Unprecedentedly High Activity and/or High Regio-/Stereoselectivity of Fluorenyl-Based CGC Allyl-Type η3:η1-tert-Butyl(dimethylfluorenylsilyl)amido Ligated Rare Earth Metal Monoalkyl Complexes in Olefin Polymerization

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Method
2.3. X-ray Crystallographic Analysis
2.4. Synthesis of (η3:η1-FluSiMe2NtBu)Ln(CH2SiMe3)(THF)2 1–3
2.5. A Typical Procedure for IP Polymerization in Table 2 Entry 4
2.6. A Typical Procedure for MY Polymerization in Table 3 Entry 3
2.7.A Typical Procedure for ST Polymerization in Table 4 Entry 9
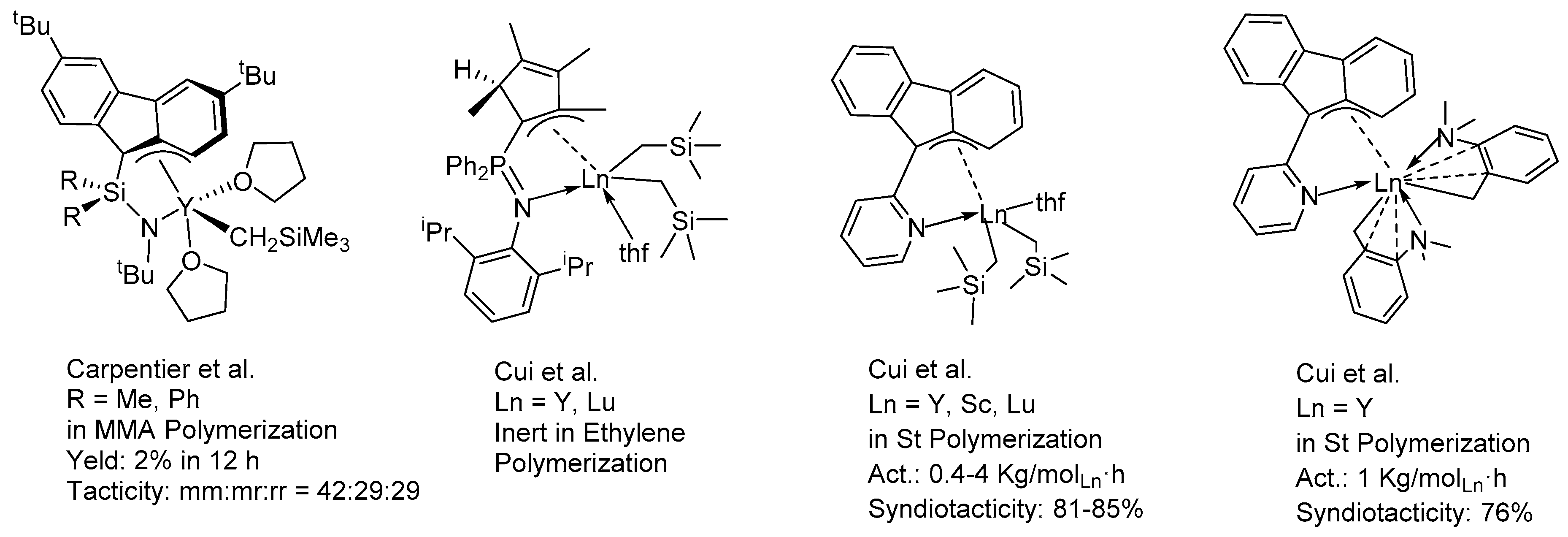
3. Results and Discussion
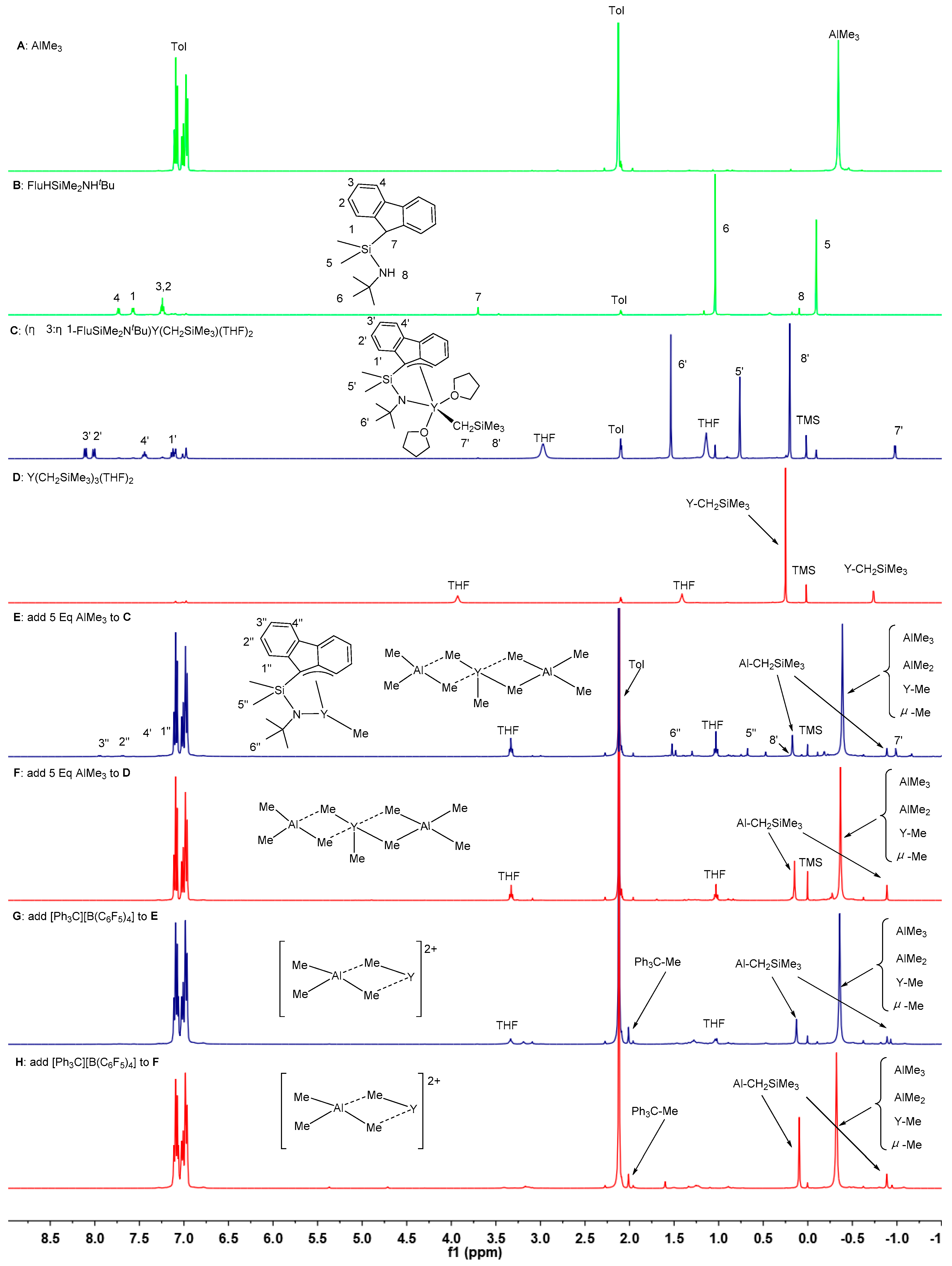
3.1. Synthesis of Flu-Based CGC Allyl-Type Rare Earth Metal Monoalkyl Complexes 1–3
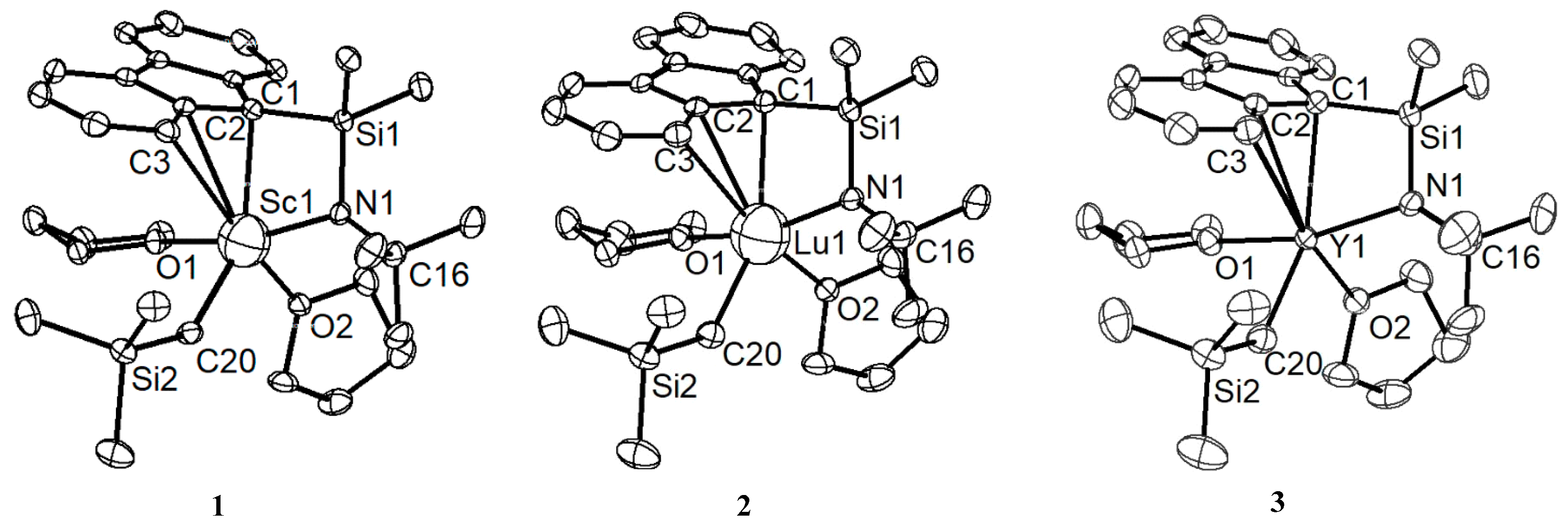
3.2. Structural Characterization of Flu-Based CGC Allyl-Type Rare Earth Metal Monoalkyl Complexes 1–3
3.3. Cis-1,4-Polymerization of Ip by the Complexes 1–3/activator/AlR3 Ternary Systems

{kind=link}
{kind=link}
{kind=link}
{kind=link}
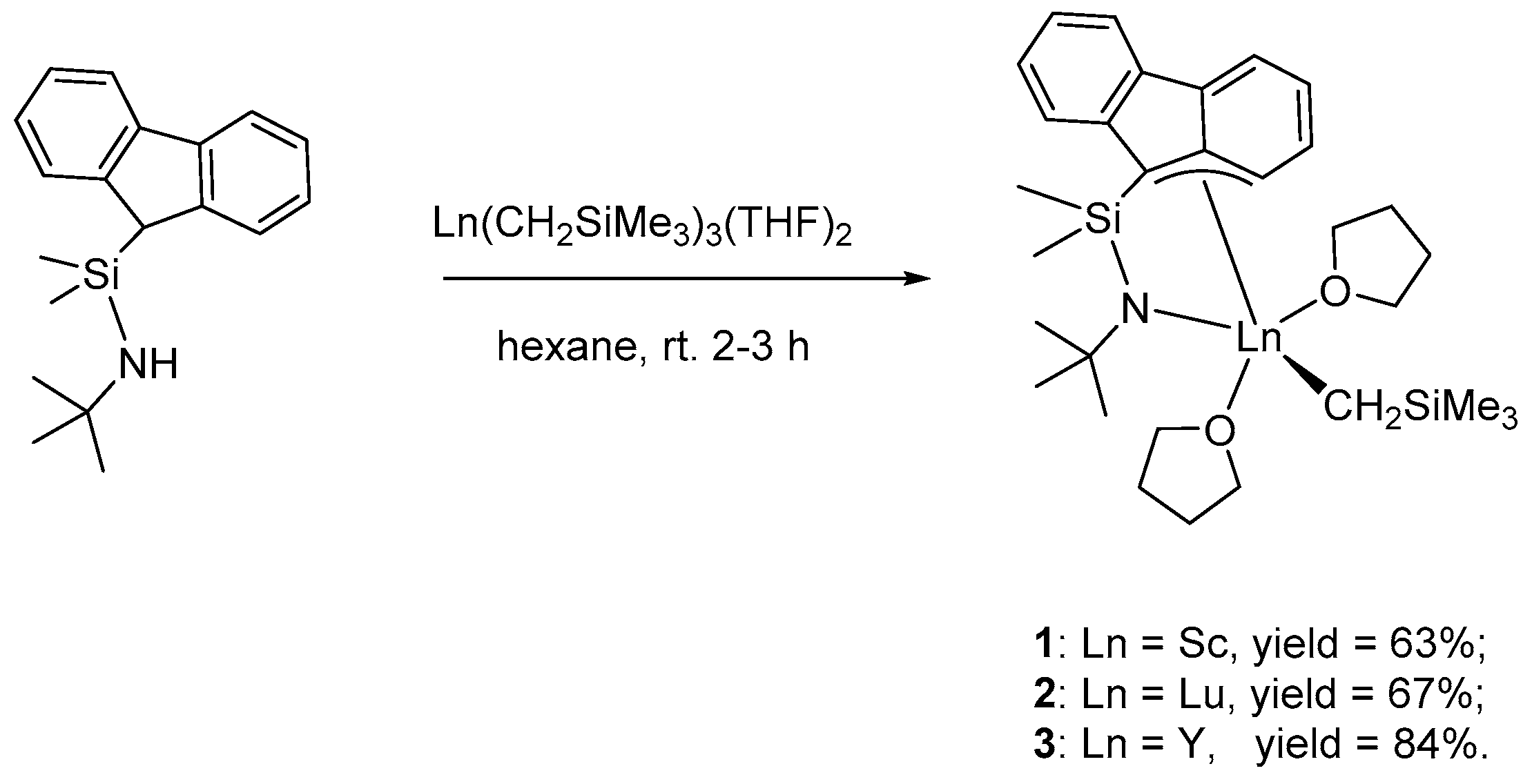{kind=link}
{kind=link}

| entry | Cat. | Ab | AlR3 | t (h) | T (°C) | Y (%) | Ac | Microstructure (%)d | Mne (105) | Mw/Mne | Tgf (°C) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [Al]/[Ln] | [IP]/[Ln] | Sol. | c-1,4 | t-1,4 | 3,4 | |||||||||||
| 1 | 3 | A | AliBu3 | 2.5 | 500 | Tol | 0.5 | 25 | 6 | 4 | 83 | 0 | 17 | 1 | 3.23 | −55 |
| 2 | 3 | B | AliBu3 | 2.5 | 500 | Tol | 0.5 | 25 | 26 | 18 | 65 | 23 | 12 | 7 | 2.31 | −52 |
| 3 | 3 | C | AliBu3 | 2.5 | 500 | Tol | 48 | 25 | - | - | - | - | - | - | - | - |
| 4 | 1 | A | AliBu3 | 10 | 500 | Tol | 0.03 | 25 | 100 | 1135 | 90 | 1 | 9 | 7 | 2.26 | −56 |
| 5 | 2 | A | AliBu3 | 10 | 500 | Tol | 1 | 25 | 51 | 17 | 90 | 0 | 10 | 3 | 3.17 | −57 |
| 6 | 3 | A | AliBu3 | 10 | 500 | Tol | 1 | 25 | 47 | 16 | 86 | 0 | 14 | 1 | 3.26 | −55 |
| 7 | 1 | A | AlMe3 | 10 | 500 | Tol | 2 | 25 | 76 | 13 | 94 | 1 | 5 | 2 | 2.55 | −62 |
| 8 | 1 | A | AlEt3 | 10 | 500 | Tol | 0.08 | 25 | 99 | 421 | 92 | 0 | 8 | 10 | 1.78 | −59 |
| 9 | 1 | A | AlMe3 | 10 | 500 | PhCl | 18 | 25 | 14 | 0.3 | 91 | 4 | 5 | 2 | 2.58 | −57 |
| 10 | 1 | A | AlMe3 | 10 | 500 | PhCl2 | 2 | 25 | 100 | 17 | 94 | 1 | 5 | 5 | 1.79 | −58 |
| 11 | 1 | A | AlMe3 | 5 | 500 | PhCl2 | 2 | 25 | 100 | 17 | 94 | 1 | 5 | 6 | 2.33 | −60 |
| 12 | 1 | A | AlMe3 | 20 | 500 | PhCl2 | 2 | 25 | 76 | 13 | 94 | 1 | 5 | 4 | 2.67 | −58 |
