Dynamic Compression Induced Solidification


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
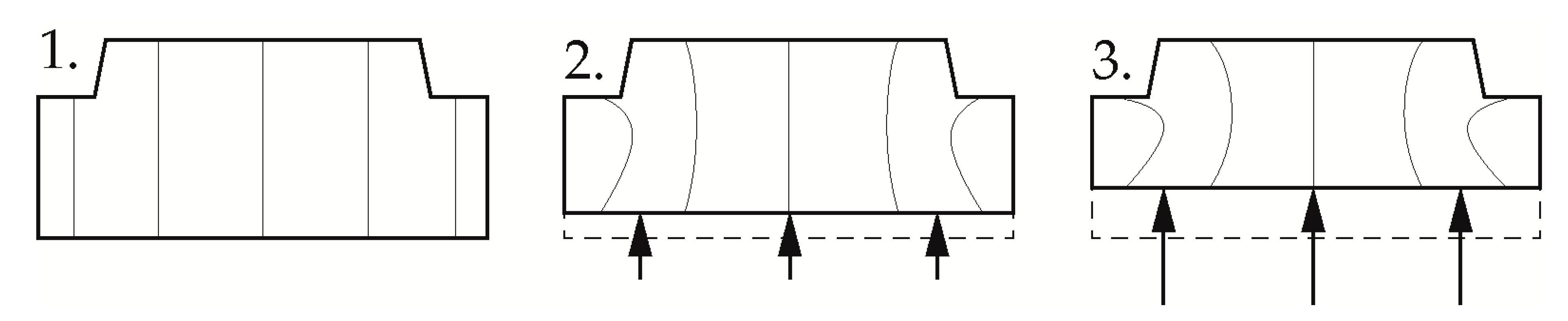
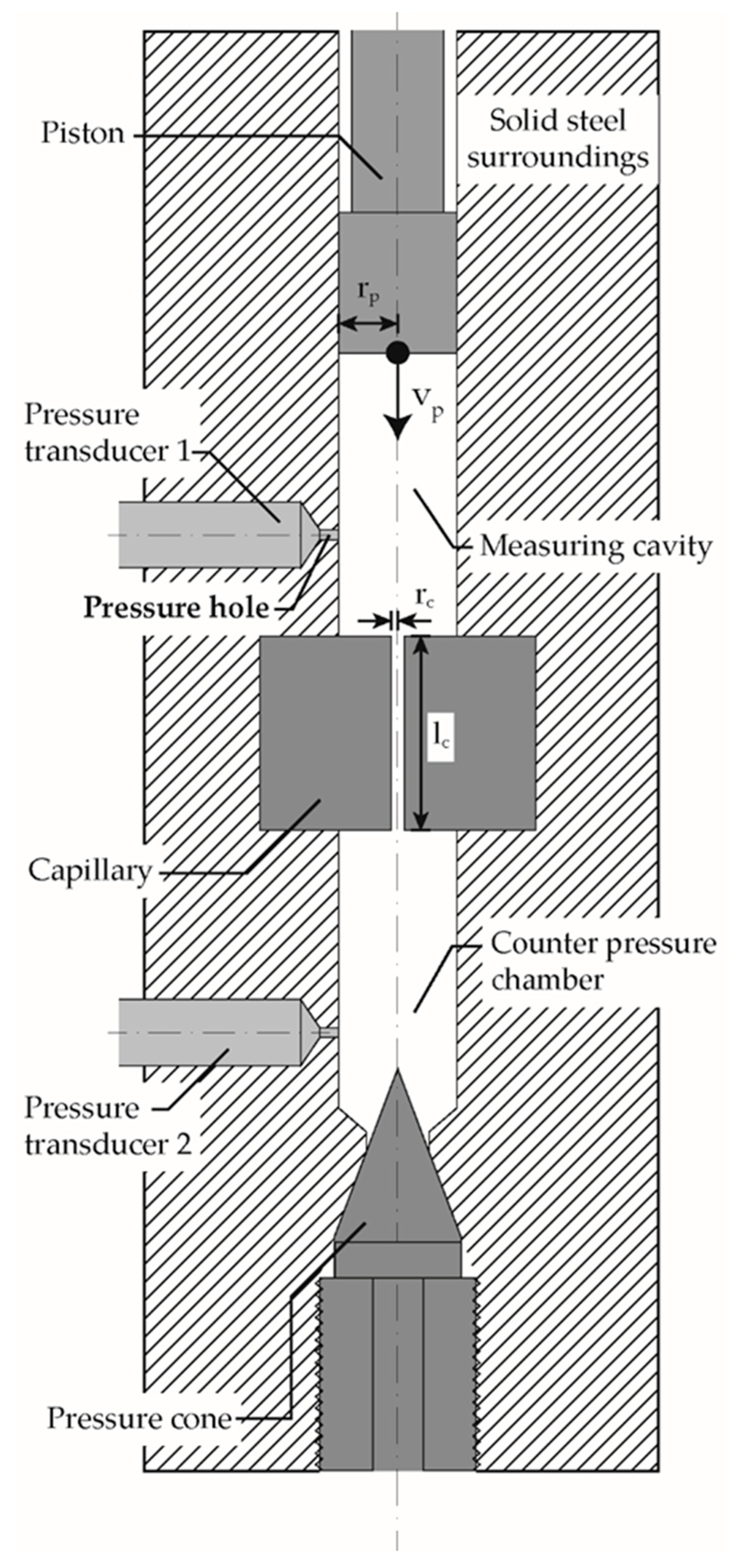
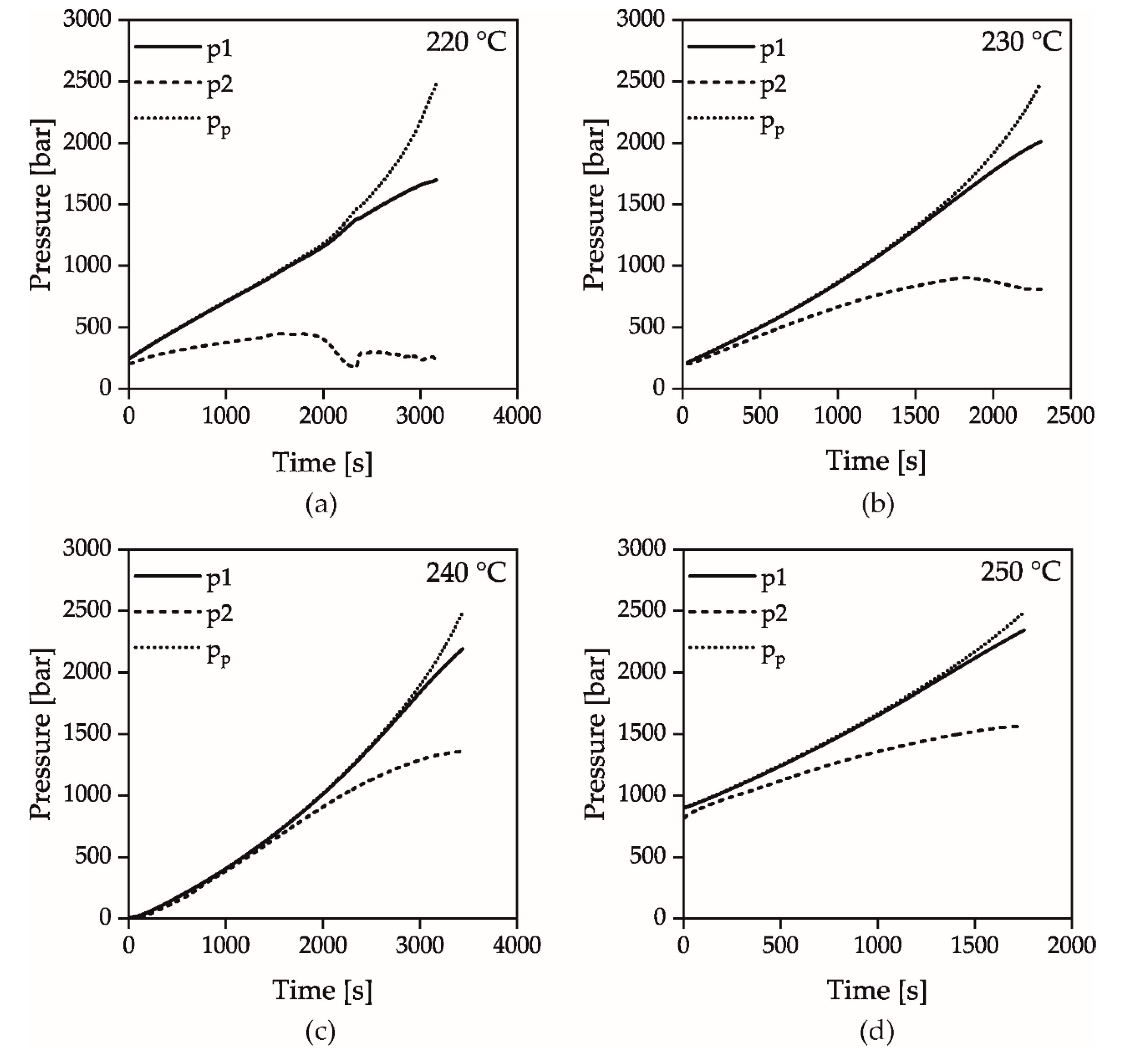
