Copolymacrolactones Grafted with l-Glutamic Acid: Synthesis, Structure, and Nanocarrier Properties


Abstract
:
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Characterization
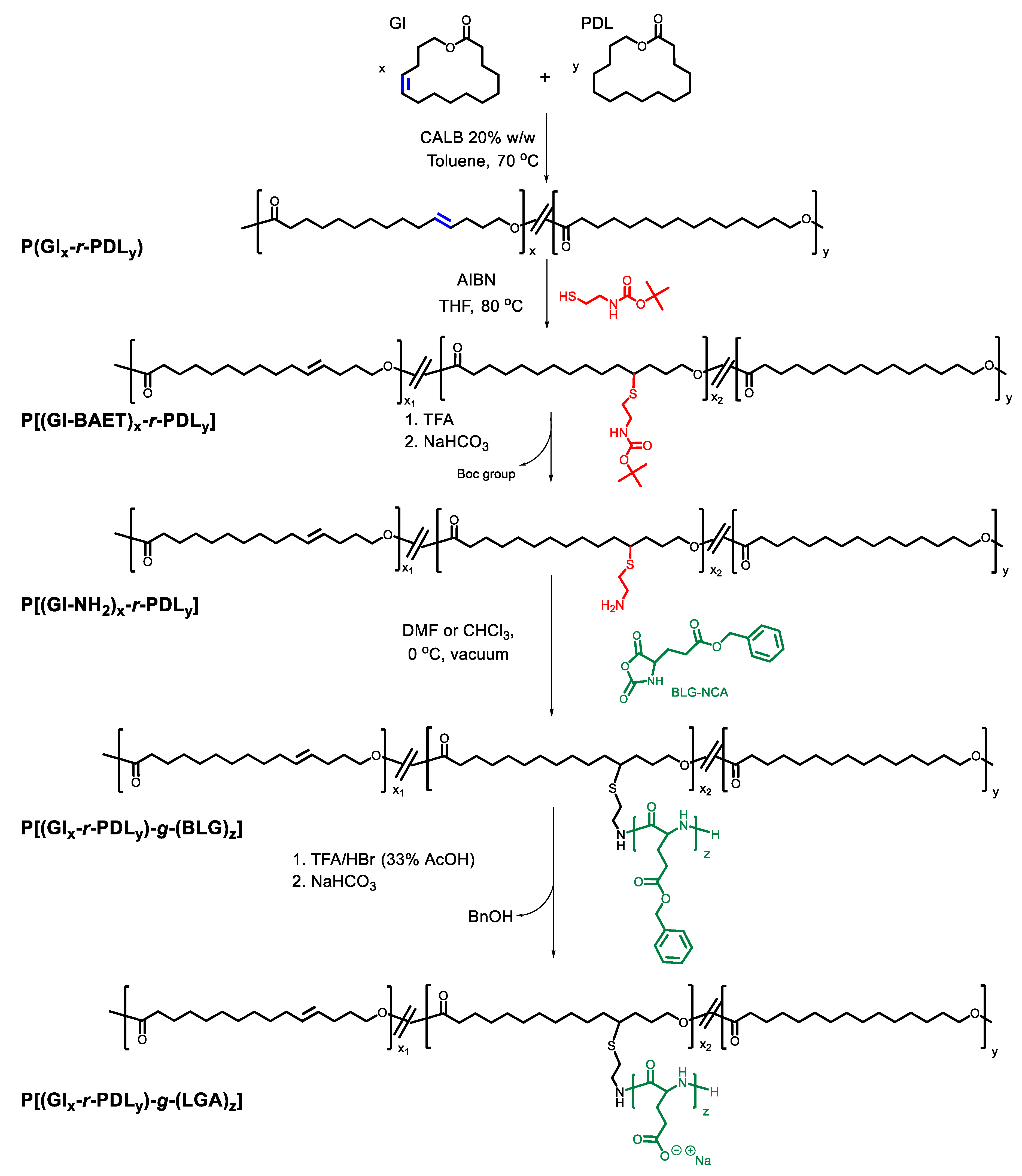
2.3. Synthesis
2.4. Graft Copolymer Self-Assembling
2.5. Doxorubicin Loading and Releasing
3. Results and Discussion
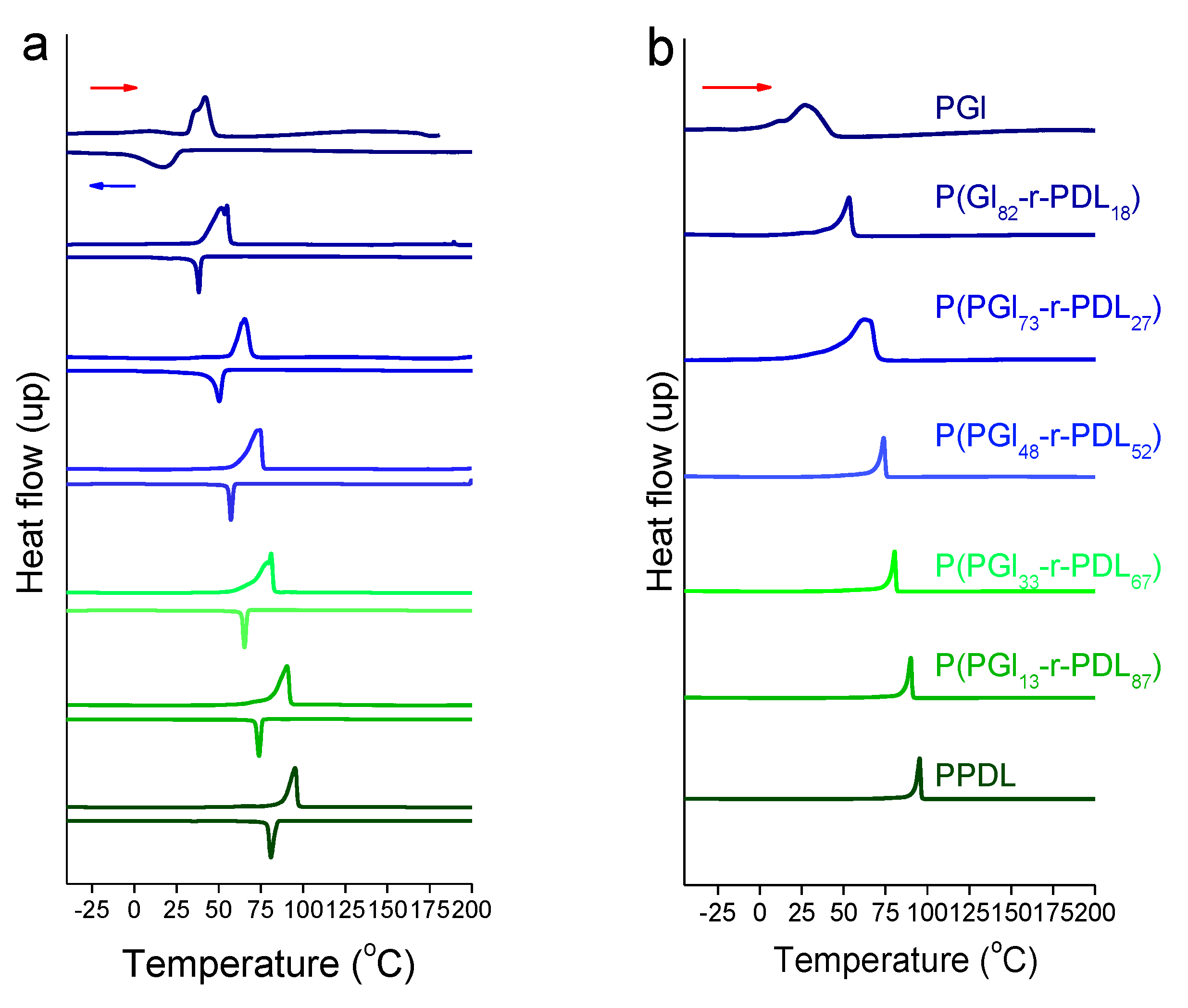
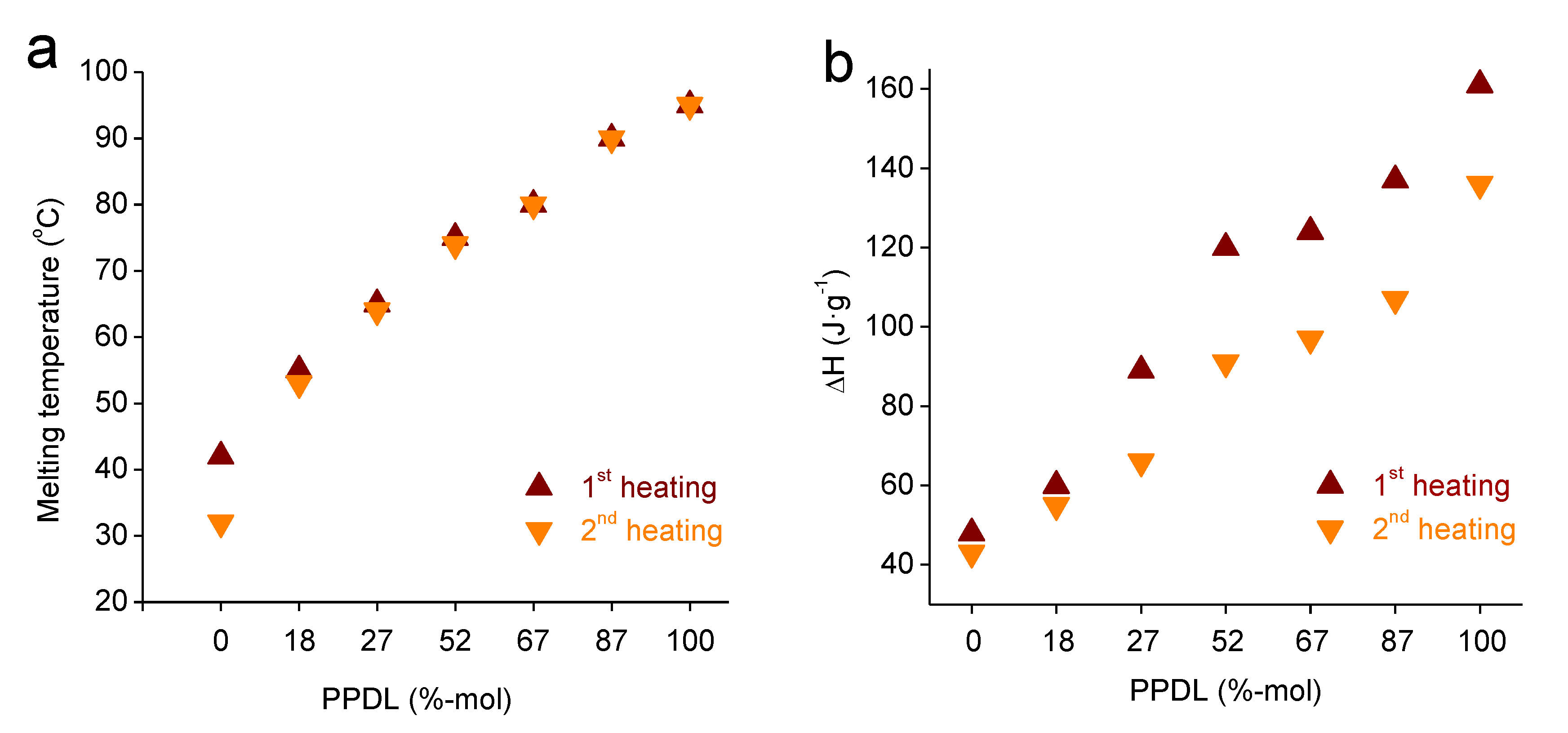
3.1. PGl-r-PDL Copolyesters
3.2. Grafting of P(Glx-r-PDLy) Copolyesters with Glutamic Acid Units: Synthesis of P[(Glx-r-PDLy)–g–(LGlu)z] Copolymers
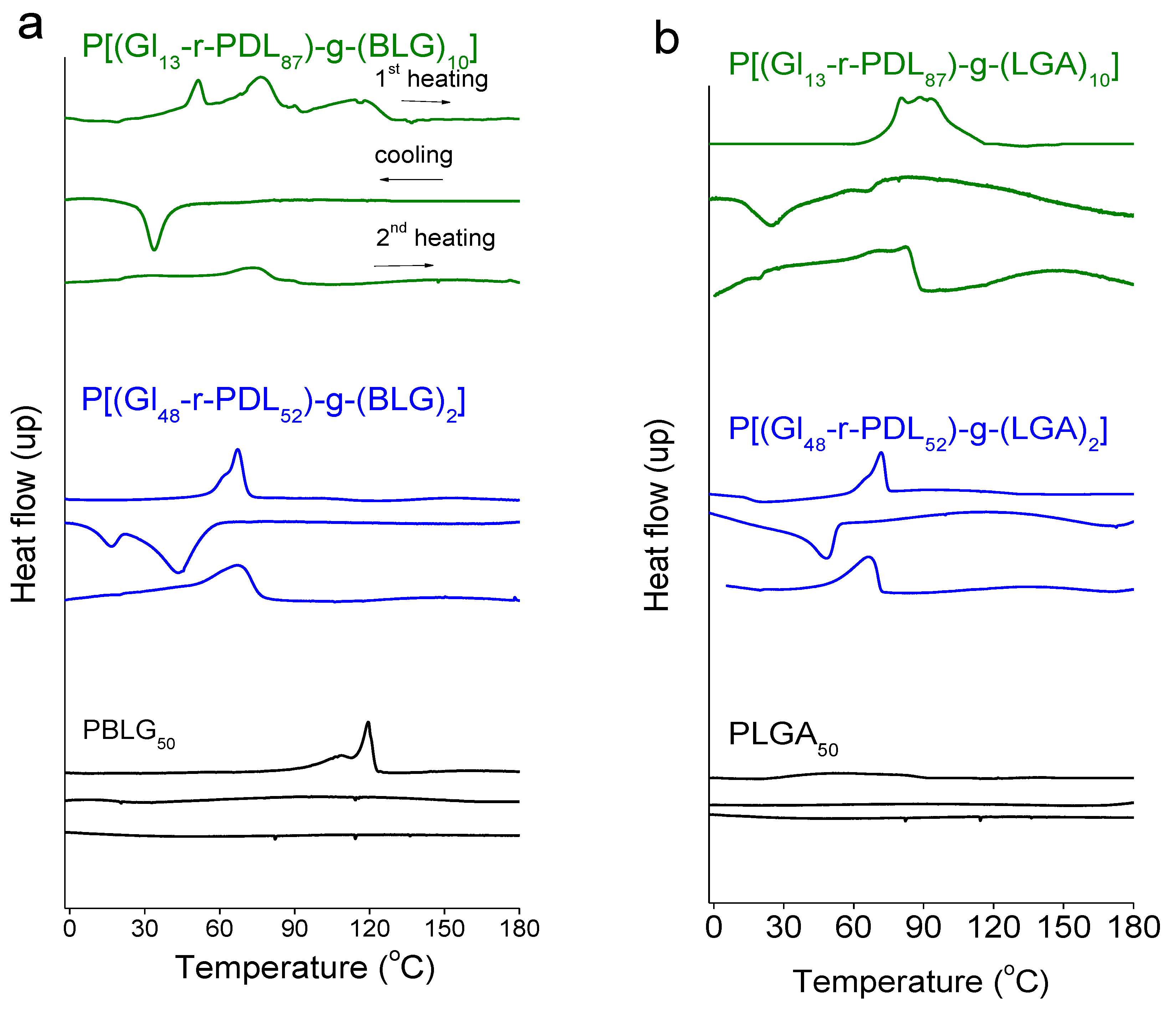
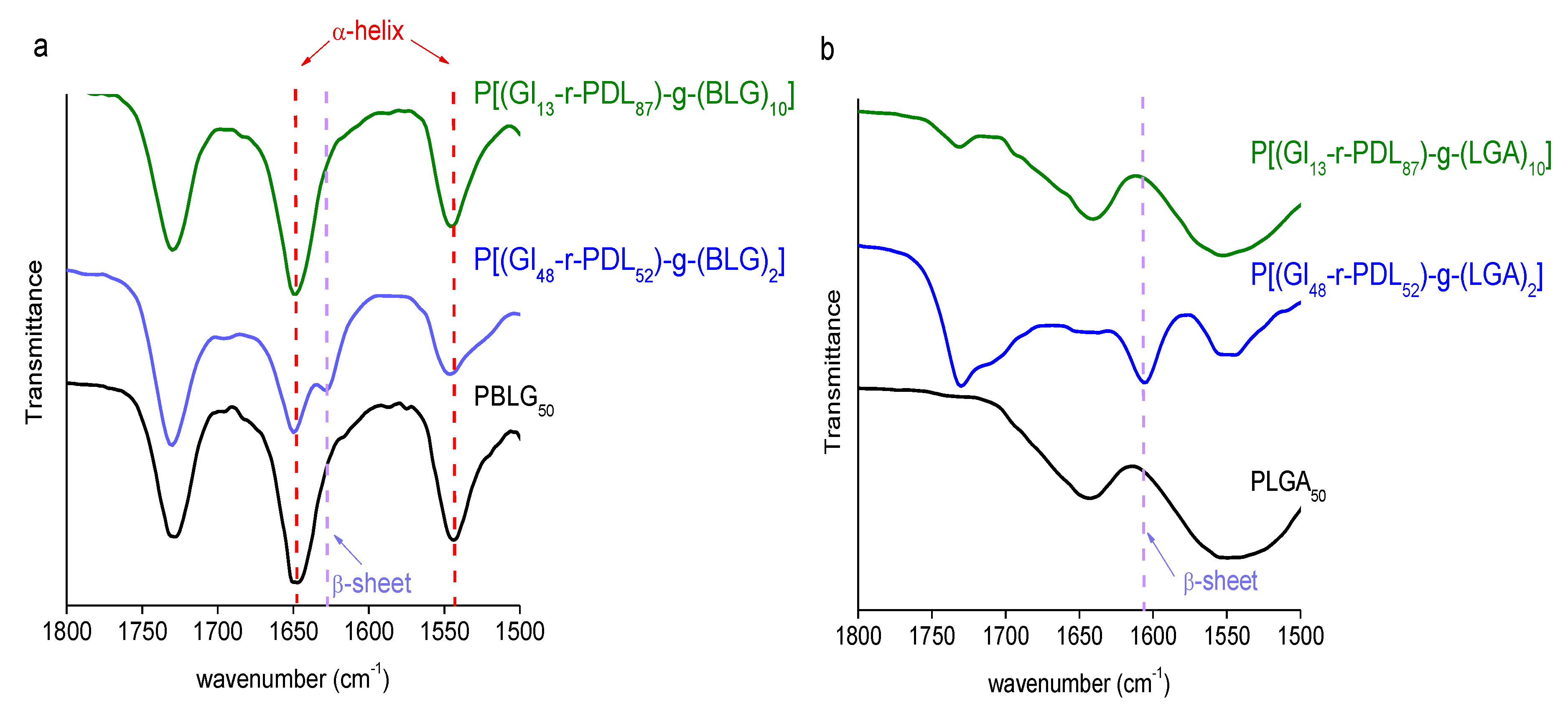
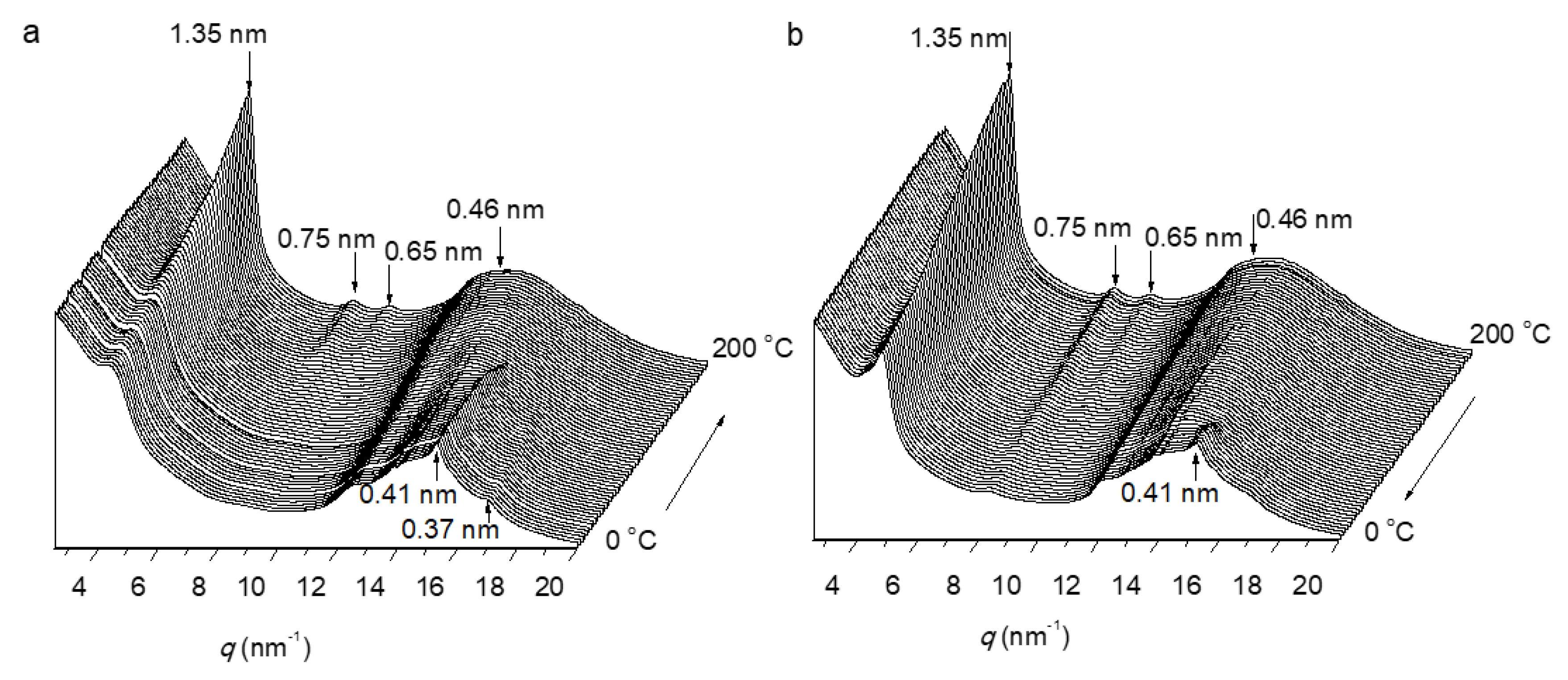
3.3. Thermal Properties and Structure of P[(Glx-r-PDLy)–g–(LGlu)z] Copolymers
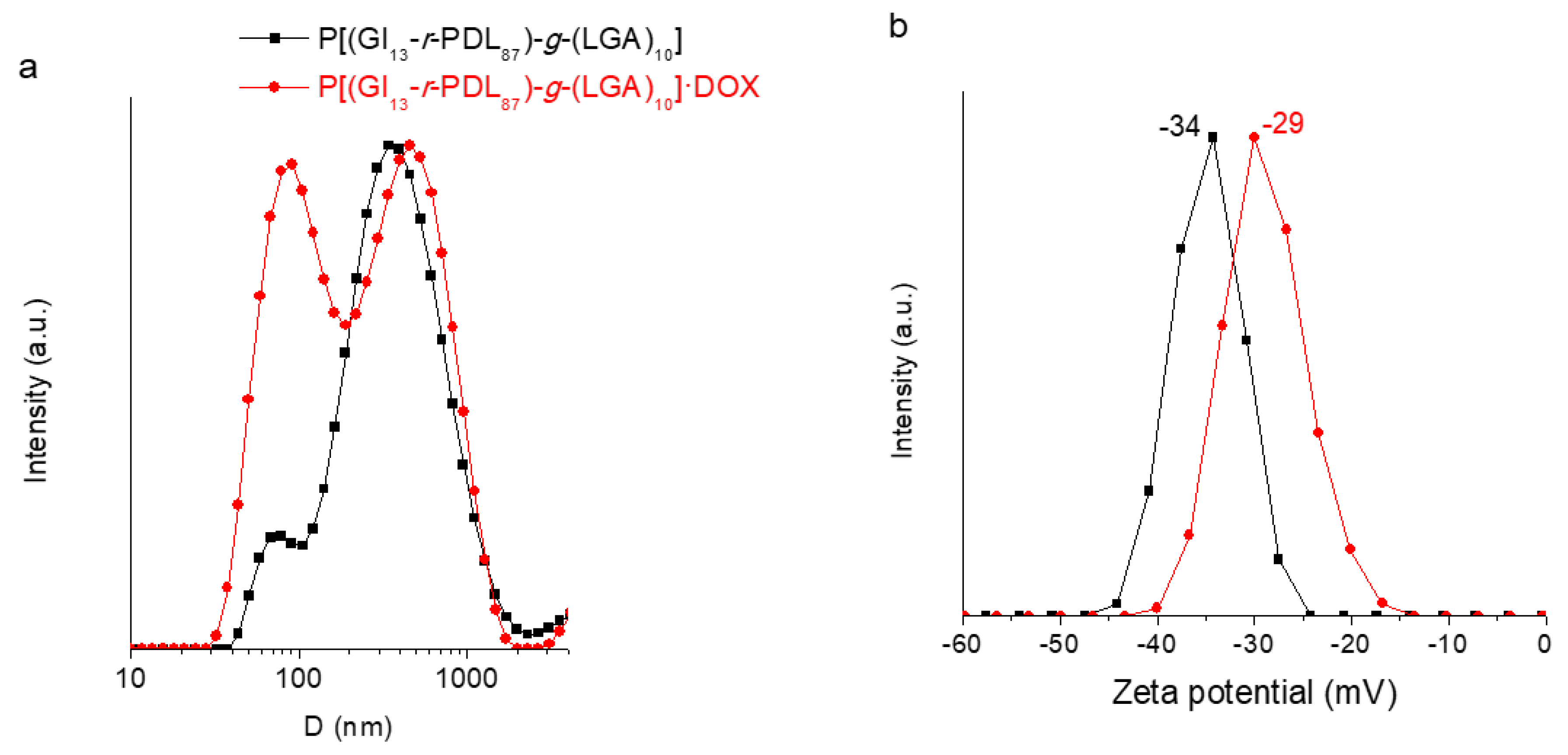
3.4. Self-Assembly of P[(Glx-r-PDLy)–g–(LGlu)z] Copolymers: DOX Loading and Releasing
4. Conclusions
Associated Information
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Deng, C.; Wu, J.; Cheng, R.; Meng, F.; Klok, H.-A.; Zhong, Z. Functional polypeptide and hybrid materials: Precision synthesis via α-amino acid N-carboxyanhydride polymerization and emerging biomedical applications. Prog. Polym. Sci. 2014, 39, 330–364. [Google Scholar] [CrossRef]