| 13 | 1 | A | AlMe3 | 10 | 500 | PhCl2 | 5 | –10 | 50 | 3 | 96 | 0 | 4 | 10 | 1.80 | −66 |
| 14 | 1 | A | AlMe3 | 10 | 500 | PhCl2 | 2 | 0 | 44 | 7 | 95 | 1 | 4 | 7 | 2.10 | −62 |
| 15 | 1 | A | AlMe3 | 10 | 500 | PhCl2 | 0.5 | 50 | 100 | 68 | 93 | 1 | 6 | 5 | 2.36 | −60 |
| 16 | 1 | A | AlMe3 | 10 | 500 | PhCl2 | 0.5 | 70 | 94 | 64 | 90 | 3 | 7 | 2 | 3.71 | −55 |
| 17 | 1 | A | AlMe3 | 10 | 100 | PhCl2 | 1 | 25 | 13 | 0.9 | 94 | 2 | 4 | 4 | 2.23 | −59 |
| 18 | 1 | A | AlMe3 | 10 | 300 | PhCl2 | 1 | 25 | 51 | 10 | 95 | 1 | 4 | 6 | 2.05 | −62 |
| 19 | 1 | A | AlMe3 | 10 | 800 | PhCl2 | 0.5 | 25 | 100 | 109 | 94 | 3 | 3 | 5 | 2.10 | −58 |
| 20 | Scg | A | AlMe3 | 10 | 500 | PhCl2 | 2 | 25 | 21 | 4 | 93 | 4 | 3 | 0.9 | 2.18 | −60 |
| 21 | Scg | Ah | AlMe3 | 10 | 500 | PhCl2 | 2 | 25 | 89 | 15 | 94 | 2 | 4 | 4 | 3.70 | −60 |
3.4. Cis-1,4-Polymerization of MY by the Complexes 1–3/Activator/AlR3 Ternary Systems

| entry | Cat. | Ab | t (h) | T (°C) | Y (%) | Ac | Microstructure (%)d | Mne (105) | Mw/Mne | Tgf (°C) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [Al]/[Ln] | [MY]/[Ln] | Sol. | c-1,4 | t-1,4 | 3,4 | 1,2 | ||||||||||
| 1 | 3 | A | 10 | 500 | Tol | 1 | 25 | 100 | 68 | 76 | 0 | 24 | 0 | 3 | 3.47 | −60 |
| 2 | 3 | B | 10 | 500 | Tol | 1 | 25 | 100 | 68 | 80 | 0 | 20 | 0 | 3 | 2.56 | −60 |
| 3 | 3 | C | 10 | 500 | Tol | 1 | 25 | 72 | 49 | >99 | 0 | 0 | 0 | 4 | 1.93 | −65 |
| 4 | 1 | C | 10 | 500 | Tol | 2 | 25 | 72 | 24 | 88 | 0 | 12 | 0 | 9 | 1.97 | −60 |
| 5 | 2 | C | 10 | 500 | Tol | 48 | 25 | 6 | 0.002 | 93 | 0 | 7 | 0 | 2 | 2.51 | −60 |
| 6 | 3 | C | 10 | 500 | PhCl | 8 | 25 | 99 | 8 | 95 | 0 | 5 | 0 | 4 | 2.24 | −61 |
| 7 | 3 | C | 10 | 500 | PhCl2 | 6 | 25 | 99 | 11 | 96 | 0 | 4 | 0 | 5 | 2.73 | −61 |
| 8 | 3 | C | 5 | 500 | Tol | 2 | 25 | 7 | 2 | 99 | 0 | 1 | 0 | 6 | 2.58 | −63 |
| 9 | 3 | C | 20 | 500 | Tol | 2 | 25 | 99 | 34 | 99 | 0 | 1 | 0 | 5 | 2.61 | −63 |
| 10 | 3 | C | 40 | 500 | Tol | 2 | 25 | 94 | 32 | 98 | 0 | 2 | 0 | 3 | 4.26 | −62 |
| 11 | 3 | C | 10 | 500 | Tol | 5 | 0 | 82 | 11 | 100 | 0 | 0 | 0 | 7 | 1.66 | −67 |
| 12 | 3 | C | 10 | 500 | Tol | 2 | 50 | 78 | 27 | 98 | 0 | 2 | 0 | 5 | 3.01 | −66 |
| 13 | 3 | C | 10 | 500 | Tol | 2 | 70 | 22 | 7 | 96 | 0 | 4 | 0 | 1 | 3.31 | −60 |
| 14 | 3 | C | 10 | 250 | Tol | 2 | 25 | 78 | 13 | 99 | 0 | 1 | 0 | 6 | 2.71 | −62 |
| 15 | 3 | C | 10 | 1000 | Tol | 2 | 25 | 75 | 51 | 100 | 0 | 0 | 0 | 5 | 1.75 | −67 |
| 16 | 3 | C | 10 | 2000 | Tol | 2 | 25 | 61 | 83 | 99 | 0 | 1 | 0 | 7 | 1.96 | −66 |
