Protein–Polyelectrolyte Complexes and Micellar Assemblies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
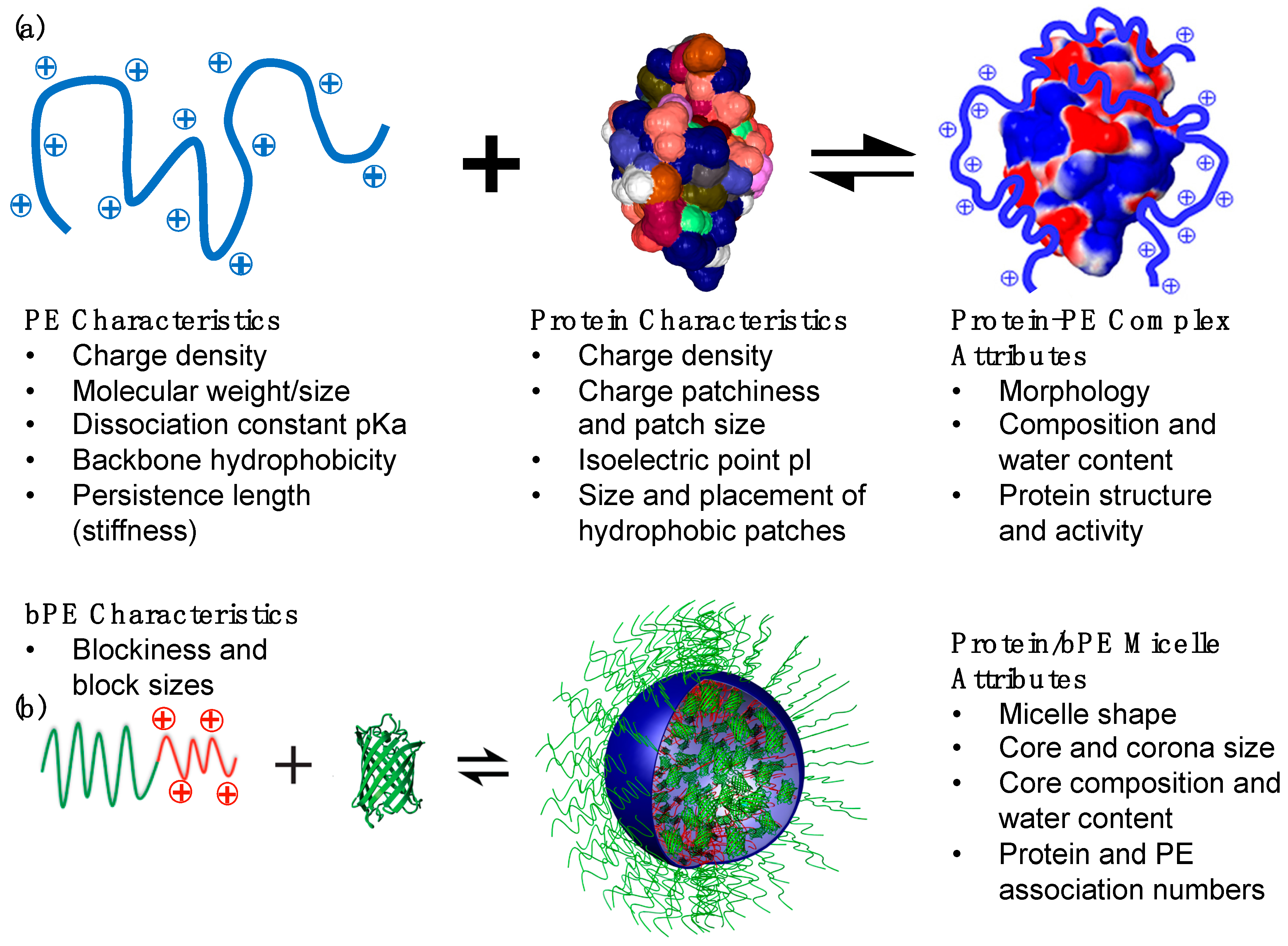
2. Proteins: Patchy Charged Nanoparticles or Amphoteric Polyelectrolytes?
3. Protein–Polyelectrolyte Bulk Complexes
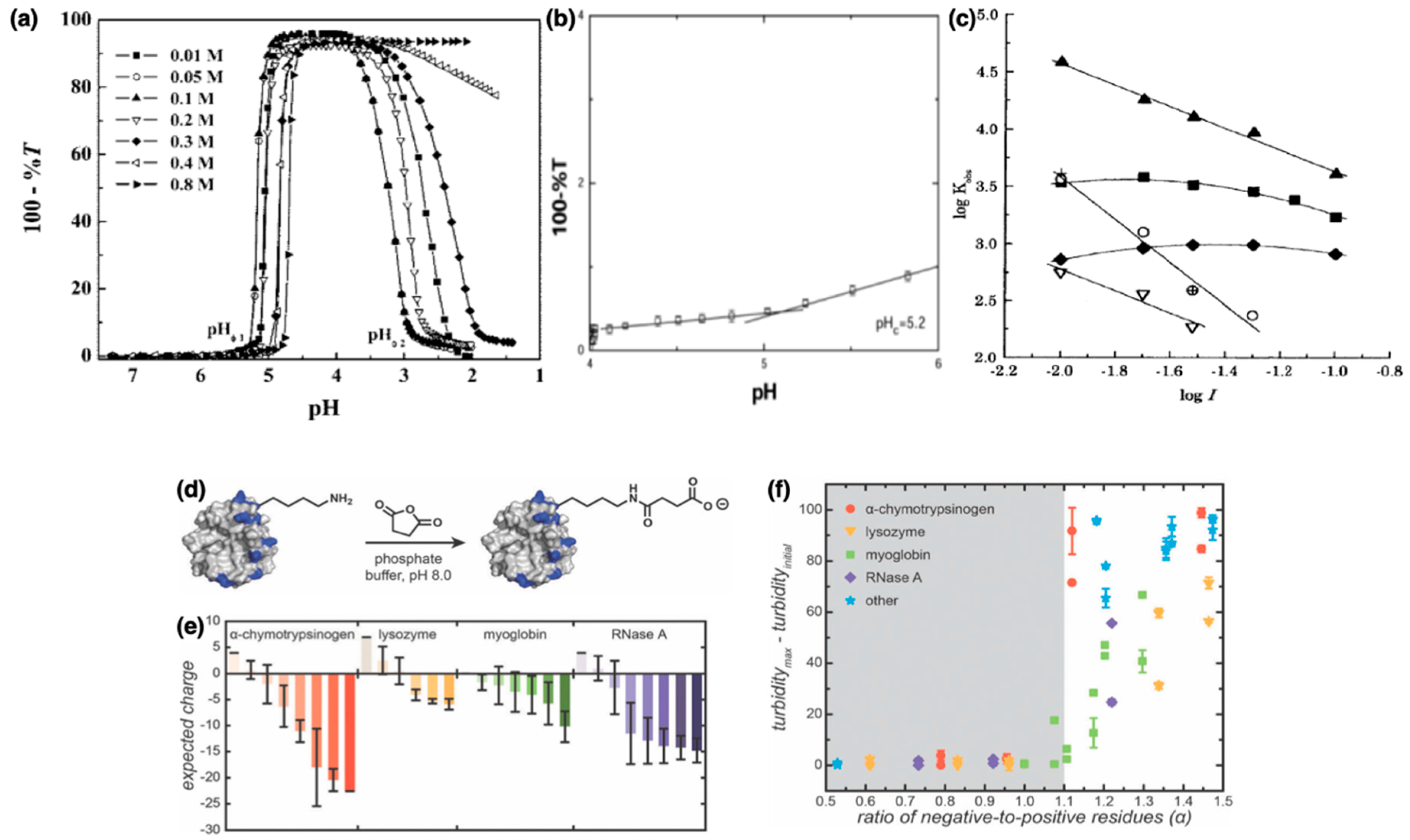
3.1. Thermodynamics and Phase Behavior of Protein–Polyelectrolyte Complexes
3.1.1. Phase Behavior
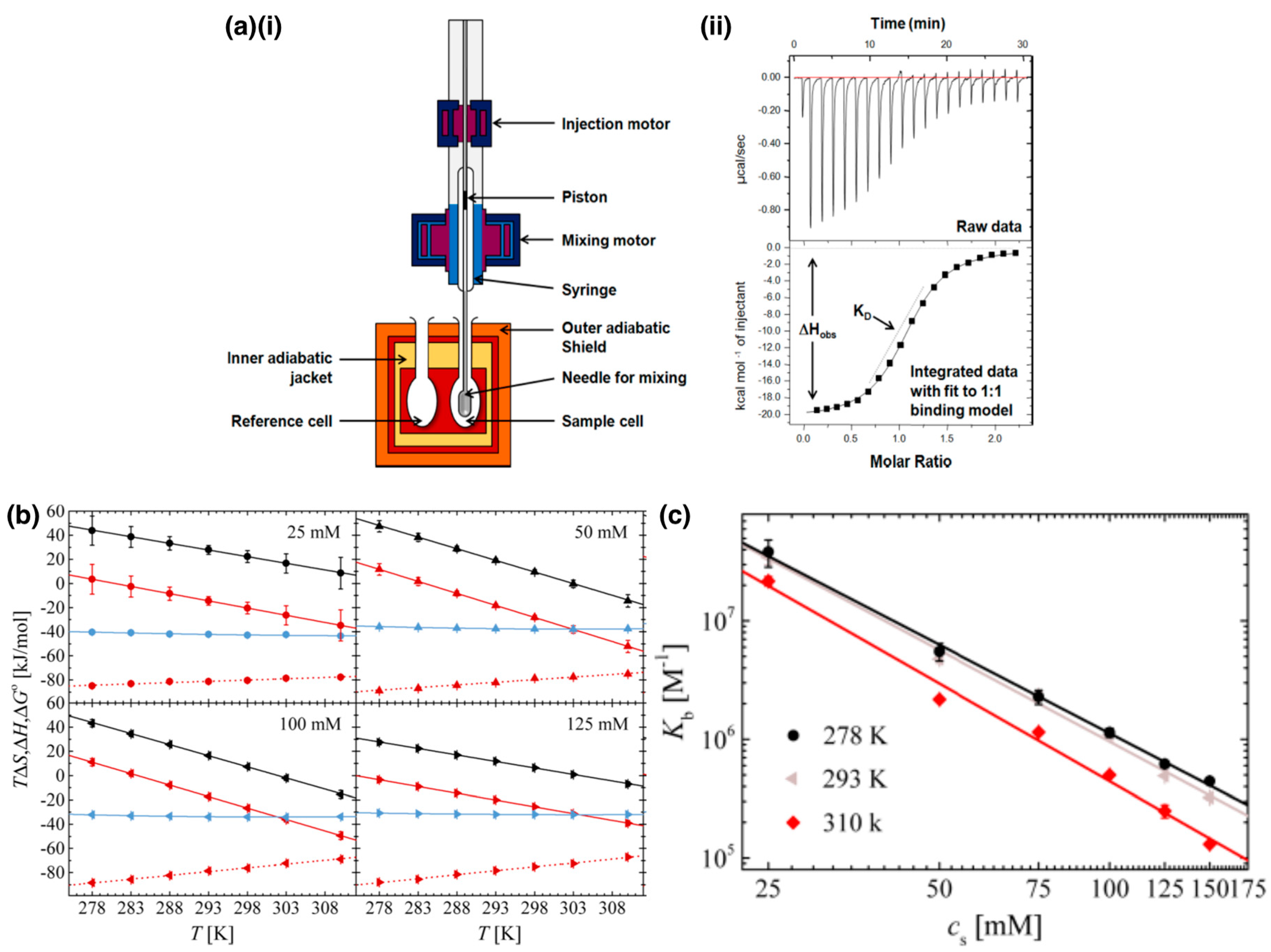
3.1.2. Thermodynamics of Complexation
3.2. Structure and Properties of Protein–Polyelectrolyte Complexes
Effect of Polyelectrolyte Stiffness
3.3. Applications of Protein–Polyelectrolyte Complexes
3.3.1. Protein Stability and Delivery
3.3.2. Protein Purification
3.3.3. Protein Encapsulation in Polyelectrolyte Complex Coacervates
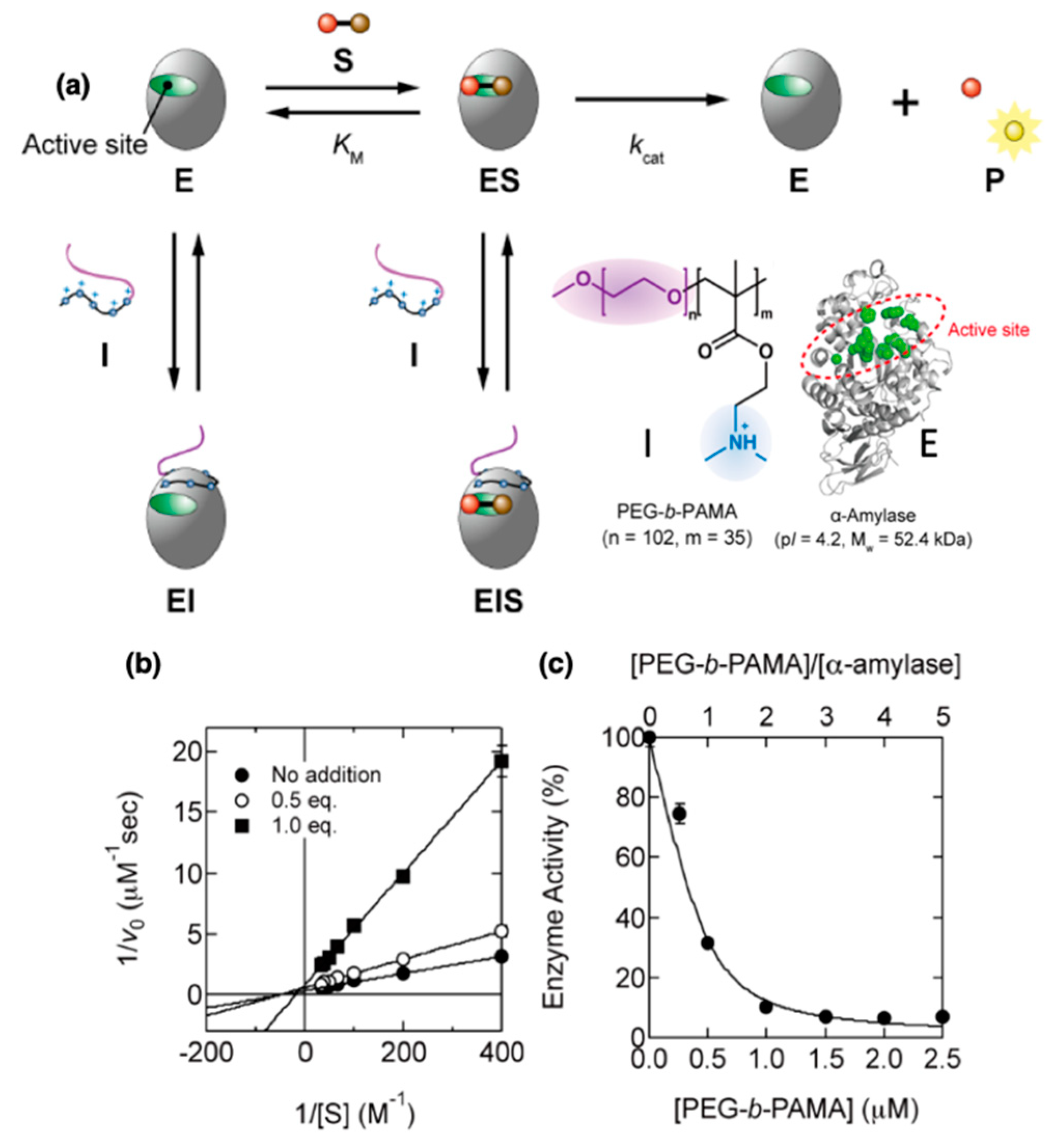
4. Protein-Block Polyelectrolyte Complex Micelles
4.1. Protein/Block Polyelectrolyte (bPE) Micelles
4.1.1. Critical Association Concentrations and Micelle Structure and Properties
4.1.2. Influence of Micellization on Protein Structure and Activity
4.1.3. Strategies for Stabilizing Protein/bPE Micelles
4.2. Protein Containing PEC Micelles
Phase Behavior and Structure
4.3. Applications of Protein–Polyelectrolyte Micelles
5. Conclusions and Future Outlook
Acknowledgments
Conflicts of Interest
References
- de Kruif, C.G.; Weinbreck, F.; de Vries, R. Complex coacervation of proteins and anionic polysaccharides. Curr. Opin. Colloid Interface Sci. 2004, 9, 340–349. [Google Scholar] [CrossRef]
- Cooper, C.L.; Dubin, P.L.; Kayitmazer, A.B.; Turksen, S. Polyelectrolyte-protein complexes. Curr. Opin. Colloid Interface Sci. 2005, 10, 52–78. [Google Scholar] [CrossRef]
- Woitovich Valetti, N.; Brassesco, M.E.; Picó, G.A. Polyelectrolytes–protein complexes: A viable platform in the downstream processes of industrial enzymes at scaling up level. J. Chem. Technol. Biotechnol. 2016, 91, 2921–2928. [Google Scholar] [CrossRef]
- Horn, J.; Kapelner, R.; Obermeyer, A. Macro- and microphase separated protein-polyelectrolyte complexes: Design parameters and current progress. Polymers 2019, 11, 578. [Google Scholar] [CrossRef] [PubMed]
- Semenyuk, P.; Muronetz, V. Protein interaction with charged macromolecules: From model polymers to unfolded proteins and post-translational modifications. Int. J. Mol. Sci. 2019, 20, 1252. [Google Scholar] [CrossRef]
- Xu, X.; Angioletti-Uberti, S.; Lu, Y.; Dzubiella, J.; Ballauff, M. Interaction of proteins with polyelectrolytes: Comparison of theory to experiment. Langmuir 2019, 35, 5373–5391. [Google Scholar] [CrossRef]
- Wang, X.; Zheng, K.; Si, Y.; Guo, X.; Xu, Y. Protein-polyelectrolyte interaction: Thermodynamic analysis based on the titration method. Polymers 2019, 11, 82. [Google Scholar] [CrossRef]
- Wang, S.; Chen, K.; Li, L.; Guo, X. Binding between proteins and cationic spherical polyelectrolyte brushes: Effect of pH, ionic strength, and stoichiometry. Biomacromolecules 2013, 14, 818–827. [Google Scholar] [CrossRef]
- Henzler, K.; Haupt, B.; Lauterbach, K.; Wittemann, A.; Borisov, O.; Ballauff, M. Adsorption of β-lactoglobulin on spherical polyelectrolyte brushes: Direct proof of counterion release by isothermal titration calorimetry. J. Am. Chem. Soc. 2010, 132, 3159–3163. [Google Scholar] [CrossRef]
- Srivastava, S.; Tirrell, M.V. Polyelectrolyte complexation. In Advances in Chemical Physics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; Volume 161, pp. 499–544. ISBN 9781119290971. [Google Scholar]
- Ulrich, S.; Seijo, M.; Stoll, S. The many facets of polyelectrolytes and oppositely charged macroions complex formation. Curr. Opin. Colloid Interface Sci. 2006, 11, 268–272. [Google Scholar] [CrossRef] [Green Version]
- Park, J.M.; Muhoberac, B.B.; Dubin, P.L.; Xia, J. Effects of protein charge heterogeneity in protein-polyelectrolyte complexation. Macromolecules 1992, 25, 290–295. [Google Scholar] [CrossRef]
- Korolev, N.; Allahverdi, A.; Lyubartsev, A.P.; Nordenskiöld, L. The polyelectrolyte properties of chromatin. Soft Matter 2012, 8, 9322–9333. [Google Scholar] [CrossRef]
- Carrivain, P.; Cournac, A.; Lavelle, C.; Lesne, A.; Mozziconacci, J.; Paillusson, F.; Signon, L.; Victor, J.M.; Barbi, M. Electrostatics of DNA compaction in viruses, bacteria and eukaryotes: Functional insights and evolutionary perspective. Soft Matter 2012, 8, 9285–9301. [Google Scholar] [CrossRef]
- Morawetz, H.; Hughes, W.L. The interaction of proteins with synthetic polyelectrolytes. I. Complexing of bovine serum albumin. J. Phys. Chem. 1952, 56, 64–69. [Google Scholar] [CrossRef]
- Berdick, M.; Morawetz, H. The interaction of catalase with synthetic polyelectrolytes. J. Biol. Chem. 1954, 206, 959–971. [Google Scholar] [PubMed]
- Sternberg, M.; Hershberger, D. Separation of proteins with polyacrylic acids. BBA-Protein Struct. 1974, 342, 195–206. [Google Scholar] [CrossRef]
- Xu, Y.; Mazzawi, M.; Chen, K.; Sun, L.; Dubin, P.L. Protein purification by polyelectrolyte coacervation: Influence of protein charge anisotropy on selectivity. Biomacromolecules 2011, 12, 1512–1522. [Google Scholar] [CrossRef]