- Canalle, L.A.; Löwik, D.W.P.M.; van Hest, J.C.M. Polypeptide–polymer bioconjugates. Chem. Soc. Rev. 2010, 39, 329–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehn, S.; Chapman, R.; Jolliffe, K.A.; Perrier, S. Synthetic strategies for the design of peptide/polymer conjugates. Polym. Rev. 2011, 51, 214–234. [Google Scholar] [CrossRef]
- Albertsson, A.-C.; Varma, I.K. Recent developments in ring opening polymerization of lactones for biomedical applications. Biomacromolecules 2003, 4, 1466–1486. [Google Scholar] [CrossRef]
- Kricheldorf, H.R. Polypeptides and 100 years of chemistry of α-amino acid N-carboxyanhydrides. Angew. Chem. Int. Ed. 2006, 45, 5752–5784. [Google Scholar] [CrossRef]
- Wilson, J.A.; Ates, Z.; Pflughaupt, R.L.; Dove, A.P.; Heise, A. Polymers from macrolactones: From pheromones to functional materials. Prog. Polym. Sci. 2019, 91, 29–50. [Google Scholar] [CrossRef]
- Williams, A.S. The synthesis of macrocyclic musks. Synthesis 1999, 10, 1707–1723. [Google Scholar] [CrossRef]
- Todd, R.; Tempelaar, S.; Lo Re, G.; Spinella, S.; McCallum, S.A.; Gross, R.A.; Raquez, J.-M.; Dubois, P. Poly(ω-pentadecalactone)-b-poly(L-lactide) block copolymers via organic-catalyzed ring opening polymerization and potential applications. ACS Macro Lett. 2015, 4, 408–411. [Google Scholar] [CrossRef]