| 17 | 3 | C | 10 | 4000 | Tol | 3 | 25 | 65 | 118 | 99 | 0 | 1 | 0 | 9 | 1.76 | −64 |
| 18 | Yg | C | 10 | 500 | Tol | 48 | 25 | 20 | 0.23 | 95 | 0 | 5 | 0 | 0.1 | 7.01 | −60 |
| 19 | Yg | Ch | 10 | 500 | Tol | 12 | 25 | 22 | 1 | 95 | 2 | 3 | 0 | 1 | 7.19 | −60 |
3.5. Syndiotactic Polymerization of ST by the Complexes 1–3/Activator/AlR3 Ternary Systems

| t | T | Y | rrrrd | Mne | Tmf | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| entry | Cat. | Ab | AlR3 | [Al]/[Ln] | [ST]/[Ln] | Sol. | (h) | (°C) | (%) | Ac | (%) | (105) | Mw/Mne | (°C) |
| 1 | 1 | A | AliBu3 | 10 | 500 | PhCl2 | 12 | 25 | 18 | 781 | >99 | 7 | 2.05 | 272 |
| 2 | 1 | B | AliBu3 | 10 | 500 | PhCl2 | 6 | 25 | 32 | 2777 | >99 | 9 | 2.38 | 271 |
| 3 | 1 | C | AliBu3 | 10 | 500 | PhCl2 | 24 | 25 | 7 | 152 | >99 | 3 | 2.36 | 274 |
| 4 | 2 | B | AliBu3 | 10 | 500 | PhCl2 | 24 | 25 | 6 | 130 | 54 | n.d. | n.d. | 260 |
| 5 | 3 | B | AliBu3 | 10 | 500 | PhCl2 | 24 | 25 | 23 | 499 | >99 | 9 | 2.21 | 271 |
| 7 | 1 | B | AlEt3 | 10 | 500 | PhCl2 | 24 | 25 | 10.4 | 225.66 | >99 | 0.07 | 6.42 | |
| 6 | 1 | B | AlEt3 | 10 | 500 | PhCl2 | 24 | 25 | 10 | 217 | >99 | 0.1 | 6.42 | 271 |
| 7 | 1 | A | AliBu3 | 10 | 500 | PhCl | 24 | 25 | 26 | 564 | 63 | n.d. | n.d. | 265 |
| 8 | 1 | B | AliBu3 | 10 | 500 | PhCl | 24 | 25 | 17 | 369 | 65 | n.d. | n.d. | 267 |
| 9 | 1 | B | AliBu3 | 10 | 500 | Tol | 20 | 25 | 43 | 1120 | >99 | 7 | 2.27 | 270 |
| 10 | 1 | B | AliBu3 | 10 | 500 | C2H2Cl4 | 48 | 25 | 17 | 184 | 61 | n.d. | n.d. | 265 |
| 11 | 1 | B | AliBu3 | 5 | 500 | PhCl2 | 12 | 25 | 5 | 217 | >99 | n.d. | n.d. | 272 |
| 12 | 1 | B | AliBu3 | 15 | 500 | PhCl2 | 12 | 25 | 42 | 1822 | >99 | 6 | 2.09 | 271 |
| 13 | 1 | B | AliBu3 | 10 | 500 | PhCl2 | 12 | 50 | 60 | 2604 | >99 | 5 | 2.00 | 273 |
| 14 | 1 | B | AliBu3 | 10 | 500 | PhCl2 | 12 | 70 | 59 | 2561 | >99 | 4 | 1.98 | 272 |
| 15 | 1 | B | AliBu3 | 10 | 500 | PhCl2 | 12 | 90 | 61 | 2647 | >99 | 0.1 | 18.78 | 271 |
| 16 | 1 | B | AliBu3 | 10 | 200 | PhCl2 | 12 | 25 | 41 | 712 | >99 | 5 | 2.02 | 275 |
| 17 | 1 | B | AliBu3 | 10 | 400 | PhCl2 | 12 | 25 | 35 | 1215 | >99 | 8 | 2.02 | 273 |
| 18 | 1 | B | AliBu3 | 10 | 700 | PhCl2 | 12 | 25 | 35 | 2126 | >99 | 13 | 1.49 | 271 |
| 19 | Scg | B | AliBu3 | 10 | 500 | PhCl2 | 48 | 25 | 7 | 76 | 78 | n.d. | n.d. | 268 |
| 20 | Scg | Bh | AliBu3 | 10 | 500 | PhCl2 | 12 | 25 | 9 | 390 | >99 | n.d. | n.d. | 272 |
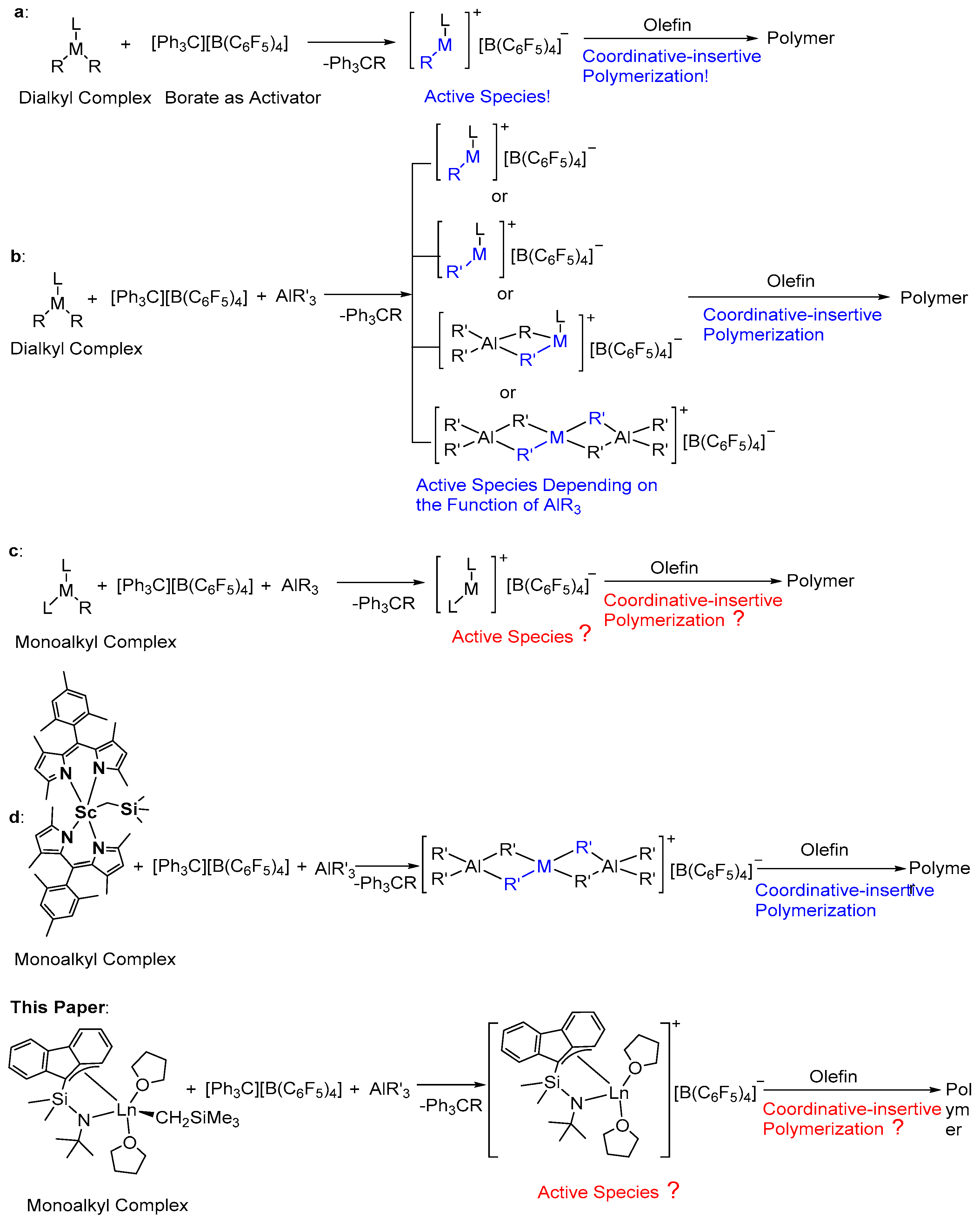
