3.1. Macromolecular Structure of PBF-Block-PEO Copolymers
Two series of PBF–block–PEO copolymers, in which PBF is defined as the rigid and PEO as the flexible segment, varied in the PEO segment’ molecular weight, and PBF to PEO ratio, as well as the PBF homopolymer, were successfully synthesized by a conventional two-step polycondensation in melt, starting from FDCA dimethyl ester and reasonable excess of bio-BD (1:2), under relatively mild process conditions. The changes in the copolymers’ composition were controlled via changes in a degree of polymerization, DP
x, of the PBF segment (
Table 1). The intrinsic viscosity numbers [η], determined at 30 °C, were: 0.87 for the homopolymer, and 0.97 to 1.27 dL/g for copolymers with increasing PEO content, regardless its molecular weight. These values correspond to the results reported in [
19]. Moreover, the materials were characterized by the following molecular weights (
Mn), i.e., 34,100 for PBF homopolymer (Ð = 1.8) as well as 56,400 (Ð = 1.9) and 69,000 (Ð = 2.0) for PBF-PEO
100050 and PBF-PEO
200050 samples, respectively. The materials are simple in processing by injection molding (
Figure 1). No shrinkage effect was observed after processing, which makes them suitable for the thermally molded precise elements.
The chemical structure and the real composition of PBF-
b-PEO copolymers were evaluated based on FTIR and
1H-NMR results. The ATR-FTIR spectra, presented in
Figure 2, show the absorption peaks characteristic for furan heterocycles and for PEO bonds with varied intensities. The PBF homopolymer reveals typical reflections at 1578–1580 cm
−1, attributed to the C=C stretching bonds, and at 3127 and 3157 cm
−1, arised from the C–H stretching bonds in the furan rings. Furthermore the breathing peaks observed at around 1019–1021 cm
−1, and the bending motions peaks at around 966, 860–861, and 763–764 cm
−1 are associated with 2,5-disubstituted furan heterocycles. Similar signals, but less intense, are observed at the copolymers’ spectra due to decreasing concentration of the furan moieties. In turn, the reflections given by the ester groups, detected near 1721 cm
−1 and 1260–1268 cm
−1 (stretching vibration of C=O and C–O, respectively), are relatively strong for all investigated materials. The signals attributed to PEO segments are recognized as the reflections at 2868–2870 cm
−1, arising from the antisymmetric and symmetric stretching of C–H in CH
2 groups of PEO chain, and near 1280 and 1362 cm
−1, attributed to C–O–C stretching vibrations. The absorption peak at 2949 cm
−1 also refers to C–H bond in CH
2 groups in bio-BD moiety. There is a lack of reflections at the 3378–3400 cm
−1 wavenumber range, attributed to the stretching vibrations of –OH bonds, present in the unreacted PEO. This fact confirms a successful effect of the polycondensation process, where the PEO segments have been fully reacted with PBF oligomeric segments.
More details about the real chemical structure and composition of the copolymers are provided by
1H-NMR analysis. The representative spectrum of the PBF-PEO
100050 copolymer sample is presented in
Figure 3, and the results for other copolymers are collected in
Table 1. The peak positions were taken from the PBF-PEO
1000 copolymers’ spectra, and positions for PBF-PEO
2000 copolymers are presented in brackets.