- Pepels, M.P.F.; Hofman, W.P.; Kleijnen, R.; Spoelstra, A.B.; Koning, C.E.; Goossens, H.; Duchateau, R. Block copolymers of “PE-Like” poly(pentadecalactone) and poly(L-lactide): Synthesis, properties, and compatibilization of polyethylene/poly(L-lactide) blends. Macromolecules 2015, 48, 6909–6921. [Google Scholar] [CrossRef] [Green Version]
- Ulker, C.; Guvenilir, Y. Enzymatic synthesis and characterization of biodegradable poly(ω-pentadecalactone-co-ε-caprolactone) copolymers. J. Renew. Mater. 2018, 6, 591–598. [Google Scholar] [CrossRef]
- Ceccorulli, G.; Scandola, M.; Kumar, A.; Kalra, B.; Gross, R.A. Cocrystallization of random copolymers of ω-pentadecalactone and ε-caprolactone synthesized by lipase catalysis. Biomacromolecules 2005, 6, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Bouyahyi, M.; Pepels, M.P.F.; Heise, A.; Duchateau, R. ω-Pentandecalactone polymerization and ω-pentadecalactone/ε-caprolactone copolymerization reactions using organic catalysts. Macromolecules 2012, 45, 3356–3366. [Google Scholar] [CrossRef]
- Jiang, Z.; Azim, H.; Gross, R.A.; Focarete, M.L.; Scandola, M. Lipase-catalyzed copolymerization of ω-pentadecalactone with p-dioxanone and characterization of copolymer thermal and crystalline properties. Biomacromolecules 2007, 8, 2262–2269. [Google Scholar] [CrossRef] [PubMed]