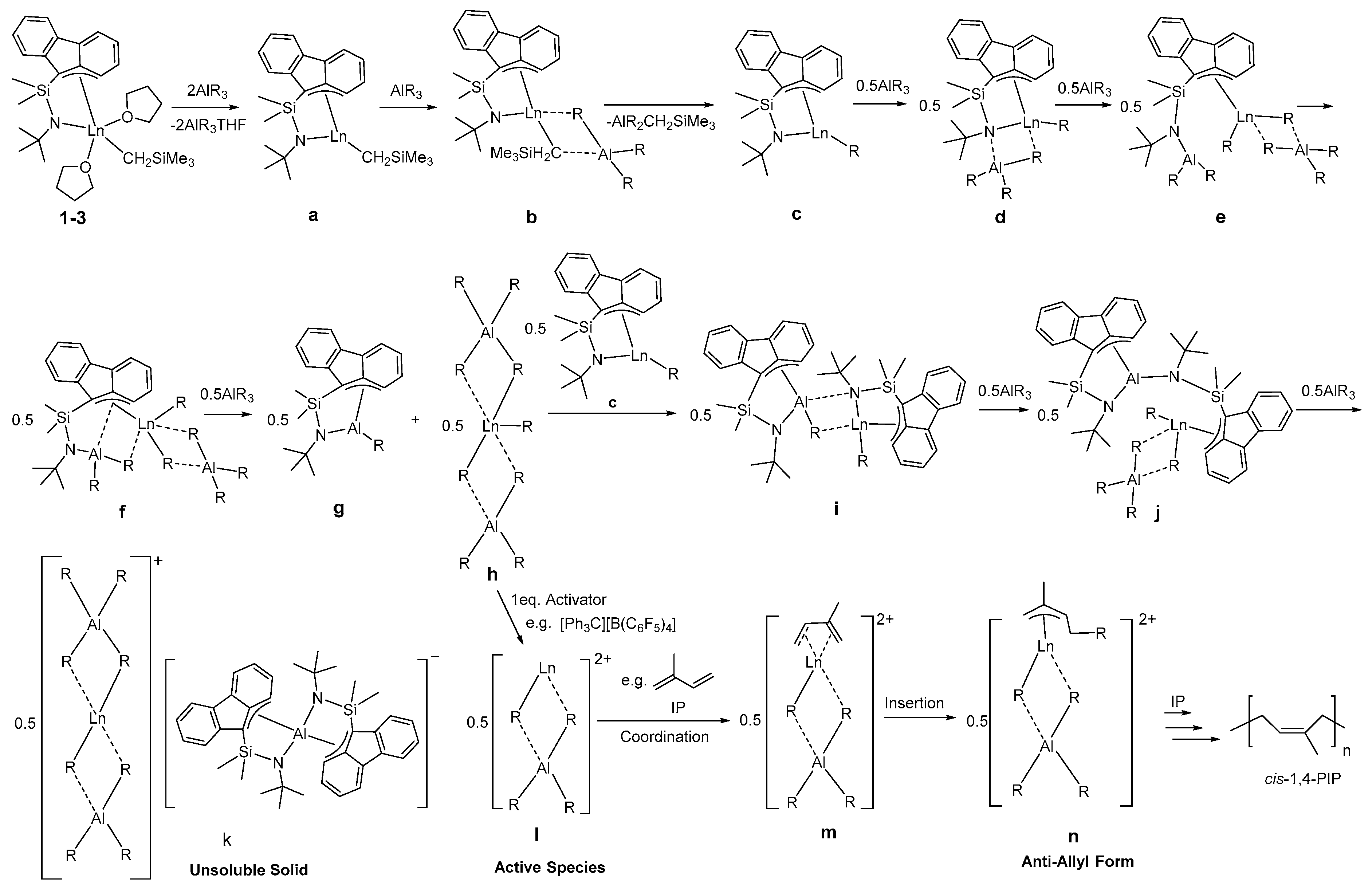
3.6. Polymerization Mechanism Study
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Edelmann, F.T. Lanthanides and Actinides: Annual Survey of their Organometallic Chemistry Covering the Year 2017. Coord. Chem. Rev. 2018, 370, 129–223. [Google Scholar] [CrossRef]
- Huang, J.; Liu, Z.; Cui, D.; Liu, X. Precisely Controlled Polymerization of Styrene and Conjugated Dienes by Group 3 Single-Site Catalysts. ChemCatChem 2018, 10, 42–61. [Google Scholar] [CrossRef]
- Soller, B.S.; Salzinger, S.; Rieger, B. Rare Earth Metal-Mediated Precision Polymerization of Vinylphosphonates and Conjugated Nitrogen-Containing Vinyl Monomers. Chem. Rev. 2016, 116, 1993–2022. [Google Scholar] [CrossRef]
- Nishiura, M.; Guo, F.; Hou, Z. Half-Sandwich Rare-Earth-Catalyzed Olefin Polymerization, Carbometalation, and Hydroarylation. Acc. Chem. Res. 2015, 48, 2209–2220. [Google Scholar] [CrossRef]
- Nishiura, M.; Hou, Z. Novel Polymerization Catalysts and Hydride Clusters from Rare-Earth Metal Dialkyls. Nat. Chem. 2010, 2, 257–268. [Google Scholar] [CrossRef]
- Zimmermann, M.; Anwander, R. Homoleptic Rare-Earth Metal Complexes Containing Ln-C σ-Bonds. Chem. Rev. 2010, 110, 6194–6259. [Google Scholar] [CrossRef] [PubMed]
- Zeimentz, P.M.; Arndt, S.; Elvidge, B.R.; Okuda, J. Cationic Organometallic Complexes of Scandium, Yttrium, and the Lanthanoids. Chem. Rev. 2006, 106, 2404–2433. [Google Scholar] [CrossRef]
- Gromada, J.; Carpentier, J.F.; Mortreux, A. Group 3 Metal Catalysts for Ethylene and α-olefin Polymerization. Coord. Chem. Rev. 2004, 248, 397–410. [Google Scholar]
- Arndt, S.; Okuda, J. Mono(cyclopentadienyl) Complexes of the Rare-Earth Metals. Chem. Rev. 2002, 102, 1953–1976. [Google Scholar] [CrossRef]
- Hou, Z.; Wakatsuki, Y. Recent Developments in Organolanthanide Polymerization Catalysts. Coord. Chem. Rev. 2002, 231, 1–22. [Google Scholar] [CrossRef]
- Piers, W.E.; Emslie, D.J. Non-Cyclopentadienyl Ancillaries in Organogroup 3 Metal Chemistry: A Fine Balance in Ligand Design. Coord. Chem. Rev. 2002, 233–234, 131–155. [Google Scholar] [CrossRef]
- Kawaoka, A.M.; Mark, T.J. Organolanthanide-Catalyzed Synthesis of Phosphine-Terminated Polyethylenes. Scope and Mechanism. J. Am. Chem. Soc. 2005, 127, 6311–6324. [Google Scholar] [CrossRef] [PubMed]
- Kirillov, E.; Lehmann, C.W.; Razavi, A.; Carpentier, J.F. Highly Syndiospecific Polymerization of Styrene Catalyzed by Allyl Lanthanide Complexes. J. Am. Chem. Soc. 2004, 126, 12240–12241. [Google Scholar] [CrossRef] [PubMed]