In general, the obtained
1H-NMR results confirm the expected multiblocked structure of the synthesized copolymers with randomly distributed rigid and flexible segments. Starting from left the resonance at 7.21 ppm (peak a (7.21)) corresponds to 2H aromatic protons of the furan ring, and the resonances detected at 4.40 ppm (peak b (4.40); triplet, 4H, –OCH
2–) and 1.91 ppm (peak c (1.91); multiplet, 4H, –(CH
2)
2–) refer to the aliphatic methylene protons in buthylene unit of the PBF segment. Additionally, the peak b indicates the CH
2 group adjacent to the carbonyl linkage of the ester group, which confirms a transesterification reaction between the hydroxyl end groups of PEO and the ester groups in PBF segment during the synthesis. In turn, the resonances at 4.48 ppm (peak f (4.47); triplet, 4H, –OCH
2–), and 3.81 ppm (peak e (3.82); triplet, 4H, –OCH
2–) may be assigned to the methylene protons of the PEO chain, wherein one in connected via the ester bond to the furanoate unit (f), and second connects the PEO backbone (e). The most intense resonance at 3.64 ppm (peak d (3.64); 84H, 21
CH
2)
2) corresponds to the backbone of the aliphatic protons of methylene groups in the PEO segment. The intensity of this peak varies depending on the PEO segments’ length in the copolymers. As seen in
Table 1 the integral intensities of signals d (
Id) and e (
Ie), corresponding to the presence of the flexible segment, are changing the most due to their different content in copolymers. The obtained results strictly correspond to the
1H-NMR results reported for the similar PBF/PEO or PBF/PPO copolymers [
18,
19,
20]. The calculated real contents of the rigid and flexible units are also very close to those theoretically calculated, what confirms once again the expected segmented structure of the copolymers.
3.2. Effect of the PEO Segment Length on Microstructure and Thermal Properties of PBF-Block-PEO Copolymers
The literature survey on the poly(ether–ester) block copolymers with poly(ethylene oxide) provided the evidence that such materials, due to a thermodynamic immiscibility of the constituents, reveal the tendency to the phase separation, in which polyester segments form a semicrystalline hard phase, whereas PEO stays amorphous forming a PEO-rich soft phase. Such microstructure results in an elastomer-like behavior, and due to its thermoreversible character, the copolymers may be processed as typical thermoplastics. The new group of furan polyesters is, however, less crystallizable than the conventional petroleum based polyesters, which also affects the effectiveness of the phase separation in copolymers. As the crystallization ability of PEO depends on its molecular weight (it is becoming crystallizable for the
Mw above 600 g/mol [
38]), it is expected that the PEO segment length should influence the crystallization ability, phase transition temperatures, and resulted microstructure of the copolymers. To study all these effects, the copolymers with PEO segments of 1000 and 2000 g/mol, and different PBF to PEO ratio have been investigated by DSC, WAXS, and DMTA techniques, and compared. The characteristic thermal and structural parameters are summarized in
Table 2, while the DSC thermograms and XRD patterns are presented in
Figure 4 and
Figure 5.
Although the PBF homopolymer reveals a clear crystallization peak,
Tc, at 104 °C as observed on the standard cooling scans of investigated materials, the PBF-PEO
1000 copolymers do not show any effects of crystallization in neither PBF nor PEO segments. In turn, relatively strong glass transition effects are observed in sub-ambient temperature range (
Figure 4a). However, when the subsequent heating scans are analyzed, the cold crystallization peaks are easily detected for all samples, as well as the melting and glass transition effects, which confirms the semicrystalline nature of the copolymers (
Figure 4b). These observations are contrary to the crystallization results reported by Hu [
19], where an incorporation of PEO1000 segments supported the melt crystallization process in all investigated copolymers, but correspond to the FSC studies by Sousa [
18]. It was concluded that the PBF/PEO copolymers’ crystallization rate is slower than that of the neat PBF, and no crystallization or melting peaks were observed for varied cooling rates. Indeed, from our DSC results one can say that, in the PBF-PEO
1000 copolymers both segments are miscible at the molecular scale, which results in existing only one (mixed) phase in the copolymers’ microstructure. It is confirmed by only one glass transition effect observed in the cooling scans of materials with different PBF to PEO segments’ ratio as well as the lack of melt crystallization effects (
Figure 4a). The
Tg values of the samples are shifted to the lower temperature range (from 4 to −34 °C), when compared to the neat PBF, along with the PEO1000 content. It provides the evidence for improved macromolecular flexibility by the incorporation of the PEO sequences within the PBF oligomers, but also that this physical transition is governed by the amount of the flexible segment. It is also confirmed by increasing values of the heat capacity at
Tg (Δ
Cp) in the copolymers (
Table 2). In the condensed state, during the subsequent heating cycle (