- Walther, P.; Naumann, S. N-Heterocyclic olefin-based (co)polymerization of a challenging monomer: Homopolymerization of ω-pentadecalactone and its copolymers with γ-butyrolactone, δ-valerolactone, and ε-caprolactone. Macromolecules 2017, 50, 8406–8416. [Google Scholar] [CrossRef]
- Xiao, Y.; Pan, J.; Wang, D.; Heise, A.; Lang, M. Chemo-enzymatic synthesis of poly(4-piperidine lactone-b- ω-pentadecalactone) block copolymers as biomaterials with antibacterial properties. Biomacromolecules 2018, 19, 2673–2681. [Google Scholar] [CrossRef]
- Kalra, B.; Kumar, A.; Gross, R.A.; Baiardo, M.; Scandola, M. Chemoenzymatic synthesis of new brush copolymers comprising poly(ω-pentadecalactone) with unusual thermal and crystalline properties. Macromolecules 2004, 37, 1243–1250. [Google Scholar] [CrossRef]
- Wilson, J.A.; Hopkins, S.A.; Wright, P.M.; Dove, A.P. Synthesis and postpolymerization modification of one-pot ω-pentadecalactone block-like copolymers. Biomacromolecules 2015, 16, 3191–3200. [Google Scholar] [CrossRef]