- Kawaoka, A.M.; Mark, T.J. Organolanthanide-Catalyzed Synthesis of Phosphine-Terminated Polyethylenes. J. Am. Chem. Soc. 2004, 126, 12764–12765. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Forrestal, K.J.; Ziller, J.W. Activity of [Sm(C5Me5)3] in Ethylene Polymerization and Synthesis of [U(C5Me5)3], the First Tris(pentamethylcyclopentadienyl) 5f-Element Complex. Angew. Chem. Int. Ed. 1997, 36, 774–776. [Google Scholar] [CrossRef]
- Lin, F.; Liu, Z.; Wang, T.; Cui, D. Highly 2,3-Selective Polymerization of Phenylallene and Its Derivatives with Rare-Earth Metal Catalysts: From Amorphous to Crystalline Products. Angew. Chem. Int. Ed. 2017, 56, 14653–14657. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Cui, D.; Tang, T. Stereo- and Temporally Controlled Coordination Polymerization Triggered by Alternating Addition of a Lewis Acid and Base. Angew. Chem. Int. Ed. 2016, 55, 11975–11978. [Google Scholar] [CrossRef]
- Arndt, S.; Beckerle, K.; Hultzsch, K.C.; Sinnema, P.J.; Voth, P.; Spaniol, T.P.; Okuda, J. Group 3 and 4 Metal Alkyl and Hydrido Complexes Containing a Linked Amido-Cyclopentadienyl Ligand: “Constrained Geometry” Polymerization Catalysts for Nonpolar and Polar Monomers. J. Mol. Catal. A-Chem. 2002, 190, 215–223. [Google Scholar] [CrossRef]
- Hultzsch, K.C.; Spaniol, T.P.; Okuda, J. Half-Sandwich Alkyl and Hydrido Complexes of Yttrium: Convenient Synthesis and Polymerization Catalysis of Polar Monomers. Angew. Chem. Int. Ed. 1999, 38, 227–230. [Google Scholar] [CrossRef]
- Chen, J.; Gao, Y.; Wang, B.; Lohr, T.L.; Marks, T.J. Scandium-Catalyzed Self-Assisted Polar Co-monomer Enchainment in Ethylene Polymerization. Angew. Chem. Int. Ed. 2017, 129, 16180–16184. [Google Scholar] [CrossRef]
- Shi, X.; Nishiura, M.; Hou, Z. Cuu Polyaddition of Dimethoxyarenes to Unconjugated Dienes by Rare Earth Catalysts. J. Am. Chem. Soc. 2016, 138, 6147–6150. [Google Scholar] [CrossRef]
- Li, X.; Baldamus, J.; Hou, Z. Alternating Ethylene–Norbornene Copolymerization Catalyzed by Cationic Half-Sandwich Scandium Complexes. Angew. Chem. Int. Ed. 2005, 44, 962–965. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Baldamus, J.; Hou, Z. Scandium Half-Metallocene-Catalyzed Syndiospecific Styrene Polymerization and Styrene-Ethylene Copolymerization: Unprecedented Incorporation of Syndiotactic Styrene-Styrene Sequences in Styrene-Ethylene Copolymers. J. Am. Chem. Soc. 2004, 126, 13910–13911. [Google Scholar] [CrossRef]