- Du, X.; Dubin, P.L.; Hoagland, D.A.; Sun, L. Protein-selective coacervation with hyaluronic acid. Biomacromolecules 2014, 15, 726–734. [Google Scholar] [CrossRef]
- Maruyama, T.; Izaki, S.; Kurinomaru, T.; Handa, K.; Kimoto, T.; Shiraki, K. Protein-poly(amino acid) precipitation stabilizes a therapeutic protein L-asparaginase against physicochemical stress. J. Biosci. Bioeng. 2015, 120, 720–724. [Google Scholar] [CrossRef]
- Izaki, S.; Kurinomaru, T.; Handa, K.; Kimoto, T.; Shiraki, K. Stress tolerance of antibody-poly(amino acid) complexes for improving the stability of high concentration antibody formulations. J. Pharm. Sci. 2015, 104, 2457–2463. [Google Scholar] [CrossRef]
- Kurinomaru, T.; Shiraki, K. Aggregative protein–polyelectrolyte complex for high-concentration formulation of protein drugs. Int. J. Biol. Macromol. 2017, 100, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Minsky, B.B.; Zheng, B.; Dubin, P.L. Inhibition of antithrombin and bovine serum albumin native state aggregation by heparin. Langmuir 2014, 30, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Klyachko, N.L.; Manickam, D.S.; Brynskikh, A.M.; Uglanova, S.V.; Li, S.; Higginbotham, S.M.; Bronich, T.K.; Batrakova, E.V.; Kabanov, A.V. Cross-linked antioxidant nanozymes for improved delivery to CNS. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manickam, D.S.; Brynskikh, A.M.; Kopanic, J.L.; Sorgen, P.L.; Klyachko, N.L.; Batrakova, E.V.; Bronich, T.K.; Kabanov, A.V. Well-defined cross-linked antioxidant nanozymes for treatment of ischemic brain injury. J. Control. Release 2012, 162, 636–645. [Google Scholar] [CrossRef] [Green Version]
- Batrakova, E.V.; Li, S.; Reynolds, A.D.; Mosley, R.L.; Bronich, T.K.; Kabanov, A.V.; Gendelman, H.E. A macrophage-nanozyme delivery system for Parkinson’s disease. Bioconjug. Chem. 2007, 18, 1498–1506. [Google Scholar] [CrossRef]
- Lim, S.; Choi, Y.S.; Kang, D.G.; Song, Y.H.; Cha, H.J. The adhesive properties of coacervated recombinant hybrid mussel adhesive proteins. Biomaterials 2010, 31, 3715–3722. [Google Scholar] [CrossRef]
- Wei, W.; Tan, Y.; Martinez Rodriguez, N.R.; Yu, J.; Israelachvili, J.N.; Waite, J.H. A mussel-derived one component adhesive coacervate. Acta Biomater. 2014, 10, 1663–1670. [Google Scholar] [CrossRef]
- Stewart, R.J.; Wang, C.S.; Shao, H. Complex coacervates as a foundation for synthetic underwater adhesives. Adv. Colloid Interface Sci. 2011, 167, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Karabiyik Acar, O.; Kayitmazer, A.B.; Torun Kose, G. Hyaluronic acid/chitosan coacervate-based scaffolds. Biomacromolecules 2018, 19, 1198–1211. [Google Scholar] [CrossRef]
- Insua, I.; Wilkinson, A.; Fernandez-Trillo, F. Polyion complex (PIC) particles: Preparation and biomedical applications. Eur. Polym. J. 2016, 81, 198–215. [Google Scholar] [CrossRef] [Green Version]
- Turgeon, S.L.; Schmitt, C.; Sanchez, C. Protein–polysaccharide complexes and coacervates. Curr. Opin. Colloid Interface Sci. 2007, 12, 166–178. [Google Scholar] [CrossRef]
- Schmitt, C.; Turgeon, S.L. Protein/polysaccharide complexes and coacervates in food systems. Adv. Colloid Interface Sci. 2011, 167, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.S.; Bradley, A.R.; Valasatava, Y.; Duarte, J.M.; Prlic, A.; Rose, P.W. NGL viewer: Web-based molecular graphics for large complexes. Bioinformatics 2018, 34, 3755–3758. [Google Scholar] [CrossRef] [PubMed]
- Nolles, A.; Westphal, A.H.; de Hoop, J.A.; Fokkink, R.G.; Kleijn, J.M.; Van Berkel, W.J.H.; Borst, J.W. Encapsulation of GFP in complex coacervate core micelles. Biomacromolecules 2015, 16, 1542–1549. [Google Scholar] [CrossRef] [PubMed]
- Kayitmazer, A.B.; Seeman, D.; Minsky, B.B.; Dubin, P.L.; Xu, Y. Protein-polyelectrolyte interactions. Soft Matter 2013, 9, 2553–2583. [Google Scholar] [CrossRef]
- Kayitmazer, A.B.; Koksal, A.F.; Kilic Iyilik, E. Complex coacervation of hyaluronic acid and chitosan: Effects of pH, ionic strength, charge density, chain length and the charge ratio. Soft Matter 2015, 11, 8605–8612. [Google Scholar] [CrossRef] [PubMed]
- Cummings, C.S.; Obermeyer, A.C. Phase separation behavior of supercharged proteins and polyelectrolytes. Biochemistry 2018, 57, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Cousin, F.; Gummel, J.; Clemens, D.; Grillo, I.; Boué, F. Multiple scale reorganization of electrostatic complexes of poly(styrenesulfonate) and lysozyme. Langmuir 2010, 26, 7078–7085. [Google Scholar] [CrossRef] [PubMed]
- Cousin, F.; Gummel, J.; Combet, S.; Boué, F. The model Lysozyme-PSSNa system for electrostatic complexation: Similarities and differences with complex coacervation. Adv. Colloid Interface Sci. 2011, 167, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Cousin, F.; Gummel, J.; Ung, D.; Boué, F. Polyelectrolyte-protein complexes: Structure and conformation of each specie revealed by SANS. Langmuir 2005, 21, 9675–9688. [Google Scholar] [CrossRef] [PubMed]
- Gummel, J.; Cousin, F.; Boue, F. Structure transition in PSS/lysozyme complexes: A chain-conformation-driven process, as directly seen by small angle neutron scattering. Macromolecules 2008, 41, 2898–2907. [Google Scholar] [CrossRef]