- Van der Meulen, I.; de Geus, M.; Antheunis, H.; Deumens, R.; Joosten, E.A.J.; Koning, C.E.; Heise, A. Polymers from functional macrolactones as potential biomaterials: Enzymatic ring opening polymerization, biodegradation, and biocompatibility. Biomacromolecules 2008, 9, 3404–3410. [Google Scholar] [CrossRef]
- Ates, Z.; Heise, A. Functional films from unsaturated poly(macrolactones) by thiol–ene cross-linking and functionalisation. Polym. Chem. 2014, 5, 2936. [Google Scholar] [CrossRef]
- De Oliveira, F.C.S.; Olvera, D.; Sawkins, M.J.; Cryan, S.-A.; Kimmins, S.D.; da Silva, T.E.; Kelly, D.J.; Duffy, G.P.; Kearney, C.; Heise, A. Direct UV-Triggered thiol–ene cross-linking of electrospun polyester fibers from unsaturated poly(macrolactone)s and their drug loading by solvent swelling. Biomacromolecules 2017, 18, 4292–4298. [Google Scholar] [CrossRef] [Green Version]
- Tinajero-Díaz, E.; Martínez de Ilarduya, A.; Cavanagh, B.; Heise, A.; Muñoz-Guerra, S. Poly(amino acid)-grafted polymacrolactones. Synthesis, self-assembling and ionic coupling properties. React. Funct. Polym. 2019, 143, 104316. [Google Scholar] [CrossRef]
- Naddeo, M.; D’Auria, I.; Viscusi, G.; Gorrasi, G.; Pellecchia, C.; Pappalardo, D. Tuning the thermal properties of poly(ethylene)-like poly(esters) by copolymerization of epsilon-caprolactone with macrolactones, in the presence of a pyridylamidozinc(II) complex. J. Polym. Sci. 2020, 58, 528–539. [Google Scholar] [CrossRef]