- Gao, W.; Cui, D. Highly cis-1,4 Selective Polymerization of Dienes with Homogeneous Ziegler-Natta Catalysts Based on NCN-Pincer Rare Earth Metal Dichloride Precursors. J. Am. Chem. Soc. 2008, 130, 4984–4991. [Google Scholar] [CrossRef]
- Zhang, L.; Suzuki, T.; Luo, Y.; Nishiura, M.; Hou, Z. Cationic Alkyl Rare-Earth Metal Complexes Bearing an Ancillary Bis(phosphinophenyl)amido Ligand: A Catalytic System for Living cis-1,4-Polymerization and Copolymerization of Isoprene and Butadiene. Angew. Chem. Int. Ed. 2007, 119, 1941–1945. [Google Scholar] [CrossRef]
- Bambirra, S.; Bouwkamp, M.W.; Meetsma, A.; Hessen, B. One Ligand Fits All: Cationic Mono(amidinate) Alkyl Catalysts over the Full Size Range of the Group 3 and Lanthanide Metals. J. Am. Chem. Soc. 2004, 126, 9182–9183. [Google Scholar] [CrossRef]
- Kirillov, E.; Toupet, L.; Lehmann, C.W.; Razavi, A.; Carpentier, J.F. “Constrained Geometry” Group 3 Metal Complexes of the Fluorenyl-Based Ligands [(3,6-tBu2Flu)SiR2NtBu]: Synthesis, Structural Characterization, and Polymerization Activity. Organometallics 2003, 22, 4467–4479. [Google Scholar] [CrossRef]
- Jian, Z.; Petrov, A.R.; Hangaly, N.K.; Li, S.; Rong, W.; Rufanov, K.A.; Harms, K.; Sundermeyer, J.; Cui, D. Phosphazene-Functionalized Cyclopentadienyl and Its Derivatives Ligated Rare-Earth Metal Alkyl Complexes: Synthesis, Structures, and Catalysis on Ethylene Polymerization. Organometallics 2012, 31, 4267–4282. [Google Scholar] [CrossRef]
- Jian, Z.; Cui, D.; Hou, Z. Rare-Earth-Metal-Hydrocarbyl Complexes Bearing Linked Cyclopentadienyl or Fluorenyl Ligands: Synthesis, Catalyzed Styrene Polymerization, and Structure-Reactivity Relationship. Chem. Eur. J. 2012, 18, 2674–2684. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, X.; Tong, X.; Zhang, H.; Chen, Y.; Liu, Y.; Liu, H.; Wang, S.; Nishiura, M.; He, H.; et al. Aluminum Effects in the Syndiospecific Copolymerization of Styrene with Ethylene by Cationic Fluorenyl Scandium Alkyl Catalysts. Organometallics 2013, 32, 1445–1458. [Google Scholar] [CrossRef]
- Du, G.; Xue, J.; Peng, D.; Yu, C.; Wang, H.; Zhou, Y.; Bi, J.; Zhang, S.; Dong, Y.; Li, X. Copolymerization of Isoprene with Ethylene Catalyzed by Cationic Half-Sandwich Fluorenyl Scandium Catalysts. J. Polym. Sci. Pol. Chem. 2015, 53, 2898–2907. [Google Scholar] [CrossRef]
