range of silanes used on carbon materials for various applications [
6,
7,
8,
9,
10,
11,
14,
15], for this study, these two simple model silanes were chosen. The inert propyl chain in these molecules minimizes intermolecular interactions as much as possible while still remaining stable in solution, allowing study of the growth mechanism with minimal interference from the functional group [
16]. PTMS contains three hydrolysable groups, while PDMMS contains only one, resulting in a difference in the structure and growth mechanism between the two types of film. PDMMS can only form one chemical bond, and thus bind to one site, either to the substrate surface or to another PDMMS molecule, whereas PTMS can form three bonds, with many combinations of surface–PTMS and PTMS–PTMS bonds possible. The mechanism of growth of the silane film has previously been found to differ when using different substrates and silanes, with some substrates showing Langmuir-like growth and others showing an oscillation in the film growth [
16,
17]. It is therefore important to investigate how the silanes grow on a carbon surface in order to obtain the best silane coverage.
In order for a silane film to grow on a surface, the silane molecules (R’Si(OR)
3) must first undergo hydrolysis to form reactive tri-silanol molecules (R‘Si(OH)
3) [
1,
2]. These tri-silanols can then undergo condensation with each other or a hydroxylated surface, resulting in film growth for both PTMS and PDMMS, and oligomerisation of PTMS in solution [
1,
2]. It is therefore important that a carbon surface is oxidized before a silane film can be grown. The carbon surface used for this work is highly oriented pyrolytic graphite (HOPG), and was chosen due to its simple, well known structure, which again simplifies the system to aid in the study of the film growth mechanism. Conventionally, wet chemical methods are used to create the necessary oxide layer on the carbon surface. This paper demonstrates the use of oxygen plasma to grow an oxide layer, which is then further functionalized with a silane film. Some work has been done previously by Schade et al. [
15] on functionalizing carbon/polymer materials with an oxygen layer prior to silane film deposition, however the plasma treatment conditions used were quite different and there was no quantitative analysis performed on the increase in oxygen content on the surface [
15]. Previous work done by Taylor has shown how the atomic percentage of oxygen present on a HOPG surface changes with several plasma parameters, including carrier gas pressure, RF-coupling power, and treatment time [
18]. The plasma conditions chosen for this work were based off this study. There are many advantages of plasma oxidation, including the very small amounts of oxygen gas used compared to the large amounts of solvents and chemicals in wet chemical methods, and the ease of controlling oxidation levels through modification of several plasma growth parameters.
The reaction of a silane with this oxidized HOPG surface is shown in
Figure 1. The number of silane molecules that will bond to the HOPG surface will depend on the number of hydroxyl groups on the silane molecule and the concentration of possible bonding sites on the HOPG, and therefore on the plasma treatment parameters. It is desirable to increase the number of bonding sites to maximize silane film formation; however, this will also increase the disruption of the HOPG lattice. It is therefore necessary to find a balance between the number of bonding sites and the modification to the morphology and bonding of the surface layer of graphite.
Silane films on carbon materials have been examined previously using atomic force microscopy (AFM) [
6,
14], Fourier transform infrared spectroscopy (FTIR) [
4,
7,
8,
9,
11,
12,
13], X-ray photoelectron spectroscopy (XPS) [
7,
8,
9,
13,
14], transmission electron microscopy (TEM) [
4,
7,
9,
12,
13], time-of-flight secondary ion mass spectroscopy (TOF-SIMS) [
14], scanning electron microscopy (SEM) [
8,
9,
14], X-ray diffraction (XRD) [
8], Auger spectroscopy [
15], and Raman spectroscopy [
8,
9]. The information from these previous studies can be combined to confirm the presence of the silane film and investigate the morphology and chemical composition of silanes on carbon materials. However, these factors have not all been investigated for one single system and thus detailed information about the film composition and morphology has not been obtained. In this investigation, silanised HOPG was characterized using XPS and scanning Auger microscopy (SAM). The use of SAM for characterization of this system is novel and has the advantage of providing detailed information on the chemical composition of the films formed with high spatial resolution and surface sensitivity, simultaneously showing nanoscale resolution SEM images. This allows for a more complete understanding of the system under study by providing additional information on the structure and morphology simultaneously.
