Highly Chlorinated Polyvinyl Chloride as a Novel Precursor for Fibrous Carbon Material

 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Raw Material
2.2. Preparation of CPVC Spun Fibre
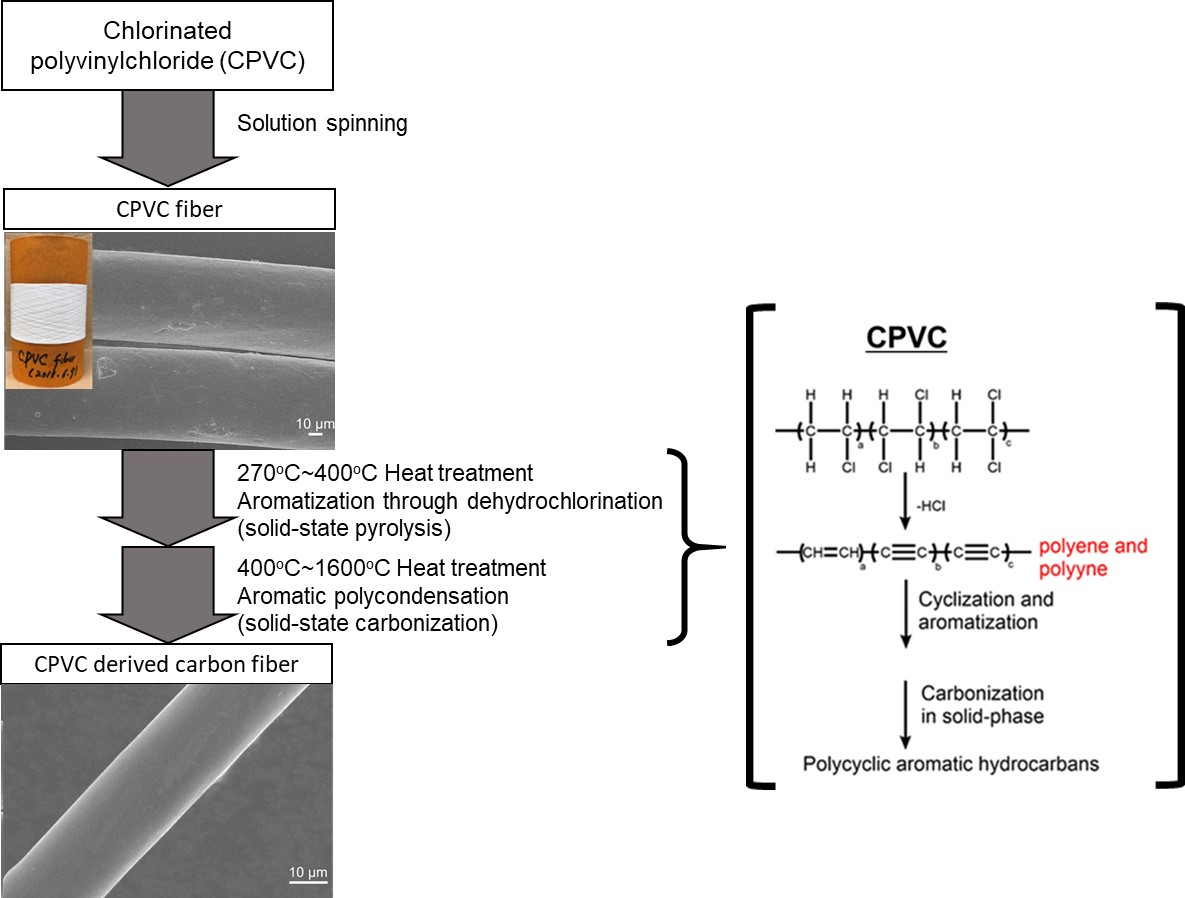
2.3. Preparation of a CPVC-Derived Carbon Fibre
2.4. Characterisation
3. Results
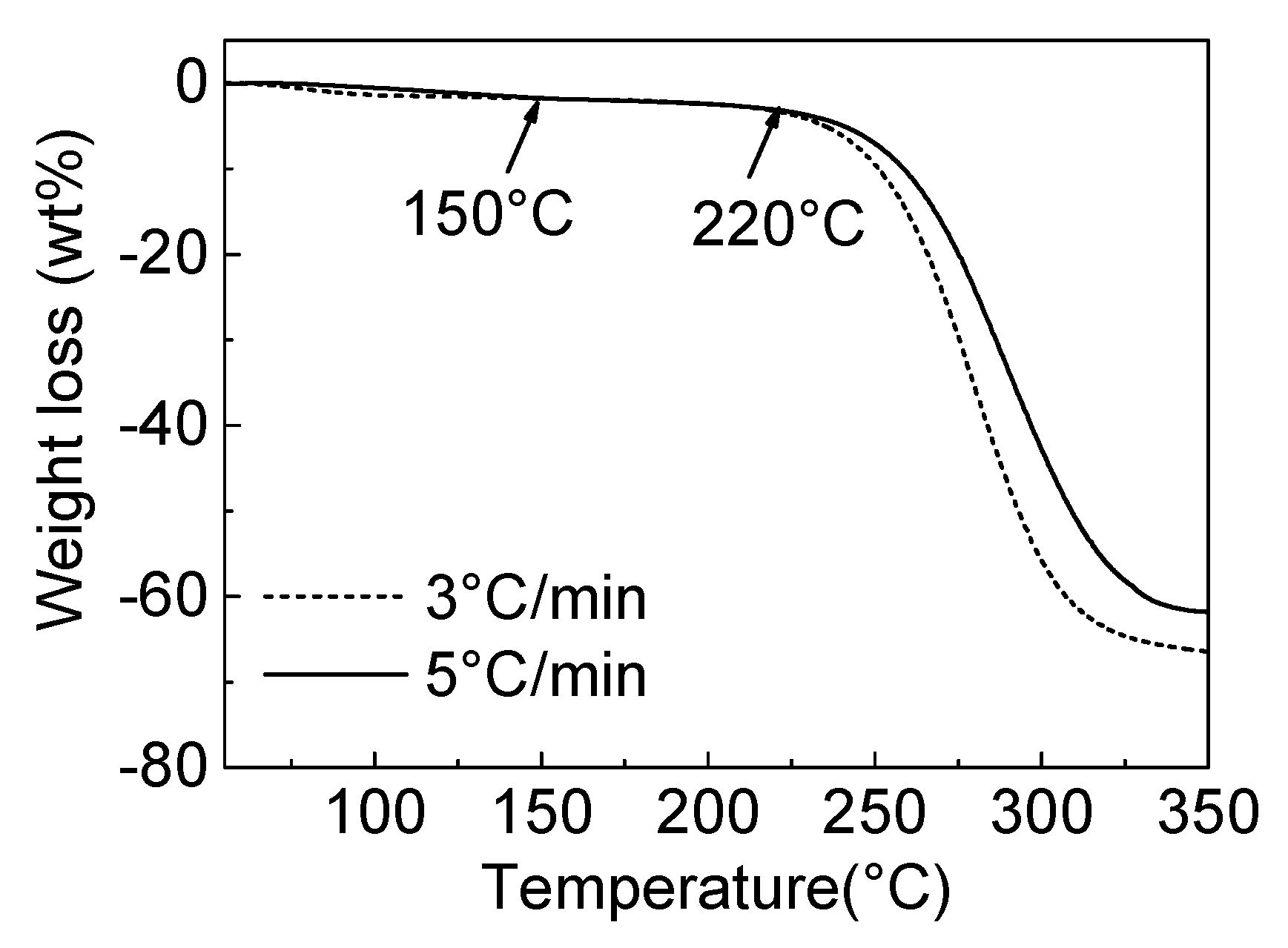
3.1. The Pyrolytic Behaviours of PVC and CPVC
3.2. The Scanning Electron Microscope (SEM) Images and the Mechanical Properties of the CPVC-Derived Carbon Fibres (CFs)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kim, B.J.; Eom, Y.E.; Kato, O.; Miyawaki, J.; Mochida, I.; Yoon, S.-H. Preparation of carbon fibers with excellent mechanical properties from isotropic pitches. Carbon 2014, 77, 747–755. [Google Scholar] [CrossRef]
- Yang, J.H.; Nakabayashi, K.; Miyawaki, J.; Yoon, S.-H. Preparation of pitch based carbon fibers using Hyper-coal as a raw material. Carbon 2016, 106, 28–36. [Google Scholar] [CrossRef]
- Kim, B.J.; Kotegawa, T.; Eom, Y.E.; An, J.C.; Hong, I.-P.; Yoon, S.-H. Enhancing the tensile strength of isotropic pitch-based carbon fibers by improving the stabilization and carbonization properties of precursor pitch. Carbon 2016, 99, 649–657. [Google Scholar] [CrossRef]
- Yoon, S.-H.; Korai, Y.; Mochida, I. Assessment and optimization of the stabilization process of mesophase pitch fibers by thermal analyses. Carbon 1994, 32, 281–287. [Google Scholar] [CrossRef]
- Ida, N.; Ylva, N.; Rickard, D.; Goran, G.; Elisabeth, S. A new method for stabilizing softwood kraft lignin fibers for carbon fiber production. J. Appl. Polym. Sci. 2013, 128, 3824–3830. [Google Scholar]
- Ota, M.; Otani, S.; Izuka, S.; Kojima, A. The Preparation of Thermostable Fiber from Condensed Polynuclear Aromatic (COPNA) Resin. J. Jpn. Chem. Soc. 1992, 1, 107–113. [Google Scholar]
