New Condensation Polymer Precursors Containing Consecutive Silicon Atoms—Decaisopropoxycyclopentasilane and Dodecaethoxyneopentasilane—And Their Sol–Gel Polymerization


Abstract
:
1. Introduction

2. Materials and Methods
2.1. Materials
2.2. Instruments and Measurements
2.3. Synthesis of Decaisopropoxycyclopentasilane (CPS, Si5(OiPr)10)
2.4. Synthesis of Dodecaethoxyneopentasilane (NPS, Si5(OEt)12)
2.5. Sol–Gel Polymerization of CPS and NPS
2.6. Preparation of CPSH
2.7. Preparation of CPSOH
2.8. Preparation of CPSN
2.9. Preparation of NPSH
2.10. Preparation of NPSOH
2.11. Preparation of NPSN
3. Results and Discussion
3.1. Synthesis of the New Condensation Polymer Precursors
3.2. Preparation of CPS and NPS Xerogels
3.3. Characterization of CPS and NPS Xerogels
3.3.1. Surface Area and Porosity
3.3.2. Solid-State CP/MAS NMR Analysis
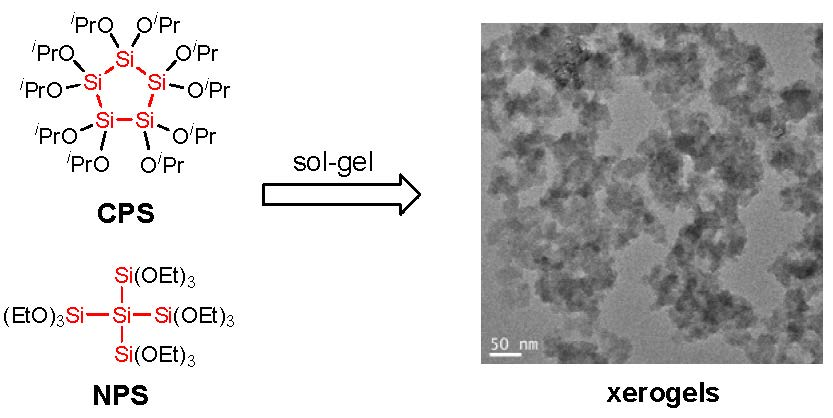
3.3.3. Microscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Brinker, C.J.; Scherer, G.W. Introduction. In Sol-Gel Science: The Physics and Chemistry of Sol-Gel Processing; Academic Press: San Diego, CA, USA, 1990; pp. 1–20. [Google Scholar]
- Brinker, C.J.; Scherer, G.W. Hydrolysis and Condensation II: Silicates. In Sol-Gel Science: The Physics and Chemistry of Sol-Gel Processing; Academic Press: San Diego, CA, USA, 1990; pp. 97–234. [Google Scholar]
- Brook, M.A. Siloxane based on T and Q units. In Silicon in Organic, Organometallic, and Polymer Chemistry; John Wiley & Sons: New York, NY, USA, 2000; pp. 309–339. [Google Scholar]
- Wilkes, G.L.; Huang, H.-H.; Glaser, R.H. New Inorganic-Organic Hybrid Materials Through Sol-Gel Approach. In Silicon-Based Polymer Science; Ziegler, J.M., Fearon, F.W.G., Eds.; American Chemical Society: Washington, DC, USA, 1990; pp. 207–226. [Google Scholar]
- Avnir, D.; Klein, L.C.; Levy, D.; Schubert, U.; Wojcik, A.B. Organo-silica sol-gel materials. In The Chemistry of Organic Silicon Compounds; Rappoport, Z., Apeloig, Y., Eds.; John Wiley & Sons: New York, NY, USA, 1998; Volume 2, pp. 2317–2362. [Google Scholar]
- Hench, L.L.; West, J.K. The Sol-Gel Process. Chem. Rev. 1990, 90, 33–72. [Google Scholar] [CrossRef]
- Schottner, G. Hybrid Sol-Gel-Derived Polymers: Applications of Multifunctional Materials. Chem. Mater. 2001, 13, 3422–3435. [Google Scholar] [CrossRef]
- Ciriminna, R.; Fidalgo, A.; Pandarus, V.; Béland, F.; Ilharco, L.M.; Pagliaro, M. The Sol–Gel Route to Advanced Silica-Based Materials and Recent Applications. Chem. Rev. 2013, 113, 6592–6620. [Google Scholar] [CrossRef] [PubMed]
- Danks, A.E.; Hall, S.R.; Schnepp, Z. The evolution of ‘sol–gel’ chemistry as a technique for materials synthesis. Mater. Horiz. 2016, 3, 91–112. [Google Scholar] [CrossRef] [Green Version]