Figure 4b), the copolymers’ microstructure remains homogeneous until the glass transition range is exceeded, and the cold crystallization of the PBF segments occurs. This transition becomes a driving force of the microstructure separation into two phases: the PBF crystalline hard phase and the soft one, being a mixture of two segments, but dominated by the PEO1000, which stays amorphous. Of course, the
Tcc values of copolymers are decreasing with the flexible segment content due to its plasticizing effect on the rigid segment folding, whereas the cold crystallization enthalpies (Δ
Hcc) depend on the amount of crystallizable PBF blocks. Under further heating the crystalline nanodomains of the hard phase are getting melt, and the observable decrease of
Tm2 values of copolymers as well as their enthalpies (Δ
Hm2) are also associated with decrease of the PBF content. However, additionally, it may result from not perfectly developed crystalline structure, disturbed by the presence of chemically bonded PEO segments or larger interfacial effects on the melting transitions. Actually, the presence of only one crystalline form in the copolymers’ microstructure is confirmed by the WAXS diffractograms displayed in
Figure 4c. When separated to the diffraction peaks and amorphous background, and then compared to the pattern of the semicrystalline PBF sample, it is clear that copolymers reveal only the characteristic crystalline peaks with the scattering angle positions very close to those of the PBF (i.e., 10.4; 18; 22.7, and 25° of 2θ). At the same time the reflections are losing their slender shape along with the PEO content, while the amorphous phase background gains in the intensity, which is a consequence of decreasing crystalline phase’s content in the copolymers. Indeed, the crystallinity degrees (
Xc), calculated from WAXS, vary from 55% for the neat PBF to 38% for the copolymer containing only 35 wt % of the furan–ester segment. Note that the similar thermal behavior and parameters were reported for PBF copolymers with dimerized fatty acid as the flexible segment [
17]. However, in the current study, the
Tg and
Tcc temperatures are slightly lower, which provides the evidence for improved chain mobility due to the higher length of the PEO segment (1000 vs. 570 g/mol of the fatty acid diol).
Considering the DSC thermograms of PBF-PEO
2000 copolymers, the differences in their thermal behavior due to increased PEO segment length become clear for materials containing 50 and 65 wt % of the flexible segment. When the PBF segment is predominant, the copolymers reveal analogous phase transitions with
Tg and
Tcc values slightly shifted to lower temperatures, whereas
Tm values are higher if compared to PBF-PEO
1000 samples (
Figure 5,
Table 2). Moreover, the very weak melt crystallization effects, in temperature range related to T
c of the neat PBF, may be detected in the cooling traces (
Figure 5a). These observations suggest that the macromolecules gain in flexibility with a longer length of the PEO segment, which initiates and drives the phase separation by forming the crystalline nanodomains. In the copolymers’ microstructure, a PBF-rich continuous phase dominates. An increase of the melting temperatures also indicates higher quality of the crystalline phase. However, when the PEO2000 content is increasing, its molecular weight enables the crystallization of the soft segments, and the evolution of the copolymers’ microstructure is evident when comparing the DSC scans of PBF-PEO
200050 and PBF-PEO
200065 samples. In case of the first one, when cooling from the melt, only one clear exothermal effect is observed, and its temperature suggests the PBF segment crystallization. However, during a subsequent heating the low-temperature cold crystallization peak is observed, and two melting temperatures
Tm1 and
Tm2 (
Figure 5b). For the copolymer with the highest PEO2000 content, in turn, two melt crystallization effects (
Tc1 and
Tc2) can be distinguished, and two melting peaks on the 2nd heating traces. There are no doubts, that all thermal effects observed in the low temperature region are strictly related to the PEO segment induced crystallization. These crystallization/melting peaks are gaining in the intensity along with the segment content. At the same time an increased molecular weight of the flexible segment entails an increase of the polymerization degree (DP
x) of the PBF segment. It results in extension of the length of the rigid segments in the polymer chains as well, which together with the improved molecular mobility and diffusion, enables their folding. That would explain the appearance of the PBF melt crystallization effects in PBF-PEO
2000 samples. Consequently, two kinds of crystalline phases with different
Tm coexist in the copolymers’ microstructure. In turn, only one glass transition region is detected with
Tg values significantly shifted to the low temperature region (−42 to −48 °C) when compared to the both PBF-PEO
1000 or other PBF-PEO
2000 copolymers. On one hand, it indicates that due to the chemical bonds between the rigid and flexible segments they are not able to separate to two neat phases given two
Tg effects. On the other hand, such a big drop in
Tg values suggests that for the materials containing 50 wt % and above of the PEO2000 a phase inversion occurs, and the PEO-rich soft amorphous phase is getting a continuous phase with dispersed crystalline nanodomains of the PBF hard phase. A decrease of the Δ
Cp values at
Tg also suggests an improved phase separation in the copolymers due to the PEO assigned crystals forming.