- Yamashita, J. The Method of Carbon Material Preparation Using Polyvinyl Alcohol as A Raw Material. Japanese Patent P2003-128407A, 8 May 2003. [Google Scholar]
- Irisawa, T. Carbon Fiber Precursor, Carbon Fiber and the Preparing Method of Carbon Fiber. Japanese Patent JP W02014/084164 A1, 5 June 2014. [Google Scholar]
- Irisawa, T. Polybenzimidazole-based Carbon Fiber and Its Preparing Method. Japanese Patent JP W02015/170623 A1, 28 September 2015. [Google Scholar]
- Li, M.; Chao, X.; Zhao, J.; Zhao, J.R.; Feng, Y.; Hu, H.Q. Preparation of anhydridized chlorinated polyvinyl chloride with enhanced properties and investigation of the factors affecting the chain structure of the graft copolymer. J. Elastomers Plast. 2015, 47, 136–152. [Google Scholar]
- Zhang, L.X.; Zhou, C.; Sun, S.L.; Liang, R.; Ma, X.L.; Zhang, H.X. Study of compatibility, morphology structure and mechanical properties of CPVC/ABS blends. J. Appl. Polym. Sci. 2016, 116, 3448–3454. [Google Scholar] [CrossRef]
- Elakesh, O.E.; Price, D.; Carty, P. Thermal decomposition of chlorinated poly (vinylchloride) (CPVC). J. Vinyl Addit. Technol. 2003, 9, 116–126. [Google Scholar] [CrossRef]
- Carty, P.; Price, D.; Milnes, G.J. Chlorinated poly (vinyl chloride) and plasticized chlorinated poly (vinyl chloride)-thermal decomposition studies. J. Vinyl Addit. Technol. 2002, 8, 227–237. [Google Scholar] [CrossRef]
- Yao, K.; Gong, J.; Zheng, J.; Wang, L.; Tan, H.Y.; Tang, T. Catalytic carbonization of chlorinated poly (vinyl chloride) microfibers into carbon microfibers with high performance in the photodegradation of congo red. J. Phys. Chem. C 2013, 117, 17016–17023. [Google Scholar] [CrossRef]