- Loy, D.A.; Shea, K.J. Bridged Polysilsesquioxanes. Highly Porous Hybrid Organic-Inorganic Materials. Chem. Rev. 1995, 95, 1431–1442. [Google Scholar] [CrossRef]
- Croissant, J.G.; Cattoën, X.; Wong Chi Man, M.; Durand, J.-O.; Khashab, N.M. Syntheses and applications of periodic mesoporous organosilica nanoparticles. Nanoscale 2015, 7, 20318–20334. [Google Scholar] [CrossRef]
- Shea, K.J.; Loy, D.A. Bridged Polysilsesquioxanes. Molecular-Engineered Hybrid Organic-Inorganic Materials. Chem. Mater. 2001, 13, 3306–3319. [Google Scholar] [CrossRef]
- Baney, R.H.; Itoh, M.; Sakakibara, A.; Suzuki, T. Silsesquioxanes. Chem. Rev. 1995, 95, 1409–1430. [Google Scholar] [CrossRef]
- Park, S.J.; Cho, H.M.; Lee, M.E.; Kim, M.; Han, G.; Hong, S.; Lim, S.; Lee, H.; Hwang, B.; Kim, S.K.; et al. Soluble polycyclosilane–polysiloxane hybrid material and silicon thin film with optical properties at 193 nm and etch selectivity. J. Mater. Chem. C 2015, 3, 239–242. [Google Scholar] [CrossRef]
- Park, S.J.; Choi, J.M.; Cho, H.M.; Kim, C.H.; Lee, M.E. N-Methylaniline-induced Si-Si bond cleavages of perchlorooligosilanes. J. Organomet. Chem. 2014, 768, 140–144. [Google Scholar] [CrossRef]
- Wilkins, C.J. The Reductions of Hexachlorodisilane with Ammonium Halides and Trimethylamine Hydrohalides. J. Chem. Soc. 1953, 3409–3412. [Google Scholar] [CrossRef]
- Kummer, D.; Köster, H. 1,4-Dihydro-1,4-bis-(trichlorosilyl)pyridine from Si2Cl6 and Pyridine. Angew. Chem. Int. Ed. Engl. 1971, 10, 412. [Google Scholar] [CrossRef]
- Tillmann, J.; Meyer-Wegner, F.; Nadj, A.; Becker-Baldus, J.; Sinke, T.; Bolte, M.; Holthausen, M.C.; Wagner, M.; Lerner, H.-W. Unexpected Disproportionation of Tetramethylethylenediamine-Supported Perchlorodisilane Cl3SiSiCl3. Inorg. Chem. 2012, 51, 8599–8606. [Google Scholar] [CrossRef]
- Zhang, J.; Xie, J.; Lee, M.E.; Zhang, L.; Zuo, Y.; Feng, S. Ionic SNi-Si Nucleophilic Substitution in N-Methylaniline-Induced Si-Si Bond Cleavages of Si2Cl6. Chem. Eur. J. 2016, 22, 5010–5016. [Google Scholar] [CrossRef]
- Schweizer, J.I.; Meyer, L.; Nadj, A.; Diefenbach, M.; Holthausen, M.C. Unraveling the Amine-Induced Disproportionation Reaction of Perchlorinated Silanes—A DFT Study. Chem. Eur. J. 2016, 22, 14328–14335. [Google Scholar] [CrossRef]
- Fernández-Sánchez, C.; Rodríguez, J.A.; Domínguez, C. Synthesis of sol-gel SiO2-based materials using alkoxydisilane precursors: mechanisms and luminescence studies. J. Sol-Gel Sci. Technol. 2015, 73, 417–427. [Google Scholar] [CrossRef]
- Rodríguez, J.A.; Fernández-Sánchez, C.; Domínguez, C.; Hernández, S.; Berencén, Y. Bulk silica-based luminescent materials by sol–gel processing of non-conventional precursors. Appl. Phys. Lett. 2012, 101, 171908. [Google Scholar] [CrossRef]
- Hengge, E.; Kovar, D. Zur reaktion der silicium—Phenyl-bindung mit HCl. J. Organomet. Chem. 1977, 125, C29–C32. [Google Scholar] [CrossRef]