The reorganization in crystalline structure of the materials should be reflected by the WAXS diffractograms. According to the literature, the PEO crystallites give two characteristic peaks at 19.2 and 23.2° scattering angles [
39]. When the PBF-PEO
2000 copolymers’ patterns are analyzed, four main peaks corresponding to PBF crystalline reflections can be identified (
Figure 5c), and two, relatively weak, and noticeable only for the copolymers, peaks at 19.7 and 20.75° (actually the 19.7° position peak is detectable only for PBF-PEO
2000 35 sample). Note that the investigated materials are quite complex multiphase systems; moreover, the WAXS measurements were performed at room temperature, whereas the melting of the PEO assigned crystalline phase was detected within 20–28 °C (
Figure 5b). This might suggest that first the low-temperature formed crystalline phase is affected/defected due to the presence of the PBF assigned crystals, which would explain why the peak positions do not exactly match the PEO related peak positions. Second, in the room temperature the same crystalline phase could partly melt, also affecting the patterns. Nevertheless, the crystallinity degree, calculated from the WAXS, is higher for the PBF-PEO
2000 copolymers with the largest PEO segment contents than the comparable PBF-PEO
1000 materials (
Table 2). Thus, it can be concluded that both DSC and WAXS results provide clear evidence that not only the PBF to PEO segment ratio, but also the molecular weight of the PEO flexible segment significantly affect the microstructure and phase separation in PBF-
block-PEO copolymers.
A thermo-mechanical behavior of the investigated materials, studied by the DMTA technique, only supports these conclusions. Changes of the storage moduli (
E’) as well as the loss factor (tanδ) within the −100 to 120 °C temperature range are presented in
Figure 6a–d. The PBF homopolymer, as the reference, reveals a clear relaxation, β
1, on tanδ curve corresponding to a drop at the storage modulus profile, and attributed to the glass transition of its amorphous phase. In the case of the PBF-PEO
1000 copolymers’ samples, the main relaxation peaks β
1 are successively shifted towards the lower temperatures with increasing flexible segment content, which is also reflected by the observed decreasing
E’ modulus values. These findings confirm once again an improved macromolecular mobility due to an incorporation of amorphous and highly flexible PEO segments. Under the heating; however, the second relaxation effect, β
2, is detected on tanδ profile within the temperature range of 40 to 70 °C, and accompanied by a visible shoulder on
E’ curves. As confirmed by the WAXS analysis, it is attributed to the thermally and mechanically induced cold crystallization of the PBF segment in the copolymers’ microstructure, also observed on the DSC heating termograms. Considering the practical aspects of the copolymers’ performance, note that if the loss factor is an indicator of the material’s ability to energy dissipation, the increasing content of the PEO1000 segment improves the copolymers’ damping capability. Particularly the PBF-PEO
100050 and 35 samples reveal the highest β
1 relaxation values and intensities, which is another advantage of their specific heterogeneous microstructure. Additionally, the same copolymers at the room temperature, are in the elastic state, which should also affect their mechanical performance.