- Coelho, J.; Pedro, G.; Miranda, D.; Gil, M.H. Characterization of suspension poly(vinyl chloride) resins and narrow polystyrene standards by size exclusion chromatography with multiple detectors: Online right angle laser-light scattering and differential viscometric detectors. Eur. Polym. J. 2006, 42, 751–763. [Google Scholar] [CrossRef] [Green Version]
- Morgan, P. Carbon Fibers and Their Composites; CRC Press: Boca Raton, FL, USA, 2005; pp. 170–340. [Google Scholar]
- Otani, S. Carbonization phenomenon. Tanso 1965, 42, 30–35. (In Japanese) [Google Scholar]
- Qiao, W.M.; Yoon, S.-H.; Korai, Y.; Mochida, I.; Inoue, S.; Sakurai, T.; Shimohara, T. Preparation of activated carbon fibers from polyvinyl chloride. Carbon 2004, 42, 1327–1331. [Google Scholar] [CrossRef]
- Miranda, R.; Yang, J.; Roy, C.; Cornelia, V. Vacuum pyrolysis of PVC I. Kinetic study. Polym. Degrad. Stab. 1999, 64, 127–144. [Google Scholar] [CrossRef]
- Ko, S.; Choi, J.-E.; Lee, C.W.; Jeon, Y.-P. Modified oxidative thermal treatment for the preparation of isotropic pitch towards cost-competitive carbon fiber. J. Ind. Eng. Chem. 2017, 54, 252–261. [Google Scholar] [CrossRef]
- Park, M.-S.; Jung, M.-J.; Lee, Y.-S. Significant reduction in stabilization temperature and improved mechanical/electrical properties of pitch-based carbon fibers by electron beam irradiation. J. Ind. Eng. Chem. 2016, 37, 227–287. [Google Scholar] [CrossRef]
- Liu, J.; Shimanoe, H.; Nakabayashi, K.; Miyawaki, J.; Choi, J.-E.; Jeon, Y.-P.; Yoon, S.-H. Enhancing the oxidative stabilization of isotropic pitch precursors prepared through the co-carbonization of ethylene bottom oil and polyvinyl chloride. J. Ind. Eng. Chem. 2018, 67, 358–364. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample 1 | Yield (wt %) | The Amount of Carbon Groups 2 | ||
|---|---|---|---|---|