2.2. Methods
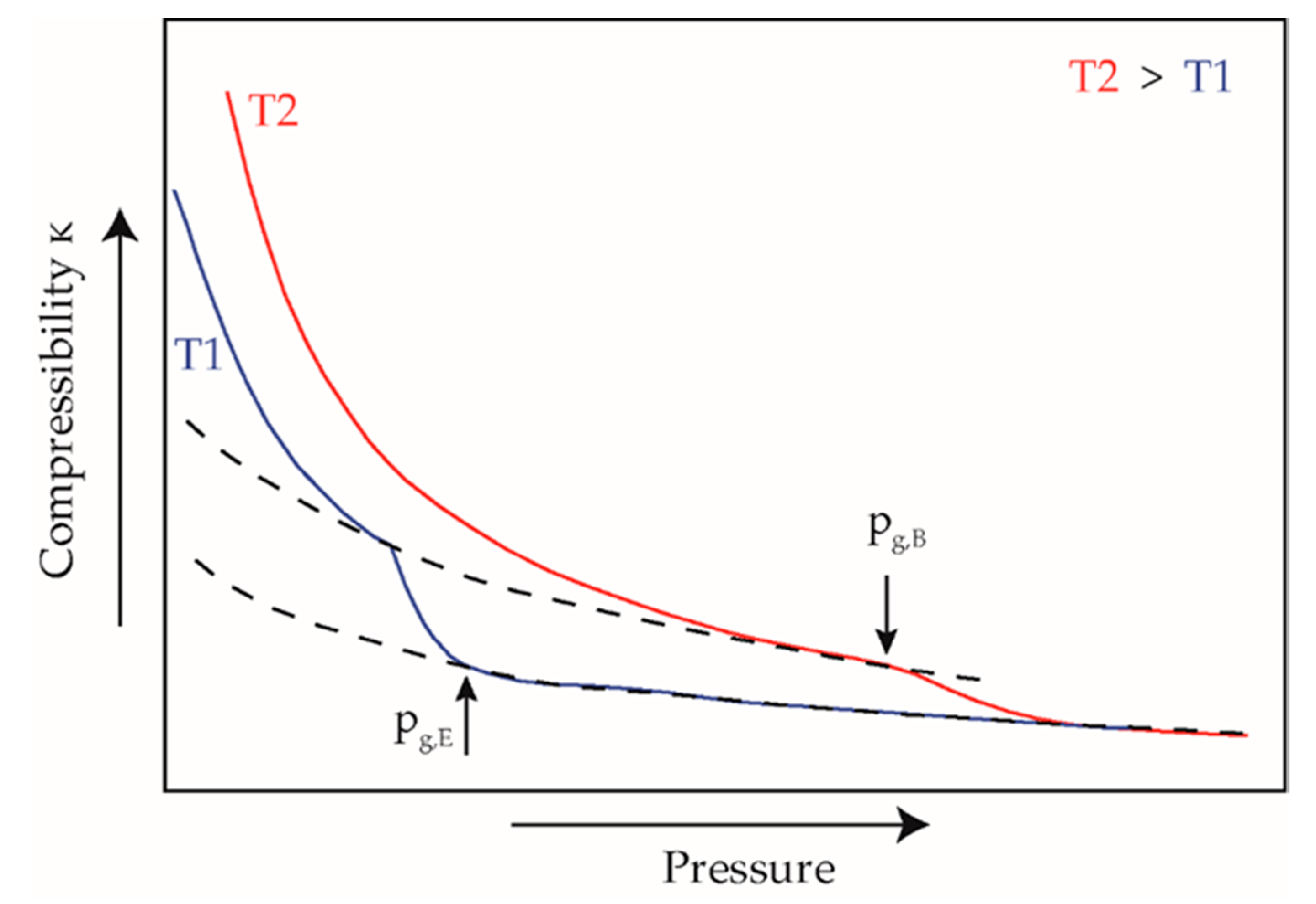
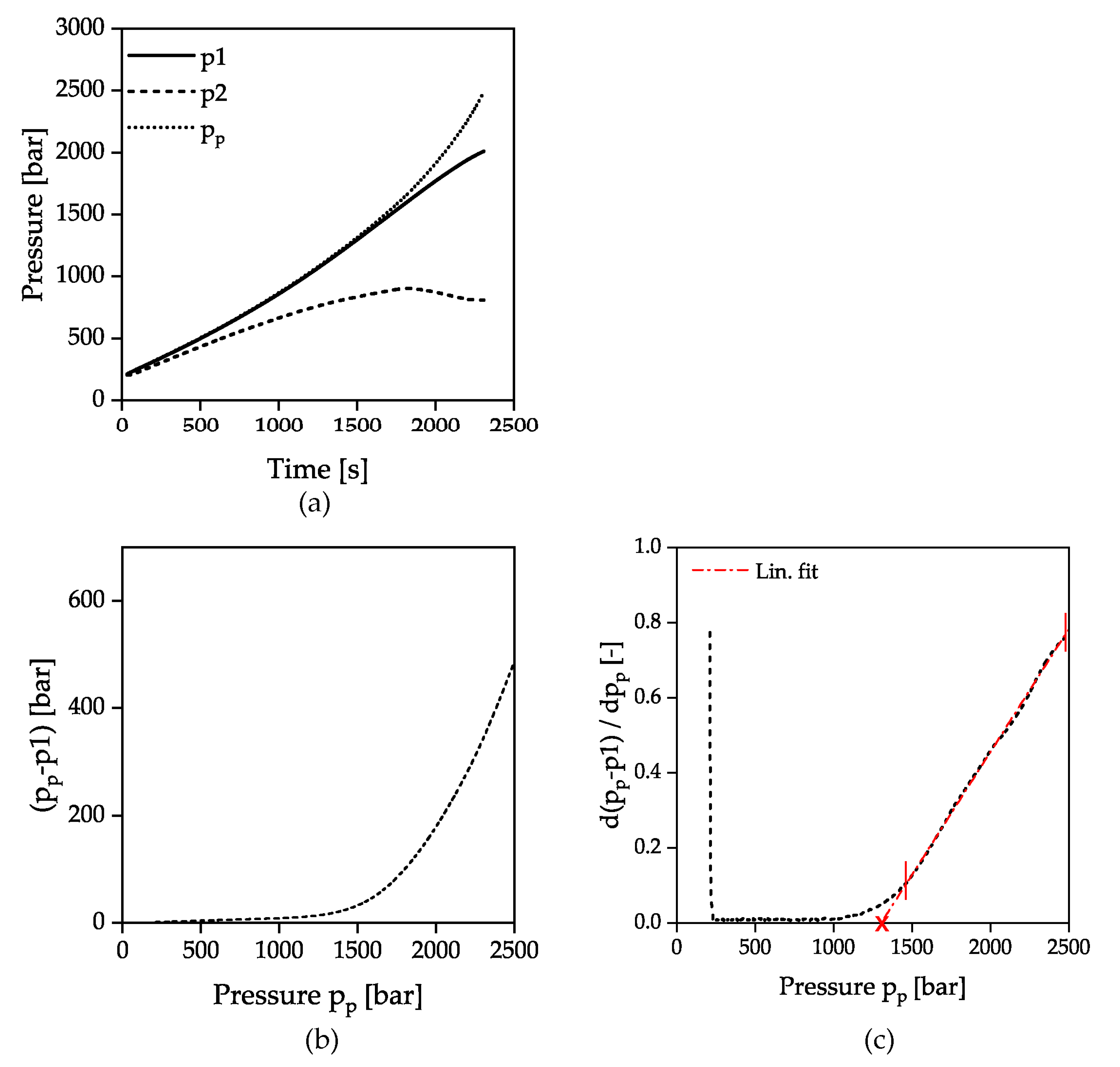
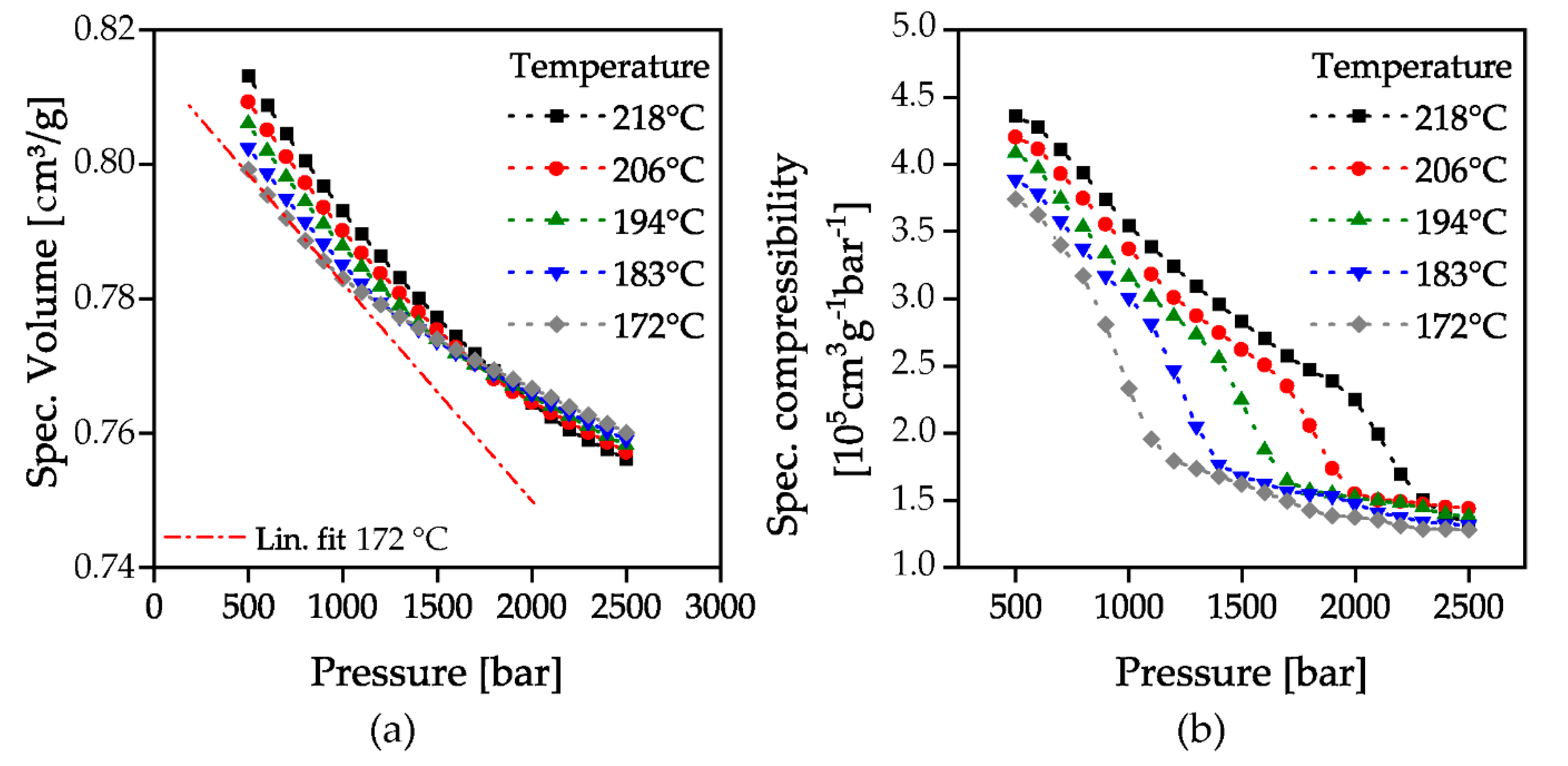
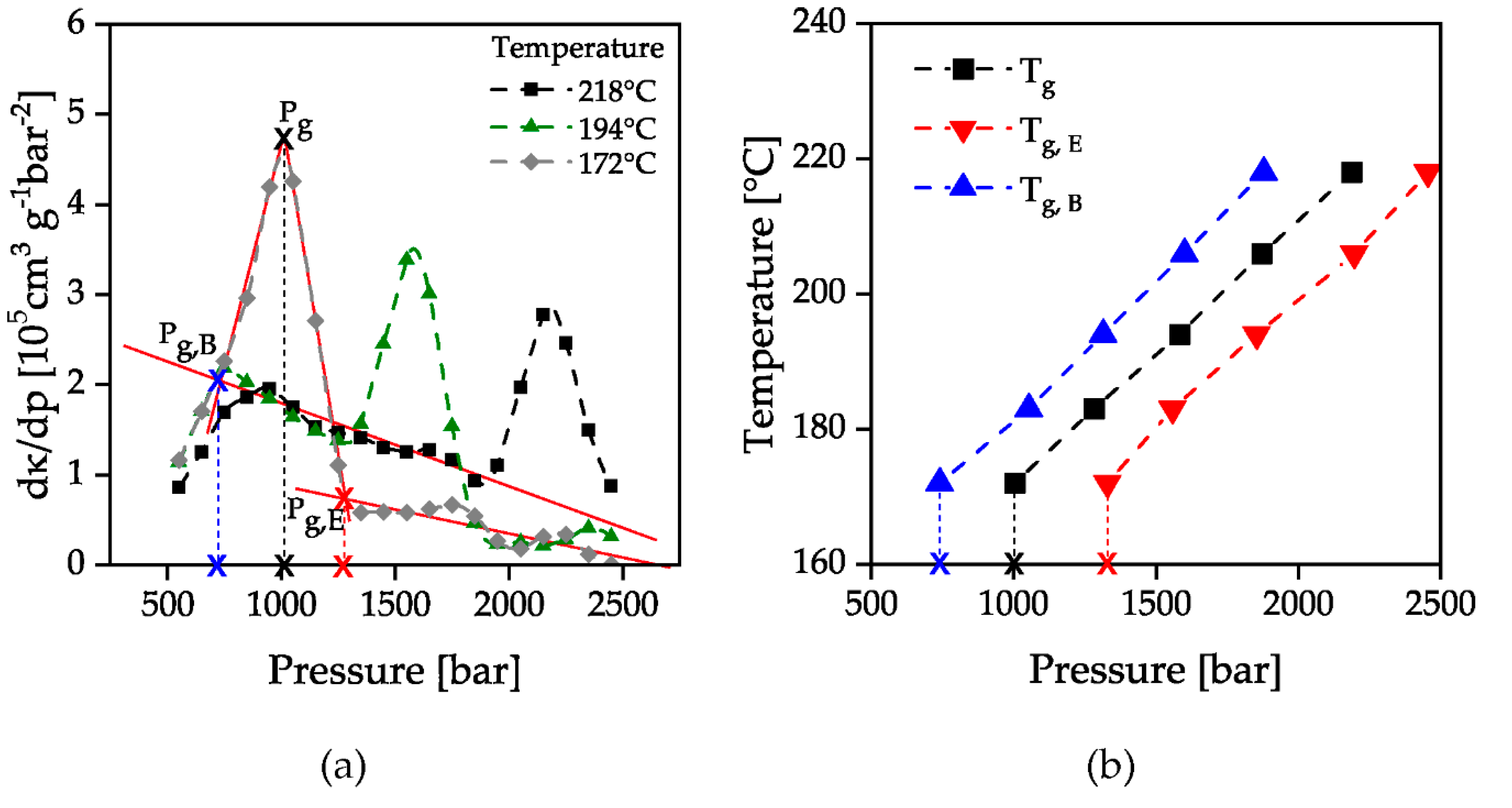
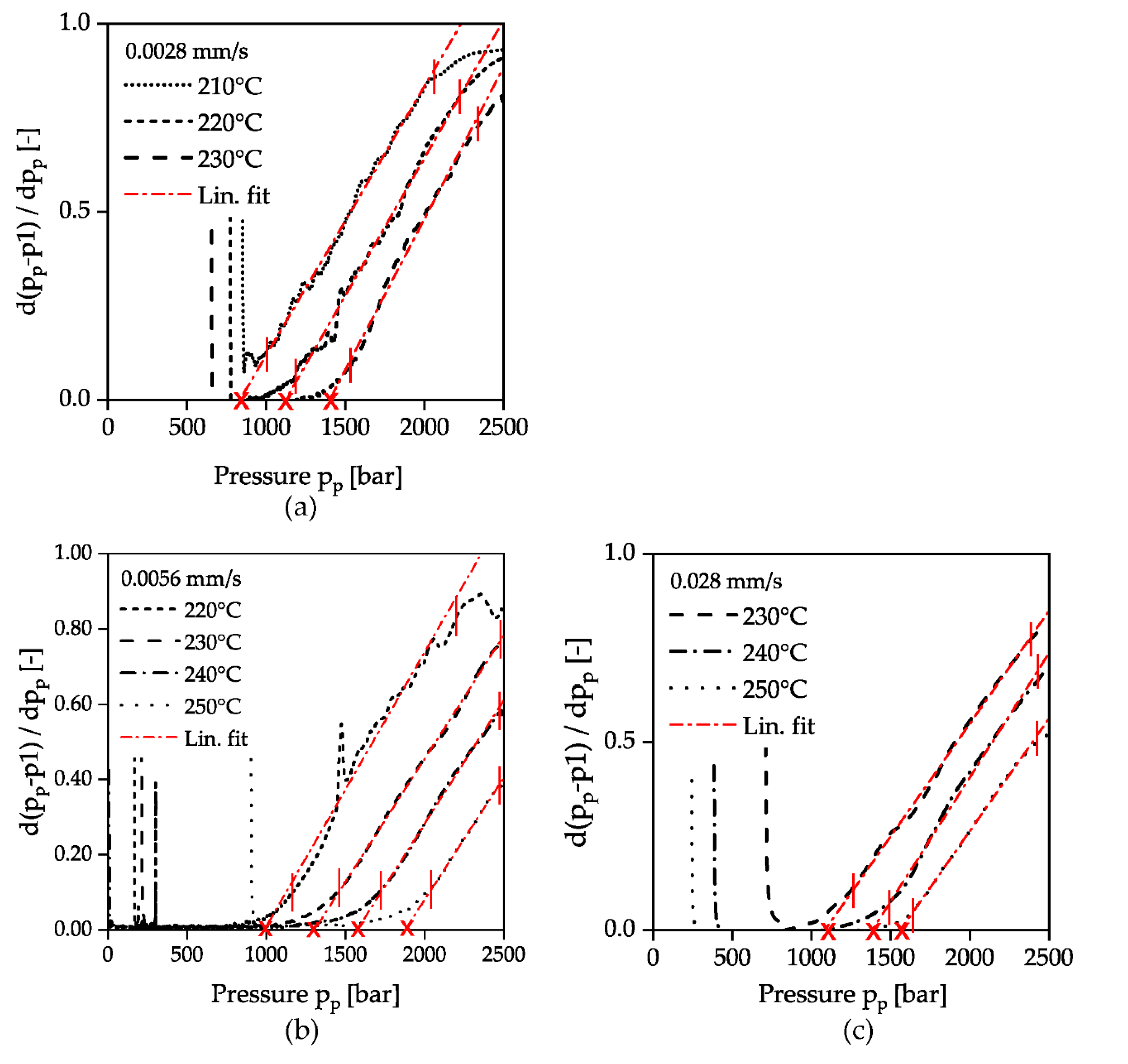