- Kovar, D.; Utvary, K.; Hengge, E. 29Si-NMR-Untersuchungen an einigen Cyclosilanderivaten. Monatsh. Chem. 1979, 110, 1295–1300. [Google Scholar] [CrossRef]
- Cannady, J.P.; Zhou, X. Composition Comprising Neopentasilane and Method of Preparing Same. WO 2008/051328 A1, 2 May 2008. [Google Scholar]
- Brunauer, S.; Emmett, P.H.; Teller, E.J. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Langmuir, I. The adsorption of gases on plane surfaces of glass, mica and platinum. J. Am. Chem. Soc. 1918, 40, 1361–1403. [Google Scholar] [CrossRef]
- Oviatt, H.W.; Shea, K.J., Jr.; Small, J.H. Alkylene-bridged silsesquioxane sol-gel synthesis and xerogel characterization. Molecular requirements for porosity. Chem. Mater. 1993, 5, 943–950. [Google Scholar] [CrossRef]
- Small, J.H.; Shea, K.J.; Loy, D.A. Arylene- and alkylene-bridged polysilsesquioxanes. J. Non-Cryst. Solids 1993, 160, 234–246. [Google Scholar] [CrossRef]
- Shea, K.J.; Loy, D.A.; Webster, O. Arylsilsesquioxane gels and related materials. New hybrids of organic and inorganic networks. J. Am. Chem. Soc. 1992, 114, 6700–6710. [Google Scholar] [CrossRef]
- Barrie, P.J.; Carr, S.W.; Ou, D.L.; Sullivan, A.C. Formation and Characterization of Novel Organodisilicate Glasses. Chem. Mater. 1995, 7, 265–270. [Google Scholar] [CrossRef]
- Birault, A.; Molina, E.; Toquer, G.; Lacroix-Desmazes, P.; Marcotte, N.; Carcel, C.; Katouli, M.; Bartlett, J.R.; Gérardin, C.; Wong Chi Man, M. Large-Pore Periodic Mesoporous Organosilicas as Advanced Bactericide Platforms. ACS Appl. Biol. Mater. 2018, 1, 1787–1792. [Google Scholar] [CrossRef]
- Engelhardt, G.; Jancke, H.; Mägi, M.; Pehk, T.; Lippmaa, E. Über die 1H-, 13C- und 29Si-NMR chemischen Verschiebungen einiger linearer, verzweigter und cyclischer Methylsiloxan-Verbindungen. J. Organomet. Chem. 1971, 28, 293–300. [Google Scholar] [CrossRef]
- Williams, E.A. NMR spectroscopy of organosilicon compounds. In The Chemistry of Organic Silicon Compounds; Patai, S., Rappoport, Z., Eds.; John Wiley & Sons: New York, NY, USA, 1989; pp. 545–547. [Google Scholar]
- Mabboux, P.-Y.; Gleason, K.K. Chemical Bonding Structure of Low Dielectric Constant Si:O:C:H Films Characterized by Solid-State NMR. J. Electrochem. Soc. 2005, 152, F7–F13. [Google Scholar] [CrossRef]
- Fyfe, C.A.; Zhang, Y.; Aroca, P. An alternative preparation of organofunctionalized silica gels and their characterization by two-dimensional high-resolution solid-state heteronuclear NMR correlation spectroscopy. J. Am. Chem. Soc. 1992, 114, 3252–3255. [Google Scholar] [CrossRef]
- Kuo, A.C.M. Poly(dimethylsiloxane). In Polymer Data Handbook; Mark, J.E., Ed.; Oxford University Press: New York, NY, USA, 1999; pp. 411–412. [Google Scholar]