2. Materials and Methods
HOPG (12 mm × 12 mm × 2 mm, ZYB grade) was obtained from Coherent Scientific Australia, (Hilton, Australia) and prepared for treatment by cleaving with adhesive tape. The oxygen gas used to create oxygen plasma was high purity research grade (99.95% purity) from BOC. PTMS (97%) was obtained from Sigma Aldrich, Sydney, Australia, and PDMMS (97%) was obtained from Silar Laboratories, Riegelwood, NC, USA.
The oxygen plasma used in this research was produced using a radio-frequency (RF) inductively coupled antenna applying a power of 30 W to the carrier gas at a pressure of 1 × 10−2 Torr, with samples exposed to the plasma for 30 minutes.
XPS characterization was carried out using a Leybold-Heraeus LHS-10 system with an EA-10/100 concentric hemispherical analyzer (CHA) electron spectrometer and a SPECS XR-50 Mg Kα (1253.6 eV) X-ray source. A base pressure of ~1 × 10−9 Torr, take-off angle of 90°, and pass energy of 20 eV were used. SAM characterization was carried out using a PHI Model 710 Scanning Auger Nanoprobe, ULVAC-PHI, Chanhassen, MN, USA. A base pressure of ~1 × 10−9 Torr was achieved in the chamber, with characterization performed using a 10 kV 10 nA electron beam.
The sample area characterized with XPS is approximately 10 mm × 5 mm, which is most of the sample surface. XPS survey scans were taken over the binding energy range of 0–1200 eV with the spectrometer in constant retarding ratio mode. High resolution spectra were taken of the C1s peak over the range of 270−305 eV with the spectrometer in constant analyzer energy mode.
The sample area characterized with SAM depended on the features of interest, but generally ranged between 5 μm and 500 μm, which is a small section of the sample surface. As a result of this small sampling area, scans were acquired at several locations on each sample, resulting in a range of elemental concentration values. In-situ SEM images were taken of the samples in areas of interest, then Auger electron spectroscopy (AES) survey scans were taken in these areas with the kinetic energy range of 30−2030 eV. After the composition of the surface had been determined using the survey scans elemental maps of carbon, oxygen, and silicon were taken of areas of interest to show the uniformity of the silane film across the surface. Depth profiles were also taken in areas where the silane film was present to determine thickness. The beam energy used was either 10 kV 10 nA or 3 kV 5 nA, with a step size of 1.0 eV and a time per step of 10 ms for survey scans. Elemental maps were created using a 3-point acquisition method with a spatial resolution of 256 × 256 pixels and step size of 1.0 eV and time per step of 10 ms. Depth profiles were created using a window acquisition mode with step size of 1.0 eV, a time per step of 10 ms, and a sputter rate of 8.5 Å.min−1.
XPS data was processed using CasaXPS Version 2.3.16 (Casa Software Ltd), where fitted peak areas from survey scans were corrected using atomic sensitivity factors to determine the atomic concentrations of elements present on the sample surface. The high resolution C1s peaks were deconvoluted using CasaXPS, but no clear trend was observed so the results are not included here.
SAM data was processed using PHI’s MultiPak software, where peak areas from differentiated survey scans were corrected using atomic sensitivity factors to determine the atomic concentrations of elements present on the sample surface. MultiPak was also used to create red-green-blue (RGB) overlays of elemental maps and to determine atomic concentrations of elements in the depth profiles.