- Focarete, M.L.; Scandola, M.; Kumar, A.; Gross, R.A. Physical characterization of poly(ω-pentadecalactone) synthesized by lipase-catalyzed ring-opening polymerization. J. Polym. Sci. Part B Polym. Phys. 2001, 39, 1721–1729. [Google Scholar] [CrossRef]
- Polloni, A.E.; Chiaradia, V.; Figura, E.M.; De Paoli, J.P.; de Oliveira, D.; de Oliveira, J.V.; de Araujo, P.H.H.; Sayer, C. Polyesters from macrolactones using commercial lipase NS 88011 and Novozym 435 as biocatalysts. Appl. Biochem. Biotechnol. 2018, 184, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Ates, Z.; Audouin, F.; Harrington, A.; O’Connor, B.; Heise, A. Functional brush-decorated poly(globalide) films by ARGET-ATRP for bioconjugation. Macromol. Biosci. 2014, 14, 1600–1608. [Google Scholar] [CrossRef]
- Hunley, M.T.; Sari, N.; Beers, K.L. Microstructure analysis and model discrimination of enzyme-catalyzed copolyesters. ACS Macro Lett. 2013, 2, 375–379. [Google Scholar] [CrossRef]
- Tinajero-Díaz, E.; Martínez de Ilarduya, A.; Muñoz-Guerra, S. Block and graft copolymers made of 16-membered macrolactones and L-alanine: A comparative study. Macromol. Chem. Phys. 2019, 220, 1900214. [Google Scholar] [CrossRef]