2.3. Measurement Evaluation
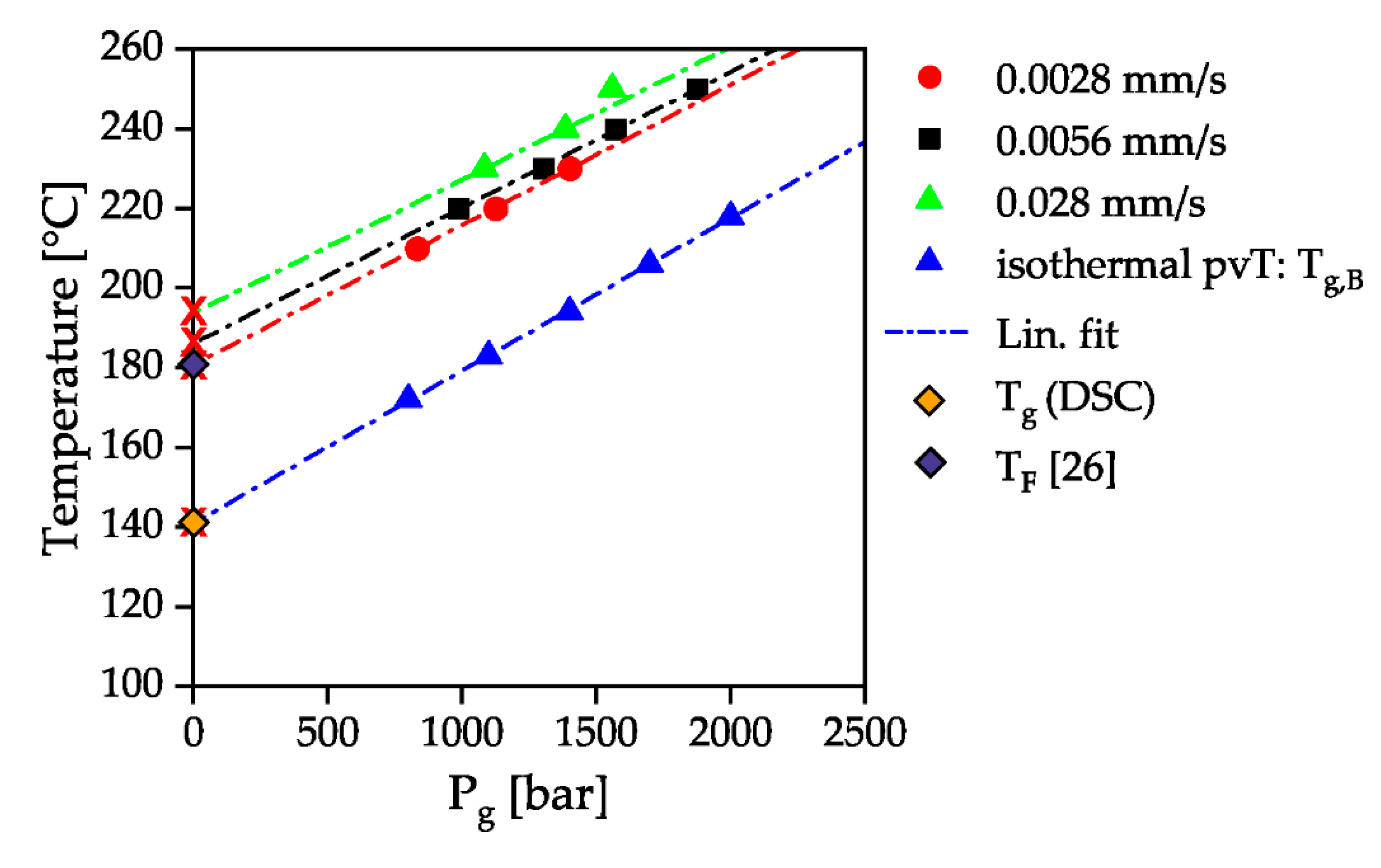
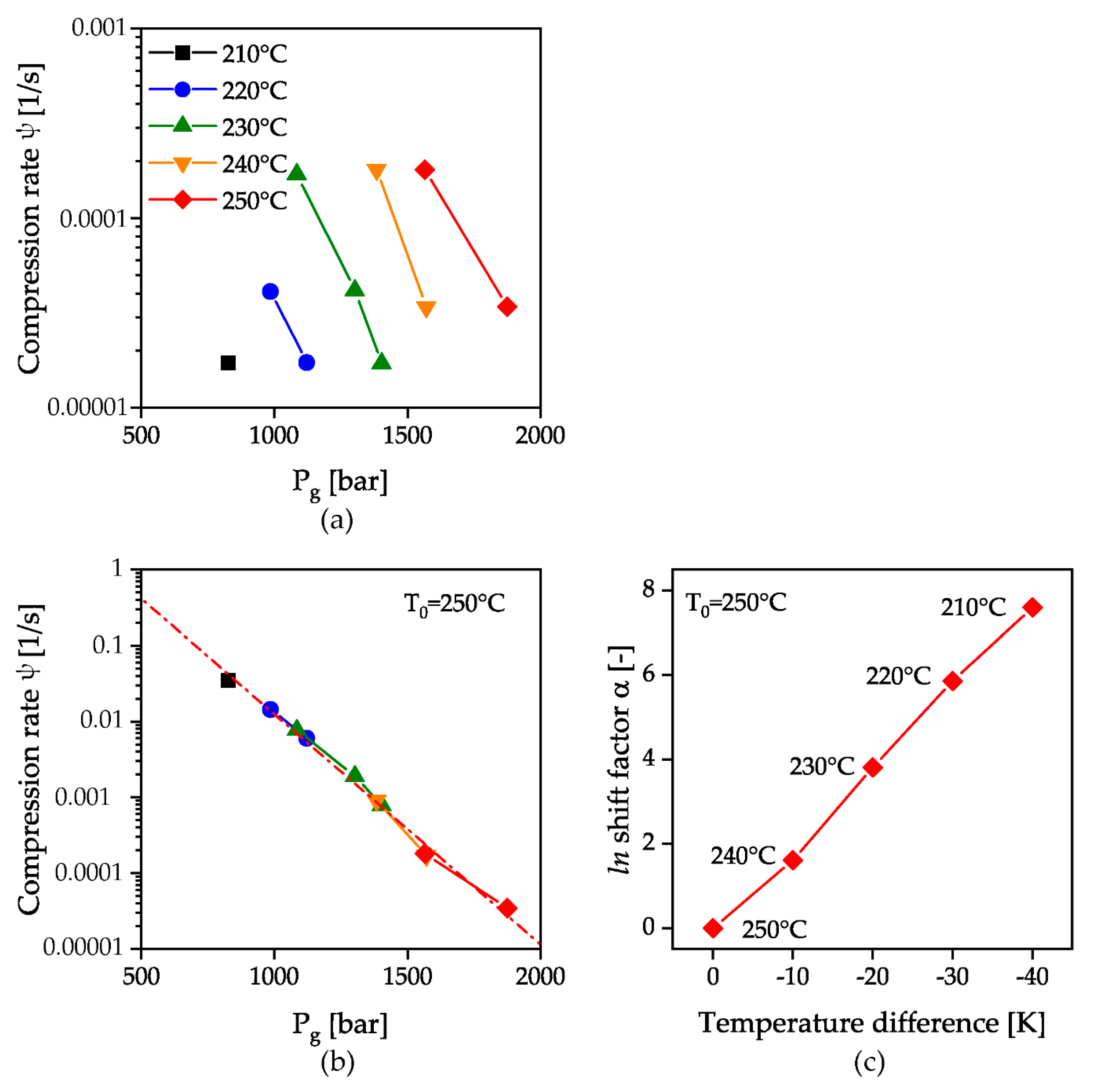
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sidiki, T.P. Mit Hochspannung unterwegs: Technische Thermoplaste begleiten die Elektrifizierung des Automobils. Kunststoffe 2019, 3, 38–44. [Google Scholar]
- Klar, R. Auslegung und Hochgenaue Fertigung Fresnelisierter Kunststoff-Freiformoptiken. Available online: https://www.photonikforschung.de/projekte/optikkomponenten/projekt/fresnelopt.html (accessed on 20 November 2019).
- Löh, D. Kunststoff ersetzt Metall: Hybrid-Bauteil in Automobil-Anwendung; Plastverarbeiter: Heidelberg, Germany, 2015. [Google Scholar]
- Maucher, T. Freiformoptiken—Universeller Einsatz Maßgeschneiderter Optikkomponenten. Available online: https://www.photonikforschung.de/projekte/optikkomponenten/projekt/autolight.html (accessed on 20 November 2019).