- Du, G.; Long, Y.; Xue, J.; Zhang, S.; Dong, Y.; Li, X. 1,4-Selective Polymerization of 1,3-Cyclohexadiene and Copolymerization with Styrene by Cationic Half-Sandwich Fluorenyl Rare Earth Metal Alkyl Catalysts. Macromolecules 2015, 48, 1627–1635. [Google Scholar] [CrossRef]
- Okuda, J.; Schattenmann, F.J.; Wocadlo, S.; Massa, W. Synthesis and Characterization of Zirconium Complexes Containing a Linked Amido-Fluorenyl Ligand. Organometallics 1995, 14, 789–795. [Google Scholar] [CrossRef]
- Bochmann, M.; Jaggar, A.J.; Nicholls, J.C. Base-Free Cationic 14-Electron Titanium and Zirconium Alkyls: In situ Generation, Solution Structures, and Olefin Polymerization Activity. Angew. Chem. Int. Ed. 1990, 29, 780–782. [Google Scholar] [CrossRef]
- Lin, Z.; Le Marechal, J.F.; Sabat, M.; Marks, T.J. Models for Organometallic Molecule-Support Complexes. Synthesis and Properties of Cationic Organoactinides. J. Am. Chem. Soc. 1987, 109, 4127–4129. [Google Scholar] [CrossRef]
- Jordan, R.F.; Dasher, W.E.; Echols, S.F. Reactive Cationic Dicyclopentadienylzirconium (IV) Complexes. J. Am. Chem. Soc. 1986, 108, 1718–1719. [Google Scholar] [CrossRef]
- Zimmermann, M.; Tornroos, K.W.; Anwander, R. Cationic Rare-Earth-Metal Half-Sandwich Complexes for the Living trans-1,4-Isoprene Polymerization. Angew. Chem. Int. Ed. 2008, 47, 775–778. [Google Scholar] [CrossRef]
- Dietrich, H.M.; Raudaschl-Sieber, G.; Anwander, R. Trimethylyttrium and Trimethyllutetium. Angew. Chem. Int. Ed. 2005, 117, 5437–5440. [Google Scholar] [CrossRef]
- Hitzbleck, J.; Beckerle, K.; Okuda, J.; Halbach, T.; Muelhaupt, R. Syndiospecific Polymerization Catalysts for Styrene Based on Rare Earth Metal Half-Sandwich Complexes. Macromol. Symp. 2006, 236, 23–29. [Google Scholar] [CrossRef]
- Yu, C.; Zhang, P.; Gao, F.; Zhang, S.; Li, X. A Displacement-Type Fluorescent Probe Reveals Active Species in the Coordinative Polymerization of Olefins. Polym. Chem. 2018, 9, 603–610. [Google Scholar] [CrossRef]
- Fischbach, A.; Klimpel, M.G.; Widenmeyer, M.; Herdtweck, E.; Scherer, W.; Anwander, R. Stereospecific Polymerization of Isoprene with Molecular and MCM-48-Grafted Lanthanide (Ш) Tetraalkylaluminates. Angew. Chem. Int. Ed. 2004, 116, 2284–2289. [Google Scholar] [CrossRef]
- Zhang, L.; Nishiura, M.; Yuki, M.; Luo, Y.; Hou, Z. Isoprene Polymerization with Yttrium Amidinate Catalysts: Switching the Regio- and Stereoselectivity by Addition of AlMe3. Angew. Chem. Int. Ed. 2008, 47, 2642–2645. [Google Scholar] [CrossRef] [PubMed]