In general, similar effects are observed for the copolymers with the PEO2000, until its content makes the flexible segment crystallizable. This is clearly visible by the variations in the
E’ and tanδ profiles of the PBF-PEO
200035 sample (
Figure 6c,d). What can be seen on the thermograms is: the relaxation related to
Tg at approximately −45 °C (β
1), then the shoulder within approximately −20 to 0 °C temperature range (β
3), which may suggest a cold crystallization of PEO segments, another drop at approximately 0 to 30 °C, corresponding to melting of PEO assigned crystalline structure, and further relaxation within 40° to 70 °C, also observed for other copolymers, and attributed to PBF segment crystallization (β
2). Not all of these relaxations were detectable on the DSC scan; however, DMTA analysis is considered as more sensitive for the phase transitions in the polymers. An increase of the crystalline nanodomains in the microstructure also disturbs its ability to the vibration energy dissipation as indicated by a drop of tan δ maximum. This is likely due to the reduction of a free volume and flexibility of the continuous soft phase. Thus, although a development in the copolymers’ microstructure induced by the crystallization of the PEO soft segments seems to be interesting from the scientific point of view, it makes this microstructure very unstable in fact, particularly within approximately −20 to 30 °C temperature range. In practice, it means that the materials’ performance will be affected by the working temperature even more than the typical thermoplastics, which limits their application profile. Alternatively additional treatment like annealing would be needed to allow the crystalline structure to get fully formed and stabilized.
The thermal stability, particularly in the thermo-oxidative atmosphere, is also an important factor in the characterization of PBF-
block-PEO copolymers due to their suitability for the thermal processing, but has not been analyzed by other authors. The TGA thermograms for both series of materials are presented in
Figure 7a,b, and the degradation temperatures were collected in
Table 2. In general, the decomposition of the furan polyesters proceeds in two steps. First, the ester linkages are subjected to the scission forming carbonyl-end groups and vinyl esters, then the oxygen atoms react with the methylene groups in diester linkage forming hydroperoxides [
40]. Such decomposition profiles were observed for the furan–polyester copolymers with the dimerized fatty acid flexible segment, regardless of which glycol was used to synthesize the rigid segment [
17,
35,
36,
41]. Also, in most cases, the presence of the fatty acids improved the stability of the copolymers. In currently investigated materials it is clear, that the PBF-
block-PEO copolymers reveal the thermo-oxidative stability at the same level as the PBF homopolymer, also an increase of 10–15 °C in
TdecMAX is observed. It means that under processing conditions undesired degradation reactions should not occur in materials structure. More detailed analysis of the TGA results shows that the decomposition temperatures vary with the PBF to PEO segment ratio. At the 5% of the weight loss (
Tdec 5%) the copolymers with the highest content of PBF segments reveal an improved stability compared to neat PBF, but along with the PEO segment content the
Tdec 5% values are slightly decreasing. Furthermore, more fluctuations, particularly in the first step of degradation profile, are observed for PBF- PEO
1000 samples. This may be related to a higher concentration of the ester/ether groups in the copolymers’ macromolecules with the PEO segments with lower molecular weight, which in turn are subjected to random scission intensified by the oxygen atoms. According to the literature PEO is also susceptible to a free radical oxidative attack, and during the degradation some volatile compounds can be released [
42]. Slightly better performance of the PBF-PEO
2000 copolymers may be explained, in turn, by enhanced interactions between PBF and PEO segments, as suggested in [
18].
3.3. The Effect of the PEO Segment Length on Mechanical and Elastic Behavior of PBF-Block-PEO Copolymers
The mechanical and elastic performance of the copolymers was investigated in the uniaxial tensile tests under the static and cyclic deformation as well as the Shore hardness measurements. The mechanical parameters together with materials’ densities are collected in
Table 3.