- Kayitmazer, A.B.; Strand, S.P.; Tribet, C.; Jaeger, W.; Dubin, P.L. Effect of polyelectrolyte structure on protein−polyelectrolyte coacervates: Coacervates of bovine serum albumin with poly(diallyldimethylammonium chloride) versus chitosan. Biomacromolecules 2007, 8, 3568–3577. [Google Scholar] [CrossRef] [PubMed]
- Sedlák, E.; Fedunová, D.; Veselá, V.; Sedláková, D.; Antalík, M. Polyanion hydrophobicity and protein basicity affect protein stability in protein-polyanion complexes. Biomacromolecules 2009, 10, 2533–2538. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, N.; Klibanov, A.M. Controling the rate of protein release from polyelectrolyte complexes. Biotechnol. Bioeng. 2003, 82, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Voets, I.K.; de Keizer, A.; Cohen Stuart, M.A. Complex coacervate core micelles. Adv. Colloid Interface Sci. 2009, 147–148, 300–318. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Miyata, K.; Nishiyama, N.; Kataoka, K. Rational design of smart supramolecular assemblies for gene delivery: Chemical challenges in the creation of artificial viruses. Chem. Soc. Rev. 2012, 41, 2718–2739. [Google Scholar]
- Cabral, H.; Miyata, K.; Osada, K.; Kataoka, K. Block copolymer micelles in nanomedicine applications. Chem. Rev. 2018, 118, 6844–6892. [Google Scholar] [CrossRef] [PubMed]
- Cohen Stuart, M.A.; Besseling, N.A.M.; Fokkink, R.G. Formation of micelles with complex coacervate cores. Langmuir 1998, 14, 6846–6849. [Google Scholar] [CrossRef]
- van der Gucht, J.; Spruijt, E.; Lemmers, M.; Cohen Stuart, M.A. Polyelectrolyte complexes: Bulk phases and colloidal systems. J. Colloid Interface Sci. 2011, 361, 407–422. [Google Scholar] [CrossRef]
- Chen, F.; Li, K.; Hart-Smith, G.; Xu, Y.D.; Jiang, Y.; Lu, H.; Fok, S.; Macmillian, A.; Pandzic, E.; Stenzel, M. Light-sheet microscopy as a tool to understanding the behaviour of Polyion complex micelles for drug delivery. Chem. Commun. 2018, 54, 12618–12621. [Google Scholar] [CrossRef]
- Harada, A.; Kataoka, K. Formation of polyion complex micelles in an aqueous milieu from a pair of oppositely-charged block copolymers with poly(ethylene glycol) segments. Macromolecules 1995, 28, 5294–5299. [Google Scholar] [CrossRef]
- Kataoka, K.; Togawa, H.; Harada, A.; Yasugi, K.; Matsumoto, T.; Katayose, S. Spontaneous formation of polyion complex micelles with narrow distribution from antisense oligonucleotide and cationic block copolymer in physiological saline. Macromolecules 1996, 29, 8556–8557. [Google Scholar] [CrossRef]
- Katayose, S.; Kataoka, K. Water-soluble polyion complex associates of DNA and poly(ethylene glycol)-poly(L-lysine) block copolymer. Bioconjug. Chem. 1997, 8, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Kim, H.J.; Mi, P.; Zheng, M.; Takemoto, H.; Toh, K.; Kim, B.S.; Hayashi, K.; Naito, M.; Matsumoto, Y.; et al. Targeted systemic delivery of siRNA to cervical cancer model using cyclic RGD-installed unimer polyion complex-assembled gold nanoparticles. J. Control. Release 2016, 244, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Barese, C.N.; Dunbar, C.E. Contributions of gene marking to cell and gene therapies. Hum. Gene Ther. 2011, 22, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Kataoka, K. Novel polyion complex micelles entrapping enzyme molecules in the core: Preparation of narrowly-distributed micelles from lysozyme and poly(ethylene glycol)-poly(aspartic acid) block copolymer in aqueous medium. Macromolecules 1998, 31, 288–294. [Google Scholar] [CrossRef]
- Harada, A.; Kataoka, K. Novel polyion complex micelles entrapping enzyme molecules in the core. 2. Characterization of the micelles prepared at nonstoichiometric mixing ratios. Langmuir 1999, 15, 4208–4212. [Google Scholar] [CrossRef]
- Harada, A.; Kataoka, K. On-off control of enzymatic activity synchronizing with reversible formation of supramolecular assembly from enzyme and charged block copolymers. J. Am. Chem. Soc. 1999, 121, 9241–9242. [Google Scholar] [CrossRef]
- Harada, A.; Kataoka, K. Pronounced activity of enzymes through the incorporation into the core of polyion complex micelles made from charged block copolymers. J. Control. Release 2001, 72, 85–91. [Google Scholar] [CrossRef]
- Harada, A.; Kataoka, K. Switching by pulse electric field of the elevated enzymatic reaction in the core of polyion complex micelles. J. Am. Chem. Soc. 2003, 125, 15306–15307. [Google Scholar] [CrossRef]
- Jaturanpinyo, M.; Harada, A.; Yuan, X.; Kataoka, K. Preparation of bionanoreactor based on core-shell structured polyion complex micelles entrapping trypsin in the core cross-linked with glutaraldehyde. Bioconjug. Chem. 2004, 15, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Harada, A.; Nagasaki, Y. Block copolymer micelles for drug delivery: Design, characterization and biological significance. Adv. Drug Deliv. Rev. 2001, 47, 113–131. [Google Scholar] [CrossRef]
- Yuan, X.; Yamasaki, Y.; Harada, A.; Kataoka, K. Characterization of stable lysozyme-entrapped polyion complex (PIC) micelles with crosslinked core by glutaraldehyde. Polymer 2005, 46, 7749–7758. [Google Scholar] [CrossRef]