- Yang, D.-Q.; Gillet, J.-N.; Meunier, M.; Sacher, E. Room temperature oxidation kinetics of Si nanoparticles in air, determined by x-ray photoelectron spectroscopy. J. Appl. Phys. 2005, 97, 024303. [Google Scholar] [CrossRef]
- Wolkin, M.V.; Jorne, J.; Fauchet, P.M.; Allan, G.; Delerue, C. Electronic States and Luminescence in Porous Silicon Quantum Dots: The Role of Oxygen. Phys. Rev. Lett. 1999, 82, 197–200. [Google Scholar] [CrossRef]
- Ritter, J.C.; Robinson, M.N.; Faraday, B.J.; Hoover, J.I. Room temperature oxidation of silicon during and after etching. J. Phys. Chem. Solids 1965, 26, 721–728. [Google Scholar] [CrossRef]
- Liao, W.-S.; Lee, S.-C. Water-induced room-temperature oxidation of Si–H and –Si–Si– bonds in silicon oxide. J. Appl. Phys. 1996, 80, 1171–1176. [Google Scholar] [CrossRef]
- Massoud, H.Z. The onset of the thermal oxidation of silicon from room temperature to 1000 °C. Microelectron. Eng. 1995, 28, 109–116. [Google Scholar] [CrossRef]
- Morita, M.; Ohmi, T. Characterization and Control of Native Oxide on Silicon. Jpn. J. Appl. Phys. 1994, 33, 370–374. [Google Scholar] [CrossRef]
- Taft, E.A. Growth of Native Oxide on Silicon. J. Electrochem. Soc. 1988, 135, 1022–1023. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Xerogels | Area S (m2/g) | Pore Volume (cm3/g) | Mean Pore Diameter (nm) |
|---|---|---|---|
| CPSH | 317 | 0.83 | 28, 50, 90 |
| CPSOH | 408 | 0.74 | 24, 43, 76 |
| CPSN | 150 | 0.24 | 32, 50, 76 |
| NPSH | No porosity | - | - |
| NPSOH | 7 | 0.02 | 66, 90, 119 |
| NPSN | 28 | 0.06 | 21, 43, 120 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.J.; Lee, M.E.; Cho, H.M.; Shim, S. New Condensation Polymer Precursors Containing Consecutive Silicon Atoms—Decaisopropoxycyclopentasilane and Dodecaethoxyneopentasilane—And Their Sol–Gel Polymerization. Polymers 2019, 11, 841. https://doi.org/10.3390/polym11050841
Park SJ, Lee ME, Cho HM, Shim S. New Condensation Polymer Precursors Containing Consecutive Silicon Atoms—Decaisopropoxycyclopentasilane and Dodecaethoxyneopentasilane—And Their Sol–Gel Polymerization. Polymers. 2019; 11(5):841. https://doi.org/10.3390/polym11050841
Chicago/Turabian StylePark, Sung Jin, Myong Euy Lee, Hyeon Mo Cho, and Sangdeok Shim. 2019. "New Condensation Polymer Precursors Containing Consecutive Silicon Atoms—Decaisopropoxycyclopentasilane and Dodecaethoxyneopentasilane—And Their Sol–Gel Polymerization" Polymers 11, no. 5: 841. https://doi.org/10.3390/polym11050841
APA StylePark, S. J., Lee, M. E., Cho, H. M., & Shim, S. (2019). New Condensation Polymer Precursors Containing Consecutive Silicon Atoms—Decaisopropoxycyclopentasilane and Dodecaethoxyneopentasilane—And Their Sol–Gel Polymerization. Polymers, 11(5), 841. https://doi.org/10.3390/polym11050841