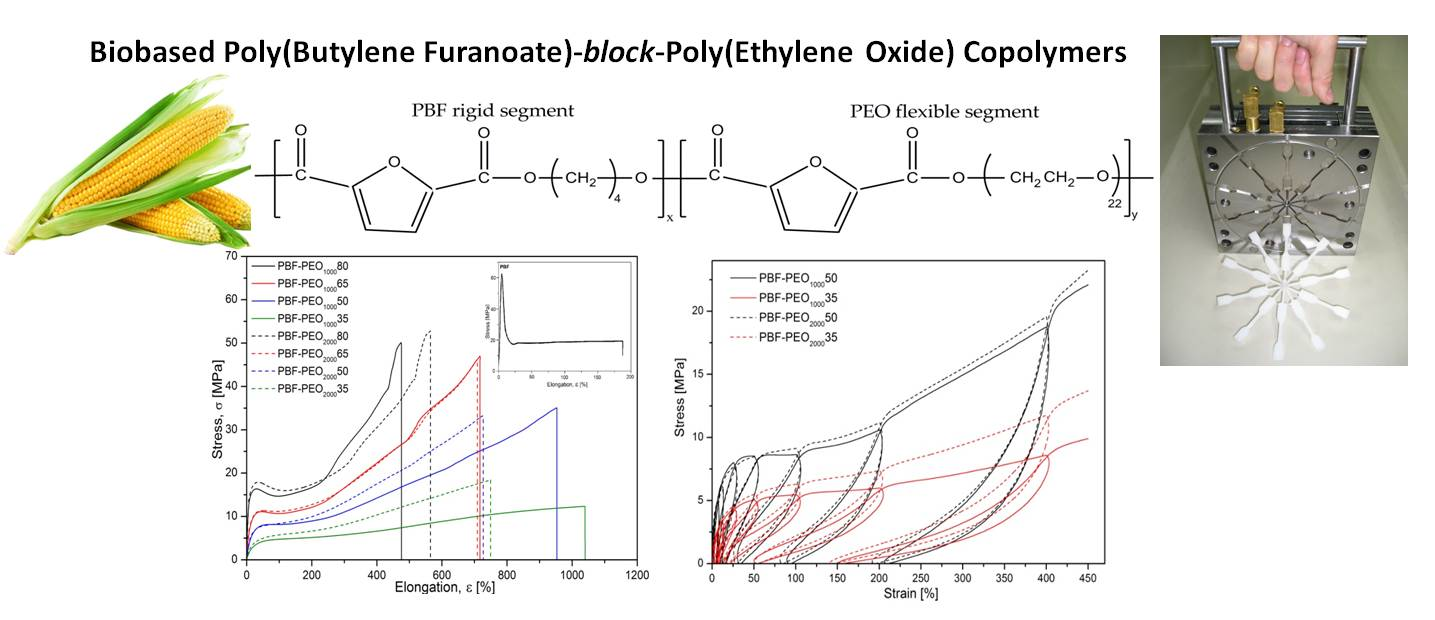
As expected, the stress–strain characteristics, presented in
Figure 8a, reveal significant changes in the mechanical behavior of the copolymers, particularly in deformation ability and stiffness, when compared to the neat PBF. It is due to the improved flexibility of the macromolecules by much more elastic PEO segments’ distribution, but also a specific microstructure formed. Such effects have been already observed for the PBF block copolymers with poly(tetramethylene glycol) [
15] or dimerized fatty acids [
17], and the mechanical parameters, namely, the tensile strength and elongation at break of the PBF-PEO
1000 samples are comparable to the results reported in [
19]. The character of deformation is also changing with the PEO segment content in both series of the copolymers, whereas the effect of the flexible segment length is mainly pronounced by the differences in elongation at break values. In general, the copolymers are characterized by distinctly reduced tensile modulus values, even for the materials containing only 20 wt % of the flexible segment (~208 MPa vs. 1460 MPa for neat PBF). However, it is in accordance with their also relatively low hardness, which is attributed to the nature of the investigated materials. Note that the E modulus values of both series show a very good fit to the exponential curve, which would be useful for estimating the E values, also for the other copolymer compositions (
Figure 8b). The copolymers with a predominance of the PBF segments demonstrate a typical for semicrystalline polymers yield effect at ~30% of elongation, subsequent necking effects and a large strengthening effect due to the strain-induced macromolecule’s orientation. At these contents of the PEO segments, the effect of their molecular weight is not obvious, because while the PBF-PEO
100065 and PBF-PEO
200065 samples behave in exactly the same way, for copolymers containing only 20 wt % of the flexible segment an improved elongation (about 100%) is observed for that containing the PEO2000. It seems to be related to an incorporation of longer and more flexible sequences of polyether between the rigid PBF blocks, which in the hard phase dominated microstructure may form larger domains of the soft phase and facilitate macromolecules’ mobility under stress. This is also confirmed by lower E modulus value of the PBF-PEO
200080 sample. With increasing content of the PEO segments the stress–strain characteristics are gaining an elastomeric character, where a gradual increase of elongation is accompanied by a relatively small but continuous increase of stress. The 50 and 65 wt % of PEO containing copolymers reveal a relatively low stress and hardness level, but a significant ability to deformation. In these cases, an improved elongation at break is observed for PEO1000 copolymerized materials. The clue lies in the differences in copolymers’ microstructure, induced by the crystallization ability of the flexible segment with higher length, as examined previously by the DSC and WAXS. Indeed, the PBF-PEO
2000 copolymers reveal improved crystallinity, which means that even though in their microstructure the PEO-rich soft phase with a significantly lower glass transition temperature dominates. Thus, under testing temperature the materials are in elastic state. In fact, the crystalline nanodomains of the PBF hard phase, together with the secondary PEO-assigned crystals, may constrain the continuous amorphous phase. Furthermore, the soft phase is depleted of the PEO segments involved in secondary crystals’ forming. As a consequence, the copolymers gain in stress transfer ability, which is observed on the characteristics by more pronounced strengthening effect during the deformation, but their ability to elongation is reduced when compared to the PEO1000 containing copolymers. These findings lead to conclusion that the phase separation improved by increasing the crystalline phase content, or more precisely by partial crystallization of the flexible segment, is not favorable for copolymers’ deformability. On the other hand, the stress–strain curves for the PBF-PEO