- Moll, D. Mikrospritzgießen—Ein Langer, Anspruchsvoller Weg; Plastverarbeiter: Heidelberg, Germany, 2018. [Google Scholar]
- Kolb, H.S. Aachen Polymer Optics Days—Expertenwissen für die Kunststoffoptik-Fertigung; Fraunhofer Institute: Aachen, Germany, 2018. [Google Scholar]
- Moynihan, C.T. Correlation between the width of the glass transition region and the temperature dependence of the viscosity of high-Tg glasses. J. Am. Ceram. Soc. 1993, 76, 1081–1087. [Google Scholar] [CrossRef]
- Curro, J.J.; Roe, R.-J. Isothermal relaxation of specific volume and density fluctuation in poly (methyl methacrylate) and polycarbonate. Polymer 1984, 25, 1424–1430. [Google Scholar] [CrossRef]
- Robertson, C.G.; Wilkes, G.L. Long-Term Volume Relaxation of Bisphenol A Polycarbonate and Atactic Polystyrene. Macromolecules 2000, 33, 3954–3955. [Google Scholar] [CrossRef]
- Hill, A.J.; Jones, P.L. Physical Aging and Isothermal Relaxation in Glassy Polycarbonate Measured by Positron Annihilation Lifetime Spectroscopy. MRS Online Proc. Libr. Arch. 1990, 215. [Google Scholar] [CrossRef]
- Napolitano, S.; Glynos, E.; Tito, N.B. Glass transition of polymers in bulk, confined geometries, and near interfaces. Rep. Prog. Phys. 2017, 80, 36602. [Google Scholar] [CrossRef] [PubMed]
- Ferry, J.D.; Stratton, R.A. The free volume interpretation of the dependence of viscosities and viscoelastic relaxation times on concentration, pressure, and tensile strain. Kolloid-Z. 1960, 171, 107–111. [Google Scholar] [CrossRef]
- Ferry, J.D. Viscoelastic Properties of Polymers; John Wiley & Sons: Hoboken, NJ, USA, 1980. [Google Scholar]
- Hill, A.J.; Katz, M.; Jones, P.L. Isothermal volume relaxation in aged polycarbonate measured by positron annihilation lifetime spectroscopy. Polym. Eng. Sci. 1990, 30, 762–768. [Google Scholar] [CrossRef]
- Hsieh, T.-T.; Tiu, C.; Simon, G.P. Correlation between molecular structure, free volume, and physical properties of a wide range of main chain thermotropic liquid crystalline polymers. J. Appl. Polym. Sci. 2001, 82, 2252–2267. [Google Scholar] [CrossRef]
- O’Connell, P.A.; McKenna, G.B. Large deformation response of polycarbonate: Time-temperature, time-aging time, and time-strain superposition. Polym. Eng. Sci. 1997, 37, 1485–1495. [Google Scholar]
- Rudolph, N.; Kühnert, I.; Schmachtenberg, E.; Ehrenstein, G.W. Pressure solidification of amorphous thermoplastics. Polym. Eng. Sci. 2009, 49, 154–161. [Google Scholar] [CrossRef]
- Jackle, J. Models of the glass transition. Rep. Prog. Phys. 1986, 49, 171–231. [Google Scholar] [CrossRef]
- Pantani, R. Pressure and cooling rate-induced densification of atactic polystyrene. J. Appl. Polym. Sci. 2003, 89, 184–190. [Google Scholar] [CrossRef]
- Zoller, P. A study of the pressure-volume-temperature relationships of four related amorphous polymers: Polycarbonate, polyarylate, phenoxy, and polysulfone. J. Polym. Sci. Polym. Phys. Ed. 1982, 20, 1453–1464. [Google Scholar] [CrossRef]
- Greener, J. Pressure-Induced densification in injection molding. Polym. Eng. Sci. 1986, 26, 534–542. [Google Scholar] [CrossRef]
- Moynihan, C.T.; Easteal, A.J.; Wilder, J.; Tucker, J. Dependence of the glass transition temperature on heating and cooling rate. J. Phys. Chem. 1974, 78, 2673–2677. [Google Scholar] [CrossRef]
- Fu, X.; Jia, W.; Li, X.; Wang, Y.; Wang, Z.; Liu, C.; Shen, C.; Shao, C. Phase transitions of the rapid-compression-induced mesomorphic isotactic polypropylene under high-pressure annealing. J. Polym. Sci. Part B Polym. Phys. 2019, 57, 651–661. [Google Scholar] [CrossRef]
- Li, X.; Jia, W.; Dong, B.; Yuan, H.; Su, F.; Wang, Z.; Wang, Y.; Liu, C.; Shen, C.; Shao, C. Structure and Mechanical Properties of Multi-Walled Carbon Nanotubes-Filled Isotactic Polypropylene Composites Treated by Pressurization at Different Rates. Polymers 2019, 11, 1294. [Google Scholar] [CrossRef] [Green Version]
- Knaapila, M.; Torkkeli, M.; Konôpková, Z.; Haase, D.; Liermann, H.-P.; Scherf, U.; Guha, S. Measuring Structural Inhomogeneity of Conjugated Polymer at High Pressures up to 30 GPa. Macromolecules 2013, 46, 8284–8288. [Google Scholar] [CrossRef]
- Bäumer, S. Handbook of Plastic Optics; Wiley Online Library: Hoboken, NJ, USA, 2010. [Google Scholar]
- Röbig, M.; Hopmann, C. Multilayer-Spritzgießen zur Produktion von Linsen; Plastverarbeiter: Heidelberg, Germany, 2017. [Google Scholar]
- Jungmeier, A.; Wildner, W.; Drummer, D.; Kühnert, I. Compression-Induced Solidification: A Novel Processing Technique for Precise Thermoplastic Optical Components with Negligible Internal Stresses. ISRN Opt. 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, N. Druckverfestigung Amorpher Thermoplaste. Ph.D. Thesis, University of Erlangen-Nürnberg, Erlangen, Germany, 2009. [Google Scholar]
- Meister, S.; Drummer, D. Investigation on the achievable flow length in injection moulding of polymeric materials with dynamic mould tempering. Sci. World J. 2013, 2013, 845916. [Google Scholar] [CrossRef] [PubMed]
- Giboz, J.; Copponnex, T.; Mélé, P. Microinjection molding of thermoplastic polymers: A review. J. Micromech. Microeng. 2007, 17, R96–R109. [Google Scholar] [CrossRef]
- Mekaru, H.; Yamada, T.; Yan, S.; Hattori, T. Microfabrication by hot embossing and injection molding at LASTI. Microsyst. Technol. 2004, 10, 682–688. [Google Scholar] [CrossRef]