- Kawamura, A.; Yoshioka, Y.; Harada, A.; Kono, K. Acceleration of enzymatic reaction of trypsin through the formation of water-soluble complexes with poly(ethylene glycol)-block-poly(α,β-aspartic acid). Biomacromolecules 2005, 6, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Kudaibergenov, S.E. Polyampholytes; Springer: Boston, MA, USA, 2002; ISBN 978-1-4613-5165-8. [Google Scholar]
- Patrickios, C.S.; Hertler, W.R.; Hatton, T.A. Protein complexation with acrylic polyampholytes. Biotechnol. Bioeng. 1994, 44, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Pathak, J.; Rawat, K.; Aswal, V.K.; Bohidar, H.B. Interactions in globular proteins with polyampholyte: Coacervation route for protein separation. RSC Adv. 2015, 5, 13579–13589. [Google Scholar] [CrossRef]
- Kudaibergenov, S.; Nuraje, N. Intra- and interpolyelectrolyte complexes of polyampholytes. Polymers 2018, 10, 1146. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Kizilay, E.; Seeman, D.; Flanagan, S.; Dubin, P.L.; Bovetto, L.; Donato, L.; Schmitt, C. Heteroprotein complex coacervation: Bovine β-lactoglobulin and lactoferrin. Langmuir 2013, 29, 15614–15623. [Google Scholar] [CrossRef] [PubMed]
- Croguennec, T.; Tavares, G.M.; Bouhallab, S. Heteroprotein complex coacervation: A generic process. Adv. Colloid Interface Sci. 2017, 239, 115–126. [Google Scholar] [CrossRef]
- Sieberz, J.; Wohlgemuth, K.; Schembecker, G. The influence of impurity proteins on the precipitation of a monoclonal antibody with an anionic polyelectrolyte. Sep. Purif. Technol. 2015, 146, 252–260. [Google Scholar] [CrossRef]
- De Vos, W.M.; Biesheuvel, P.M.; De Keizer, A.; Kleijn, J.M.; Stuart, M.A.C. Adsorption of the protein bovine serum albumin in a planar poly(acrylic acid) brush layer as measured by optical reflectometry. Langmuir 2008, 24, 6575–6584. [Google Scholar] [CrossRef] [PubMed]
- Ladam, G.; Gergely, C.; Senger, B.; Decher, G.; Voegel, J.C.; Schaaf, P.; Cuisinier, F.J.G. Protein interactions with polyelectrolyte multilayers: Interactions between human serum albumin and polystyrene sulfonate/polyallylamine multilayers. Biomacromolecules 2000, 1, 674–687. [Google Scholar] [CrossRef] [PubMed]
- Czeslik, C.; Jackler, G.; Steitz, R.; Von Grünberg, H.H. Protein binding to like-charged polyelectrolyte brushes by counterion evaporation. J. Phys. Chem. B 2004, 108, 13395–13402. [Google Scholar] [CrossRef]
- Seyrek, E.; Dubin, P.L.; Tribet, C.; Gamble, E.A. Ionic strength dependence of protein–polyelectrolyte interactions. Biomacromolecules 2003, 4, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Wittemann, A.; Ballauff, M. Interaction of proteins with linear polyelectrolytes and spherical polyelectrolyte brushes in aqueous solution. Phys. Chem. Chem. Phys. 2006, 8, 5269–5275. [Google Scholar] [CrossRef] [PubMed]
- Biesheuvel, P.M.; Wittemann, A. A modified box model including charge regulation for protein adsorption in a spherical polyelectrolyte brush. J. Phys. Chem. B 2005, 109, 4209–4214. [Google Scholar] [CrossRef] [PubMed]
- Biesheuvel, P.M.; Leermakers, F.A.M.; Stuart, M.A.C. Self-consistent field theory of protein adsorption in a non-Gaussian polyelectrolyte brush. Phys. Rev. E 2006, 73, 011802. [Google Scholar] [CrossRef] [Green Version]
- Brangwynne, C.P.; Eckmann, C.R.; Courson, D.S.; Rybarska, A.; Hoege, C.; Gharakhani, J.; Juelicher, F.; Hyman, A.A. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 2009, 324, 1729–1732. [Google Scholar] [CrossRef] [PubMed]
- Elbaum-Garfinkle, S.; Kim, Y.; Szczepaniak, K.; Chen, C.C.-H.; Eckmann, C.R.; Myong, S.; Brangwynne, C.P. The disordered P granule protein LAF-1 drives phase separation into droplets with tunable viscosity and dynamics. Proc. Natl. Acad. Sci. USA 2015, 112, 7189–7194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brangwynne, C.P.; Tompa, P.; Pappu, R.V. Polymer physics of intracellular phase transitions. Nat. Phys. 2015, 11, 899–904. [Google Scholar] [CrossRef]
- Girard, M.; Turgeon, S.L.; Gauthier, S.F. Thermodynamic parameters of β-lactoglobulin−pectin complexes assessed by isothermal titration calorimetry. J. Agric. Food Chem. 2003, 51, 4450–4455. [Google Scholar] [CrossRef] [PubMed]
- Capito, F.; Bauer, J.; Rapp, A.; Schröter, C.; Kolmar, H.; Stanislawski, B. Feasibility study of semi-selective protein precipitation with salt-tolerant copolymers for industrial purification of therapeutic antibodies. Biotechnol. Bioeng. 2013, 110, 2915–2927. [Google Scholar] [CrossRef] [PubMed]
- Turgeon, S.L.; Beaulieu, M.; Schmitt, C.; Sanchez, C. Protein–polysaccharide interactions: Phase-ordering kinetics, thermodynamic and structural aspects. Curr. Opin. Colloid Interface Sci. 2003, 8, 401–414. [Google Scholar] [CrossRef]
- Comert, F.; Dubin, P.L. Liquid-liquid and liquid-solid phase separation in protein–polyelectrolyte systems. Adv. Colloid Interface Sci. 2017, 239, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Comert, F.; Malanowski, A.J.; Azarikia, F.; Dubin, P.L. Coacervation and precipitation in polysaccharide-protein systems. Soft Matter 2016, 12, 4154–4161. [Google Scholar] [CrossRef] [PubMed]