- Watanabe, J.; Uematsu, I. Anomalous properties of poly(γ-benzyl L-glutamate) film composed of unusual 7/2 helices. Polymer 1984, 25, 1711–1717. [Google Scholar] [CrossRef]
- Rong, G.; Deng, M.; Deng, C.; Tang, Z.; Piao, L.; Chen, X.; Jing, X. Synthesis of poly(ε-caprolactone)-b-poly(γ-benzyl L-glutamic acid) block copolymer using amino organic calcium catalyst. Biomacromolecules 2003, 4, 1800–1804. [Google Scholar] [CrossRef]
- Schappacher, M.; Soum, A.; Guillaume, S.M. Synthesis of polyester−polypeptide diblock and triblock copolymers using amino poly(ε-caprolactone) macroinitiators. Biomacromolecules 2006, 7, 1373–1379. [Google Scholar] [CrossRef]
- Babin, J.; Rodriguez-Hernandez, J.; Lecommandoux, S.; Klok, H.-A.; Achard, M.-F. Self-assembled nanostructures from peptide–synthetic hybrid block copolymers: Complex, stimuli-responsive rod–coil architectures. Faraday Discuss. 2005, 128, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, P.; Floudas, G.; Klok, H.-A.; Schnell, I.; Pakula, T. Self-assembly and dynamics of poly(γ-benzyl L-glutamate) peptides. Biomacromolecules 2004, 5, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Klok, H.-A.; Langenwalter, J.F.; Lecommandoux, S. Self-assembly of peptide-based diblock oligomers. Macromolecules 2000, 33, 7819–7826. [Google Scholar] [CrossRef]