200050 and PBF-PEO
200035 samples retain their elastomeric character. Therefore, a deeper insight into elastic deformability and reversibility of the copolymers seems to be useful in evaluating the effect of the segment length on the materials elastic performance.
For this reason, the samples were subjected to the cyclic loading/unloading tests under tension mode, and the elastic recovery after every cycle of a defined strain level was evaluated by determining a permanent set value. The smaller permanent set the better elastic recovery or higher elasticity of the material in other words. Both the stress–strain characteristics of cyclic tensile tests as well as the level of the permanent set for 5, 10, 25, 50, 100, 200, and 400% of the attained strain in the following cycle are presented in
Figure 9. In general, the presented characteristics of all investigated materials reflect typical behavior of block copolymers with the features of thermoplastic elastomers: well detectable hysteresis–like curves (loops) with constantly increasing stress, and permanent set level after every loading/unloading cycle (
Figure 9a). The contour made by the loops for a single material strongly corresponds to the characteristics obtained under the static tensile tests with comparable level of stress. As mentioned, the permanent set values increase with every cycle of deformation, which is particularly visible when the maximum attained strain exceeds 100%. The differences in the copolymers’ behavior resulting from the PEO segment content are getting more pronounced, and it is obvious that materials containing more rigid segments reveal higher values of the residual strain (
Figure 9b). However, for the assessment of the materials’ elasticity, the most meaningful are the initial cycles of deformation. Thus, up to 10% of max. attained strain, all copolymers reveal comparable and relatively high ability to elastic recovery (permanent set values less than 5%), and it can be said that at this level of deformation, the effect of the PEO segment content and length on the copolymers’ elastic behavior is minimal. With the increase of the attained strain to 25%, the differences between copolymers containing 50 and 65 wt % of the PEO are becoming noticeable, but the effects resulting from the PEO molecular weight can be distinguished too. The elastic recovery is still relatively high with the permanent set values less than 10% for both series, however the copolymers with PEO2000 reveal slightly better elasticity, and this trend is observed in subsequent loading/unloading cycles up to the sample break. These findings are consistent with the theory about the thermoplastic elastomers, in which semicrystalline nanodomains of the hard phase, formed due to the induced phase separation, act as the physical crosslinks (knots), similar to the chemical cross-linking in the rubber. Under the tensile stress, when the continuous soft phase undergoes the deformation, the crystalline knots prevent conformational changes of the macromolecules, and ensure their return to the initial state when the force is released. Formation of the secondary crystals due to the cold crystallization of the PEO2000 segment leads to increase of the number of such nanodomains, dispersed in the continuous phase, which, in fact, “keep” the microstructure. In the PEO1000 containing copolymers, in which the effect of the phase separation is poor, the macromolecules are more liable to orientation and flow under the force, which results in larger permanent deformation observed. It is evident that the better developed phase separation in the PBF-
block-PEO copolymers reduces the ability of samples to large elongations, but is beneficial if the elastic behavior of the materials is required.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}