- Döring, C.; Kempe, R. Synthesis and Structure of Aminopyridinato-Stabilized Yttrium and Lanthanum Amides and Their Reactivity towards Alkylaluminium Compounds. Eur. J. Inorg. Chem. 2009, 2009, 412–418. [Google Scholar] [CrossRef]






| 1 | 2 | 3 | |
|---|---|---|---|
| Ln−N1 | 2.065(3) | 2.180(3) | 2.213(3) |
| Ln−C1 | 2.395(3) | 2.504(4) | 2.566(3) |
| Ln−C2 | 2.730(3) | 2.746(4) | 2.769(3) |
| Ln−C3 | 3.067(3) | 3.044(4) | 3.036(4) |
| Ln−C20 | 2.226(4) | 2.338(4) | 2.392(4) |
| N1−Ln−C20 | 109.34(13) | 111.36(14) | 112.29(13) |
| N1−Ln−C1 | 75.05(11) | 71.80(13) | 70.57(11) |
| C20−Ln−C1 | 144.95(13) | 148.23(15) | 149.02(13) |
| N1−Ln−C2 | 91.58(11) | 88.71(12) | 87.82(11) |
| C20−Ln−C2 | 113.15(12) | 117.13(14) | 118.36(13) |
| C1−Ln−C2 | 32.52(11) | 31.92(13) | 31.58(11) |
| N1−Ln−C3 | 86.93(11) | 85.21(13) | 84.56(11) |
| C20−Ln−C3 | 89.52(11) | 93.24(14) | 94.39(12) |
| C1−Ln−C3 | 55.57(11) | 55.06(13) | 54.68(11) |
| C2−Ln−C3 | 27.22(10) | 27.32(12) | 27.47(11) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, G.; Wu, X.; Yan, X.; Yan, L.; Li, X.; Zhang, S.; Qiu, N. Unprecedentedly High Activity and/or High Regio-/Stereoselectivity of Fluorenyl-Based CGC Allyl-Type η3:η1-tert-Butyl(dimethylfluorenylsilyl)amido Ligated Rare Earth Metal Monoalkyl Complexes in Olefin Polymerization. Polymers 2019, 11, 836. https://doi.org/10.3390/polym11050836
Guo G, Wu X, Yan X, Yan L, Li X, Zhang S, Qiu N. Unprecedentedly High Activity and/or High Regio-/Stereoselectivity of Fluorenyl-Based CGC Allyl-Type η3:η1-tert-Butyl(dimethylfluorenylsilyl)amido Ligated Rare Earth Metal Monoalkyl Complexes in Olefin Polymerization. Polymers. 2019; 11(5):836. https://doi.org/10.3390/polym11050836
Chicago/Turabian StyleGuo, Ge, Xiaolu Wu, Xiangqian Yan, Li Yan, Xiaofang Li, Shaowen Zhang, and Nannan Qiu. 2019. "Unprecedentedly High Activity and/or High Regio-/Stereoselectivity of Fluorenyl-Based CGC Allyl-Type η3:η1-tert-Butyl(dimethylfluorenylsilyl)amido Ligated Rare Earth Metal Monoalkyl Complexes in Olefin Polymerization" Polymers 11, no. 5: 836. https://doi.org/10.3390/polym11050836
APA StyleGuo, G., Wu, X., Yan, X., Yan, L., Li, X., Zhang, S., & Qiu, N. (2019). Unprecedentedly High Activity and/or High Regio-/Stereoselectivity of Fluorenyl-Based CGC Allyl-Type η3:η1-tert-Butyl(dimethylfluorenylsilyl)amido Ligated Rare Earth Metal Monoalkyl Complexes in Olefin Polymerization. Polymers, 11(5), 836. https://doi.org/10.3390/polym11050836