- Souza, C.J.F.; da Costa, A.R.; Souza, C.F.; Tosin, F.F.S.; Garcia-Rojas, E.E. Complex coacervation between lysozyme and pectin: Effect of pH, salt, and biopolymer ratio. Int. J. Biol. Macromol. 2018, 107, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Souza, C.J.F.; Garcia-Rojas, E.E. Effects of salt and protein concentrations on the association and dissociation of ovalbumin-pectin complexes. Food Hydrocoll. 2015, 47, 124–129. [Google Scholar] [CrossRef]
- Zhang, R.; Shklovskii, B.I.T. Phase diagram of solution of oppositely charged polyelectrolytes. Phys. A 2005, 352, 216–238. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, Y.W.; Ruengruglikit, C.; Huang, Q. Effects of salt concentration on formation and dissociation of β-lactoglobulin/pectin complexes. J. Agric. Food Chem. 2007, 55, 10432–10436. [Google Scholar] [CrossRef] [PubMed]
- Antonov, M.; Mazzawi, M.; Dubin, P.L. Entering and exiting the protein–polyelectrolyte coacervate phase via nonmonotonic salt dependence of critical conditions. Biomacromolecules 2010, 11, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Obermeyer, A.C.; Mills, C.E.; Dong, X.-H.H.; Flores, R.J.; Olsen, B.D. Complex coacervation of supercharged proteins with polyelectrolytes. Soft Matter 2016, 12, 3570–3581. [Google Scholar] [CrossRef] [PubMed]
- Kapelner, R.A.; Obermeyer, A.C. Ionic polypeptide tags for protein phase separation. Chem. Sci. 2019, 10, 2700–2707. [Google Scholar] [CrossRef] [Green Version]
- Thilakarathne, V.K.; Briand, V.A.; Kasi, R.M.; Kumar, C.V. Tuning hemoglobin-poly(acrylic acid) interactions by controlled chemical modification with triethylenetetramine. J. Phys. Chem. B 2012, 116, 12783–12792. [Google Scholar] [CrossRef]
- Lenormand, H.; Deschrevel, B.; Vincent, J.-C. pH effects on the hyaluronan hydrolysis catalysed by hyaluronidase in the presence of proteins: Part I. Dual aspect of the pH-dependence. Matrix Biol. 2010, 29, 330–337. [Google Scholar] [CrossRef]
- Ran, Q.; Xu, X.; Dzubiella, J.; Haag, R.; Ballauff, M. Thermodynamics of the Binding of Lysozyme to a Dendritic Polyelectrolyte: Electrostatics Versus Hydration. ACS Omega 2018, 3, 9086–9095. [Google Scholar] [CrossRef]
- Aberkane, L.; Jasniewski, J.; Gaiani, C.; Scher, J.; Sanchez, C. Thermodynamic characterization of acacia gum−β-lactoglobulin complex coacervation. Langmuir 2010, 26, 12523–12533. [Google Scholar] [CrossRef]
- Geschwindner, S.; Ulander, J.; Johansson, P. Ligand binding thermodynamics in drug discovery: Still a hot tip? J. Med. Chem. 2015, 58, 6321–6335. [Google Scholar] [CrossRef]
- Ball, V.; Winterhalter, M.; Schwinte, P.; Lavalle, P.; Voegel, J.-C.C.; Schaaf, P. Complexation mechanism of bovine serum albumin and poly(allylamine hydrochloride). J. Phys. Chem. B 2002, 106, 2357–2364. [Google Scholar] [CrossRef]
- Yu, S.; Xu, X.; Yigit, C.; Van Der Giet, M.; Zidek, W.; Jankowski, J.; Dzubiella, J.; Ballauff, M. Interaction of human serum albumin with short polyelectrolytes: A study by calorimetry and computer simulations. Soft Matter 2015, 11, 4630–4639. [Google Scholar] [CrossRef]
- Gummel, J.; Cousin, F.; Boué, F. Counterions release from electrostatic complexes of polyelectrolytes and proteins of opposite charge: A direct measurement. J. Am. Chem. Soc. 2007, 129, 5806–5807. [Google Scholar] [CrossRef]
- Xu, X.; Ran, Q.; Dey, P.; Nikam, R.; Haag, R.; Ballauff, M.; Dzubiella, J. Counterion-release entropy governs the inhibition of serum proteins by polyelectrolyte drugs. Biomacromolecules 2018, 19, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Hattori, T.; Bat-Aldar, S.; Kato, R.; Bohidar, H.B.; Dubin, P.L. Characterization of polyanion-protein complexes by frontal analysis continuous capillary electrophoresis and small angle neutron scattering: Effect of polyanion flexibility. Anal. Biochem. 2005, 342, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G. Applying thermodynamic profiling in lead finding and optimization. Nat. Rev. Drug Discov. 2015, 14, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.; Johnson, C.M.; Lakey, J.H.; Nöllmann, M. Heat does not come in different colours: Entropy-enthalpy compensation, free energy windows, quantum confinement, pressure perturbation calorimetry, solvation and the multiple causes of heat capacity effects in biomolecular interactions. Biophys. Chem. 2001, 93, 215–230. [Google Scholar] [CrossRef]
- Mattison, K.W.; Brittain, I.J.; Dubin, P.L. Protein—Polyelectrolyte phase boundaries. Biotechnol. Prog. 1995, 11, 632–637. [Google Scholar] [CrossRef]
- Chodankar, S.; Aswal, V.K.; Kohlbrecher, J.; Vavrin, R.; Wagh, A.G. Structural study of coacervation in protein–polyelectrolyte complexes. Phys. Rev. E 2008, 78, 031913. [Google Scholar] [CrossRef] [PubMed]
- Gummel, J.; Boué, F.; Clemens, D.; Cousin, F. Finite size and inner structure controlled by electrostatic screening in globular complexes of proteins and polyelectrolytes. Soft Matter 2008, 4, 1653–1664. [Google Scholar] [CrossRef] [Green Version]