The various treatments applied are described in
Table 1. Samples were prepared by drop-casting either neat silane (50 μL) or a drop-wise mixture of silane and Milli Q water (50:50, 50 μL total volume) to determine if the addition of water is necessary for film growth. For the 25 μL case, the total number of silane molecules introduced to the surface is estimated to be ~ 10
20 and with a typical surface atomic density of ~10
15 atoms per square centimeter exhibited by most materials, a multi-layer film is expected. The samples were left for three minutes and then either rinsed with excess water or dried with nitrogen before curing at 80 °C for two hours, before a final rinse to examine the impact of rinsing on the film pre-cure. The condensation reaction is an equilibrium process, thus the curing step is needed to force this reaction to completion. Some samples were also treated with PTMS without oxygen plasma to confirm the necessity of a hydroxylated surface.
4. Discussion
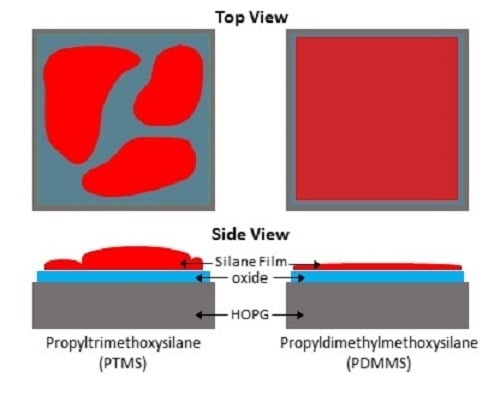
The formation of a silane film on HOPG appears to be strongly dependent on the chemical structure of the particular silane used, thus the two silane cases studied in this work will be considered separately. PTMS has the ability to bond to both the surface and to other PTMS molecules via each of its three methoxy groups once they are hydrolysed to form silanols (Si–OH)
3. This can lead to large oligomers that are bound to the surface by only a few covalent bonds, as well as single PTMS molecules bonded to the surface by substitution of all three methoxy groups with covalent bonds, and anything in between. These possible structures make it unlikely that there will be a complete, even film across the entire surface, as the distribution and structure of silane molecules and oligomers will not be uniform. The addition of water during PTMS deposition seems to encourage the preferential bonding of PTMS molecules to each other over bonding to the surface, as the coverage of silicon is less uniform in PTMS 3 than in PTMS 1, with some areas having almost no silicon and others exceeding 20%. These areas also appear different in SEM images, with darker areas containing little to no silicon and lighter areas containing large amounts of silicon. This is demonstrated in
Figure 2, which shows that in darker areas of the SEM there is very little silicon and oxygen, while there is a large amount of carbon. The thickness of the silane film also varies across the surface. One area of PTMS 3 has a silane layer that is almost 4 nm thick (
Figure 4), however this is not the same across the entire surface. The XPS results verify this. For non-hydrolysed silane molecules the carbon:oxygen ratio will be 6:3 (or 2:1), and for fully hydrolysed it would be 3:3 (or 1:1), whereas the observed ratio for samples PTMS 1 and PTMS 4 is ~3.3:1 due to the large cracks that occur upon curing. In order to obtain a covalently bonded film on a HOPG surface the results demonstrate that it is necessary to perform the oxygen plasma treatment first, and it is preferable to rinse the sample after curing to remove physisorbed species. Further studies on the addition of water to PTMS need to be undertaken to attempt to control the oligomerisation, however it has been observed that the addition of water encourages the formation of oligomers and should be avoided if a thinner, more uniform film is desired.
Unlike the tri-methoxy silane PTMS, PDMMS contains only one hydrolysable methoxy group and therefore can bond only to either the surface or another PDMMS molecule to form a rather stable dimer in solution, resulting in a single uniform layer expected across the surface. The addition of water to the PDMMS results in many of the molecules bonding to each other rather than the surface, as shown by the decrease in silicon content on the surface by XPS and the very low amount present in SAM. This low concentration found with SAM suggests that there is non-uniform coverage of PDMMS across the sample, as there were very few areas where silicon was observed. It is thus suggested that to obtain a uniform film with the best film coverage water should not be added to the system with the silane, but rinsing should be performed after curing to remove physisorbed species.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}