| Aromatic Carbon (wt %) 3 | Aliphatic Carbon (wt %) 4 | |||
| Chlorinated Carbon (–CCl2–, –CHCl–) | Methylene Carbon (–CH2–) | |||
| PVC | -- | -- | 48.6 | 51.4 |
| CPVC | -- | -- | 74.0 | 26.0 |
| 200 | 38.3 | 21.2 | 54.2 | 24.6 |
| 300 | 29.7 | 96.2 | 1.9 | 1.9 |
| 400 | 28.0 | 98.8 | ~0 | 1.2 |
| 1000 | 26.2 | 99.2 | 0 | 0.8 |
| CPVC Derived Carbon Fibres 1 | Diameter (μm) | Tensile Strength (MPa) | Young’s Modulus (GPa) | Elongation (%) |
|---|---|---|---|---|
| 1000 °C | 20.4 ± 1.8 | 480 ± 58 | 39 ± 6 | 1.3 ± 0.2 |
| 1600 °C | 19.4 ± 2.3 | 590 ± 84 | 49 ± 8 | 1.2 ± 0.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Shimanoe, H.; Ko, S.; Lee, H.; Jo, C.; Lee, J.; Hong, S.-H.; Lee, H.; Jeon, Y.-P.; Nakabayashi, K.; et al. Highly Chlorinated Polyvinyl Chloride as a Novel Precursor for Fibrous Carbon Material. Polymers 2020, 12, 328. https://doi.org/10.3390/polym12020328
Liu J, Shimanoe H, Ko S, Lee H, Jo C, Lee J, Hong S-H, Lee H, Jeon Y-P, Nakabayashi K, et al. Highly Chlorinated Polyvinyl Chloride as a Novel Precursor for Fibrous Carbon Material. Polymers. 2020; 12(2):328. https://doi.org/10.3390/polym12020328
Chicago/Turabian StyleLiu, Jinchang, Hiroki Shimanoe, Seunghyun Ko, Hansong Lee, Chaehyun Jo, Jaewoong Lee, Seong-Hwa Hong, Hyunchul Lee, Young-Pyo Jeon, Koji Nakabayashi, and et al. 2020. "Highly Chlorinated Polyvinyl Chloride as a Novel Precursor for Fibrous Carbon Material" Polymers 12, no. 2: 328. https://doi.org/10.3390/polym12020328
APA StyleLiu, J., Shimanoe, H., Ko, S., Lee, H., Jo, C., Lee, J., Hong, S. -H., Lee, H., Jeon, Y. -P., Nakabayashi, K., Miyawaki, J., & Yoon, S. -H. (2020). Highly Chlorinated Polyvinyl Chloride as a Novel Precursor for Fibrous Carbon Material. Polymers, 12(2), 328. https://doi.org/10.3390/polym12020328