- Xu, A.Y.; Melton, L.D.; Ryan, T.M.; Mata, J.P.; Rekas, A.; Williams, M.A.K.; McGillivray, D.J. Effects of polysaccharide charge pattern on the microstructures of β-lactoglobulin-pectin complex coacervates, studied by SAXS and SANS. Food Hydrocoll. 2018, 77, 952–963. [Google Scholar] [CrossRef]
- Boué, F.; Cousin, F.; Gummel, J.; Oberdisse, J.; Carrot, G.; El Harrak, A. Small angle scattering from soft matter-application to complex mixed systems. Comptes Rendus Phys. 2007, 8, 821–844. [Google Scholar] [CrossRef]
- Schmidt, I.; Cousin, F.; Huchon, C.; Boué, F.; Axelos, M.A.V. Spatial structure and composition of polysaccharide-protein complexes from small angle neutron scattering. Biomacromolecules 2009, 10, 1346–1357. [Google Scholar] [CrossRef]
- Cooper, C.L.; Goulding, A.; Kayitmazer, A.B.; Ulrich, S.; Stoll, S.; Turksen, S.; Yusa, S.; Kumar, A.; Dubin, P.L. Effects of polyelectrolyte chain stiffness, charge mobility, and charge sequences on binding to proteins and micelles. Biomacromolecules 2006, 7, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Kurinomaru, T.; Maruyama, T.; Izaki, S.; Handa, K.; Kimoto, T.; Shiraki, K. Protein-poly(amino acid) complex precipitation for high-concentration protein formulation. J. Pharm. Sci. 2014, 103, 2248–2254. [Google Scholar] [CrossRef] [PubMed]
- McCall, P.M.; Srivastava, S.; Perry, S.L.; Kovar, D.R.; Gardel, M.L.; Tirrell, M.V. Partitioning and enhanced self-assembly of actin in polypeptide coacervates. Biophys. J. 2018, 114, 1636–1645. [Google Scholar] [CrossRef] [PubMed]
- Bourganis, V.; Karamanidou, T.; Kammona, O.; Kiparissides, C. Polyelectrolyte complexes as prospective carriers for the oral delivery of protein therapeutics. Eur. J. Pharm. Biopharm. 2017, 111, 44–60. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.M.; Chess, R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003, 2, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Tang, Y.; Zhou, Z.H.; Yan, M.; Priceman, S.; Segura, T.; Liang, M.; Zhang, W.; Lu, Y.; Du, J.; et al. A novel intracellular protein delivery platform based on single-protein nanocapsules. Nat. Nanotechnol. 2009, 5, 48–53. [Google Scholar]
- Liang, S.; Liu, Y.; Jin, X.; Liu, G.; Wen, J.; Zhang, L.; Li, J.; Yuan, X.; Chen, I.S.Y.; Chen, W.; et al. Phosphorylcholine polymer nanocapsules prolong the circulation time and reduce the immunogenicity of therapeutic proteins. Nano Res. 2016, 9, 1022–1031. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, W.; Zhu, X.; Lu, Y. Encapsulating therapeutic proteins with polyzwitterions for lower macrophage nonspecific uptake and longer circulation time. ACS Appl. Mater. Interfaces 2017, 9, 7972–7978. [Google Scholar] [CrossRef]
- Izaki, S.; Kurinomaru, T.; Maruyama, T.; Uchida, T.; Handa, K.; Kimoto, T.; Shiraki, K. Feasibility of antibody-poly(glutamic acid) complexes: Preparation of high-concentration antibody formulations and their pharmaceutical properties. J. Pharm. Sci. 2015, 104, 1929–1937. [Google Scholar] [CrossRef]
- Xia, J.; Mattison, K.; Romano, V.; Dubin, P.L.; Muhoberac, B.B. Complexation of trypsin and alcohol dehydrogenase with poly(diallyldimethylammonium chloride). Biopolymers 1997, 41, 359–365. [Google Scholar] [CrossRef]
- Lindhoud, S.; Claessens, M.M.A.E. Accumulation of small protein molecules in a macroscopic complex coacervate. Soft Matter 2015, 12, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Black, K.A.; Priftis, D.; Perry, S.L.; Yip, J.; Byun, W.Y.; Tirrell, M. Protein encapsulation via polypeptide complex coacervation. ACS Macro Lett. 2014, 3, 1088–1091. [Google Scholar] [CrossRef]
- Zhao, M.; Zacharia, N.S. Protein encapsulation via polyelectrolyte complex coacervation: Protection against protein denaturation. J. Chem. Phys. 2018, 149, 163326. [Google Scholar] [CrossRef] [PubMed]
- Blocher McTigue, W.C.; Perry, S.L. Design rules for encapsulating proteins into complex coacervates. Soft Matter 2019, 15, 3089–3103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Srivastava, S.; Andreev, M.; Marciel, A.B.; De Pablo, J.J.; Tirrell, M.V. Phase behavior and salt partitioning in polyelectrolyte complex coacervates. Macromolecules 2018, 51, 2988–2995. [Google Scholar] [CrossRef]
- Gu, Z.; Biswas, A.; Zhao, M.; Tang, Y. Tailoring nanocarriers for intracellular protein delivery. Chem. Soc. Rev. 2011, 40, 3638–3655. [Google Scholar] [CrossRef]
- Gombotz, W.R.; Pettit, D.K. Biodegradable polymers for protein and peptide drug delivery. Bioconjug. Chem. 1995, 6, 332–351. [Google Scholar] [CrossRef]
- Harada, A.; Kataoka, K. Formation of stable and monodispersive polyion complex micelles in aqueous medium from poly(l-lysine) and poly(ethylene glycol)-poly(aspartic acid) block copolymer. J. Macromol. Sci. Part A 1997, 34, 2119–2133. [Google Scholar] [CrossRef]
- Kataoka, A.; Harada, A. Chain length recognition: Core-shell supramolecular assembly from oppositely charged block copolymers. Science 1999, 283, 65–67. [Google Scholar]
- Katayose, S.; Kataoka, K. Remarkable increase in nuclease resistance of plasmid dna through supramolecular assembly with poly(ethylene glycol)–poly(l-lysine) block copolymer. J. Pharm. Sci. 1998, 87, 160–163. [Google Scholar] [CrossRef]