- Yokoi, H.; Han, X.; Takahashi, T.; Kim, W.K. Effects of molding conditions on transcription molding of microscale prism patterns using ultra-high-speed injection molding. Polym. Eng. Sci. 2006, 46, 1140–1146. [Google Scholar] [CrossRef]
- Greiner, R.; Schwarzl, F.R. Thermal contraction and volume relaxation of amorphous polymers. Rheol. Acta 1984, 23, 378–395. [Google Scholar] [CrossRef]
- Delbreilh, L.; Dargent, E.; Grenet, J.; Saiter, J.-M.; Bernès, A.; Lacabanne, C. Study of poly(bisphenol A carbonate) relaxation kinetics at the glass transition temperature. Eur. Polym. J. 2007, 43, 249–254. [Google Scholar] [CrossRef] [Green Version]
- Doolittle, A.K. Studies in Newtonian Flow. II. The Dependence of the Viscosity of Liquids on Free-Space. J. Appl. Phys. 1951, 22, 1471–1475. [Google Scholar]
- Semjonow, V. Über ein rotationsviskosimeter zur messung der druckabhängigkeit der viskosität hochpolymerer schmelzen. Rheol. Acta 1962, 2, 138–143. [Google Scholar] [CrossRef]
- Sedlacek, T.; Zatloukal, M.; Filip, P.; Boldizar, A.; Saha, P. On the effect of pressure on the shear and elongational viscosities of polymer melts. Polym. Eng. Sci. 2004, 44, 1328–1337. [Google Scholar] [CrossRef]
- Rudolph, N.M.; Agudelo, A.C.; Granada, J.C.; Park, H.E.; Osswald, T.A. WLF model for the pressure dependence of zero shear viscosity of polycarbonate. Rheol. Acta 2016, 55, 673–681. [Google Scholar] [CrossRef]
- Maxwell, B.; Jung, A. Hydrostatic pressure effect on polymer melt viscosity. Mod. Plast. 1957, 35, 174–182. [Google Scholar]
- Mackley, M.R.; Spitteler, P.H.J. Experimental observations on the pressure-dependent polymer melt rheology of linear low density polyethylene, using a multi-pass rheometer. Rheol. Acta 1996, 35, 202–209. [Google Scholar] [CrossRef]
- Lord, H.A. Flow of polymers with pressure-dependent viscosity in injection molding dies. Polym. Eng. Sci. 1979, 19, 469–473. [Google Scholar] [CrossRef]
- Kadijk, S.E.; van den Brule, B.H.A.A. On the pressure dependency of the viscosity of molten polymers. Polym. Eng. Sci. 1994, 34, 1535–1546. [Google Scholar] [CrossRef]
- Driscoll, P.D.; Bogue, D.C. Pressure effects in polymer melt rheology. J. Appl. Polym. Sci. 1990, 39, 1755–1768. [Google Scholar] [CrossRef]
- Aho, J.; Syrjälä, S. Measurement of the pressure dependence of viscosity of polymer melts using a back pressure-regulated capillary rheometer. J. Appl. Polym. Sci. 2010, 117, 1076–1084. [Google Scholar] [CrossRef]
- Binding, D.M.; Couch, M.A.; Walters, K. The pressure dependence of the shear and elongational properties of polymer melts1Dedicated to Professor Marcel, J. Crochet on the occasion of his 60th birthday.1. J. Non-Newton. Fluid Mech. 1998, 79, 137–155. [Google Scholar] [CrossRef]
- Liang, J.-Z. Pressure effect of viscosity for polymer fluids in die flow. Polymer 2001, 42, 3709–3712. [Google Scholar] [CrossRef]
- Yamada, M.; Porter, R.S. Compressional effects in the capillary flow of polycarbonate. J. Appl. Polym. Sci. 1974, 18, 1711–1724. [Google Scholar] [CrossRef]
- Bueche, F. Influence of Rate of Shear on the Apparent Viscosity of A—Dilute Polymer Solutions, and B—Bulk Polymers. J. Chem. Phys. 1954, 22, 1570–1576. [Google Scholar] [CrossRef]
- Casale, A.; Penwell, R.C.; Porter, B.S. The influence of pressure on the capillary flow of poly(methyl methacrylate). Rheol. Acta 1971, 10, 412–417. [Google Scholar] [CrossRef]
- Karl, V.-H. Über die druckabhängigkeit der viskoelastischen und physikalisch-chemischen eigenschaften von polymeren, 8. Die viskosität von polyethylen bis 5000 bar. Angew. Makromol. Chem. 1979, 79, 11–19. [Google Scholar] [CrossRef]
- Datasheet Makrolon LQ2647. Covestro AG 2017. Available online: https://solutions.covestro.com/en/products/makrolon/makrolon-lq2647_56979771-00009631?SelectedCountry=US (accessed on 19 February 2020).
- Mattner, T.; Drummer, D. Influence of pressure on end corrections in capillary rheometry. Rheol. Acta 2016, 55, 823–832. [Google Scholar] [CrossRef]
- Breuer, H.; Rehage, G. Zur Thermodynamik der glasigen Erstarrung. Kolloid-Z. 1967, 216, 159–179. [Google Scholar]
- Quach, A.; Simha, R. Pressure-Volume-Temperature Properties and Transitions of Amorphous Polymers; Polystyrene and Poly (orthomethylstyrene). J. Appl. Phys. 1971, 42, 4592–4606. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature [°C] | Pressure Range [bar] | Pressure Increment Δp [bar] |
|---|---|---|
| 172 °C | 500–2500 | 100 |
| 183 °C | 500–2500 | 100 |
| 194 °C | 500–2500 | 100 |
| 206 °C | 500–2500 | 100 |
| 218 °C | 500–2500 | 100 |
| Piston Velocity [mm/s] | Temperature [°C] |
|---|---|
| 0.0028 | 210 |
| 220 | |
| 230 | |
| 0.0056 | 220 |
| 230 | |
| 240 | |
| 250 | |
| 0.028 | 230 |
| 240 | |
| 250 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roth, B.; Wildner, W.; Drummer, D. Dynamic Compression Induced Solidification. Polymers 2020, 12, 488. https://doi.org/10.3390/polym12020488
Roth B, Wildner W, Drummer D. Dynamic Compression Induced Solidification. Polymers. 2020; 12(2):488. https://doi.org/10.3390/polym12020488
Chicago/Turabian StyleRoth, Benedikt, Wolfgang Wildner, and Dietmar Drummer. 2020. "Dynamic Compression Induced Solidification" Polymers 12, no. 2: 488. https://doi.org/10.3390/polym12020488
APA StyleRoth, B., Wildner, W., & Drummer, D. (2020). Dynamic Compression Induced Solidification. Polymers, 12(2), 488. https://doi.org/10.3390/polym12020488