- Miyata, K.; Christie, R.J.; Kataoka, K. Polymeric micelles for nano-scale drug delivery. React. Funct. Polym. 2011, 71, 227–234. [Google Scholar] [CrossRef]
- Pippa, N.; Pispas, S.; Demetzos, C. Recent advances in micellar-like polyelectrolyte/protein complexes. In Design and Development of New Nanocarriers; Elsevier: Amsterdam, The Netherlands, 2018; pp. 57–88. ISBN 9780128136270. [Google Scholar]
- Chen, F.; Stenzel, M.H. Polyion complex micelles for protein delivery. Aust. J. Chem. 2018, 71, 768. [Google Scholar] [CrossRef]
- Lee, Y.; Fukushima, S.; Bae, Y.; Hiki, S.; Ishii, T.; Kataoka, K. A protein nanocarrier from charge-conversion polymer in response to endosomal pH. J. Am. Chem. Soc. 2007, 129, 5362–5363. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Orellana, A.; Di Antonio, M.; Conte, C.; Falcone, F.H.; Bosquillon, C.; Childerhouse, N.; Mantovani, G.; Stolnik, S. Effect of polymer topology on non-covalent polymer–protein complexation: Miktoarm versus linear mPEG-poly(glutamic acid) copolymers. Polym. Chem. 2017, 8, 2210–2220. [Google Scholar] [CrossRef]
- Zhang, Y.; Han, K.; Lu, D.; Liu, Z. Reversible encapsulation of lysozyme within mPEG-b-PMAA: Experimental observation and molecular dynamics simulation. Soft Matter 2013, 9, 8723–8729. [Google Scholar] [CrossRef]
- Pispas, S. Complexes of lysozyme with sodium (sulfamate-carboxylate)isoprene/ethylene oxide double hydrophilic block copolymers. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 509–520. [Google Scholar] [CrossRef]
- Gao, G.; Yan, Y.; Pispas, S.; Yao, P. Sustained and extended release with structural and activity recovery of lysozyme from complexes with sodium (sulfamate carboxylate) isoprene/ethylene oxide block copolymer. Macromol. Biosci. 2010, 10, 139–146. [Google Scholar] [CrossRef]
- Sotiropoulou, M.; Bokias, G.; Staikos, G. Water-soluble complexes through Coulombic interactions between bovine serum albumin and anionic polyelectrolytes grafted with hydrophilic nonionic side chains. Biomacromolecules 2005, 6, 1835–1838. [Google Scholar] [CrossRef]
- Kawamura, A.; Kojima, C.; Iijima, M.; Harada, A.; Kono, K. Polyion complex micelles formed from glucose oxidase and comb-type polyelectrolyte with poly(ethylene glycol) grafts. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 3842–3852. [Google Scholar] [CrossRef]
- De Vos, W.M.; Leermakers, F.A.M.; De Keizer, A.; Stuart, M.A.C.; Kleijn, J.M. Field theoretical analysis of driving forces for the uptake of proteins by like-charged polyelectrolyte brushes: Effects of charge regulation and patchiness. Langmuir 2010, 26, 249–259. [Google Scholar] [CrossRef]
- Nolles, A.; Hooiveld, E.; Westphal, A.H.; van Berkel, W.J.H.; Kleijn, J.M.; Borst, J.W. FRET reveals the formation and exchange dynamics of protein-containing complex coacervate core micelles. Langmuir 2018, 34, 12083–12092. [Google Scholar] [CrossRef] [PubMed]
- Danial, M.; Klok, H.A.; Norde, W.; Cohen Stuart, M.A.; Stuart, M.A.C. Complex coacervate core micelles with a lysozyme-modified corona. Langmuir 2007, 23, 8003–8009. [Google Scholar] [CrossRef] [PubMed]
- Kurinomaru, T.; Tomita, S.; Kudo, S.; Ganguli, S.; Nagasaki, Y.; Shiraki, K. Improved complementary polymer pair system: Switching for enzyme activity by pegylated polymers. Langmuir 2012, 28, 4334–4338. [Google Scholar] [CrossRef] [PubMed]
- Kuwada, K.; Kurinomaru, T.; Tomita, S.; Shiraki, K. Noncovalent PEGylation-based enzyme switch in physiological saline conditions using quaternized polyamines. Colloid Polym. Sci. 2016, 294, 1551–1556. [Google Scholar] [CrossRef]
- Kurinomaru, T.; Kuwada, K.; Tomita, S.; Kameda, T.; Shiraki, K. Noncovalent PEGylation through protein-polyelectrolyte interaction: Kinetic experiment and molecular dynamics simulation. J. Phys. Chem. B 2017, 121, 6785–6791. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.G.; Jiang, Y.W.; Chen, Z.; Yu, Z.W. Folding behaviors of protein (lysozyme) confined in polyelectrolyte complex micelle. Langmuir 2016, 32, 3655–3664. [Google Scholar] [CrossRef]
- Nolles, A.; van Dongen, N.J.E.; Westphal, A.H.; Visser, A.J.W.G.; Kleijn, J.M.; van Berkel, W.J.H.; Borst, J.W. Encapsulation into complex coacervate core micelles promotes EGFP dimerization. Phys. Chem. Chem. Phys. 2017, 19, 11380–11389. [Google Scholar] [CrossRef]
- Nolles, A.; Westphal, A.; Kleijn, J.; van Berkel, W.; Borst, J. Colorful packages: Encapsulation of fluorescent proteins in complex coacervate core micelles. Int. J. Mol. Sci. 2017, 18, 1557. [Google Scholar] [CrossRef]
- Harada, A.; Yoshioka, Y.; Kawamura, A.; Kojima, C.; Kono, K. Effect of polycarboxylate blocks on the amidase activity of trypsin through complexation with PEG/polycarboxylate block ionomers. Macromol. Biosci. 2007, 7, 339–343. [Google Scholar] [CrossRef]
- Solomatin, S.V.; Bronich, T.K.; Bargar, T.W.; Eisenberg, A.; Kabanov, V.A.; Kabanov, A.V. Environmentally responsive nanoparticles from block ionomer complexes: Effects of ph and ionic strength. Langmuir 2003, 19, 8069–8076. [Google Scholar] [CrossRef]
- Yuan, X.; Harada, A.; Yamasaki, Y.; Kataoka, K. Stabilization of lysozyme-incorporated polyion complex micelles by the ω-end derivatization of poly(ethylene glycol)−poly(α,β-aspartic acid) block copolymers with hydrophobic groups. Langmuir 2005, 21, 2668–2674. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ishii, T.; Cabral, H.; Kim, H.J.; Seo, J.-H.H.; Nishiyama, N.; Oshima, H.; Osada, K.; Kataoka, K. Charge-conversional polyionic complex micelles-efficient nanocarriers for protein delivery into cytoplasm. Angew. Chem. Int. Ed. 2009, 48, 5309–5312. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ishii, T.; Kim, H.J.; Nishiyama, N.; Hayakawa, Y.; Itaka, K.; Kataoka, K. Efficient Delivery of Bioactive Antibodies into the Cytoplasm of Living Cells by Charge-Conversional Polyion Complex Micelles. Angew. Chem. Int. Ed. 2010, 49, 2552–2555. [Google Scholar] [CrossRef] [PubMed]
- Lindhoud, S.; de Vries, R.; Norde, W.; Stuart, M.A.C.; Cohen Stuart, M.A.; Stuart, M.A.C. Structure and stability of complex coacervate core micelles with lysozyme. Biomacromolecules 2007, 8, 2219–2227. [Google Scholar] [CrossRef] [PubMed]
- Lindhoud, S. Polyelectrolyte Complex Micelles as Wrapping for Enzymes; Wageningen University: Wageningen, The Netherlands, 2009. [Google Scholar]
- Lindhoud, S.; Norde, W.; Stuart, M.A.C.; Cohen Stuart, M.A. Reversibility and relaxation behavior of polyelectrolyte complex micelle formation. J. Phys. Chem. B 2009, 113, 5431–5439. [Google Scholar] [CrossRef] [PubMed]
- Lindhoud, S.; Voorhaar, L.; de Vries, R.; Schweins, R.; Cohen Stuart, M.A.; Norde, W. Salt-induced disintegration of lysozyme-containing polyelectrolyte complex micelles. Langmuir 2009, 25, 11425–11430. [Google Scholar] [CrossRef]
- Lindhoud, S.; De Vries, R.; Schweins, R.; Cohen Stuart, M.A.; Norde, W. Salt-induced release of lipase from polyelectrolyte complex micelles. Soft Matter 2009, 5, 242–250. [Google Scholar] [CrossRef]
- Lindhoud, S.; Norde, W.; Cohen Stuart, M.A.; Stuart, M.A.C.; Cohen Stuart, M.A.; Stuart, M.A.C. Effects of polyelectrolyte complex micelles and their components on the enzymatic activity of lipase. Langmuir 2010, 26, 9802–9808. [Google Scholar] [CrossRef]
- Kim, J.O.; Ramasamy, T.; Yong, C.S.; Nukolov, N.V.; Bronich, T.K.; Kabanov, A.V. Cross-linked polymeric micelles based on block ionomer complexes. Mendeleev Commun. 2013, 23, 179–186. [Google Scholar] [CrossRef]
- Pippa, N.; Pispas, S.; Demetzos, C. Polymer self-assembled nanostructures as innovative drug nanocarrier platforms. Curr. Pharm. Des. 2016, 22, 2788–2795. [Google Scholar] [CrossRef]
- Hofs, B.; Voets, I.K.; De Keizer, A.; Cohen Stuart, M.A. Comparison of complex coacervate core micelles from two diblock copolymers or a single diblock copolymer with a polyelectrolyte. Phys. Chem. Chem. Phys. 2006, 8, 4242–4251. [Google Scholar] [CrossRef] [PubMed]
- de Vries, R.; Cohen Stuart, M. Theory and simulations of macroion complexation. Curr. Opin. Colloid Interface Sci. 2006, 11, 295–301. [Google Scholar] [CrossRef]
- Jiang, Y.; Fay, J.M.; Poon, C.D.; Vinod, N.; Zhao, Y.; Bullock, K.; Qin, S.; Manickam, D.S.; Yi, X.; Banks, W.A.; et al. Nanoformulation of brain-derived neurotrophic factor with target receptor-triggered-release in the central nervous system. Adv. Funct. Mater. 2018, 28, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Jeong, J.H.; Joe, C.O.; Park, T.G. Folate receptor mediated intracellular protein delivery using PLL-PEG-FOL conjugate. J. Control. Release 2005, 103, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Miura, Y.; Ishii, T.; Mutaf, O.F.; Nishiyama, N.; Cabral, H.; Kataoka, K. Intracellular delivery of charge-converted monoclonal antibodies by combinatorial design of block/homo polyion complex micelles. Biomacromolecules 2016, 17, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhang, Y.; Zhang, J.; Gao, H.; Liu, G.; Ma, R.; An, Y.; Kong, D.; Shi, L. pH/sugar dual responsive core-cross-linked pic micelles for enhanced intracellular protein delivery. Biomacromolecules 2013, 14, 3434–3443. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, L.; Yang, S.; Zhao, H.; Liu, L. Multi-responsive protein nanocarriers from an anionic dynamic covalent copolymer. Polym. Chem. 2014, 5, 4797–4804. [Google Scholar] [CrossRef]
- Kawamura, A.; Harada, A.; Kono, K.; Kataoka, K. Self-assembled nano-bioreactor from block ionomers with elevated and stabilized enzymatic function. Bioconjug. Chem. 2007, 18, 1555–1559. [Google Scholar] [CrossRef]












© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, S.; Holkar, A.; Srivastava, S. Protein–Polyelectrolyte Complexes and Micellar Assemblies. Polymers 2019, 11, 1097. https://doi.org/10.3390/polym11071097
Gao S, Holkar A, Srivastava S. Protein–Polyelectrolyte Complexes and Micellar Assemblies. Polymers. 2019; 11(7):1097. https://doi.org/10.3390/polym11071097
Chicago/Turabian StyleGao, Shang, Advait Holkar, and Samanvaya Srivastava. 2019. "Protein–Polyelectrolyte Complexes and Micellar Assemblies" Polymers 11, no. 7: 1097. https://doi.org/10.3390/polym11071097
APA StyleGao, S., Holkar, A., & Srivastava, S. (2019). Protein–Polyelectrolyte Complexes and Micellar Assemblies. Polymers, 11(7), 1097. https://doi.org/10.3390/polym11071097