- Lu, H.; Wang, J.; Bai, Y.; Lang, J.W.; Liu, S.; Lin, Y.; Cheng, J. Ionic polypeptides with unusual helical stability. Nat. Commun. 2011, 2, 206. [Google Scholar] [CrossRef] [Green Version]
- Blout, E.R.; Asadourian, A. Polypeptides. V. The infrared spectra of polypeptides derived from γ-benzyl L-glutamate. J. Am. Chem. Soc. 1956, 78, 955–961. [Google Scholar] [CrossRef]
- Lecommandoux, S.; Achard, M.; Langenwalter, J.F.; Klok, H. Self-assembly of rod−coil diblock oligomers based on α-helical peptides. Macromolecules 2001, 34, 9100–9111. [Google Scholar] [CrossRef]
- Tinajero-Díaz, E.; Martínez de Ilarduya, A.; Muñoz-Guerra, S. Synthesis and properties of diblock copolymers of ω-pentadecalactone and α-amino acids. Eur. Polym. J. 2019, 116, 169–179. [Google Scholar] [CrossRef]
- Minich, E.A.; Nowak, A.P.; Deming, T.J.; Pochan, D.J. Rod–rod and rod–coil self-assembly and phase behavior of polypeptide diblock copolymers. Polymer 2004, 45, 1951–1957. [Google Scholar] [CrossRef]
- Caillol, S.; Lecommandoux, S.; Mingotaud, A.-F.; Schappacher, M.; Soum, A.; Bryson, N.; Meyrueix, R. Synthesis and self-assembly properties of peptide−polylactide block copolymers. Macromolecules 2003, 36, 1118–1124. [Google Scholar] [CrossRef]
- Rivankar, S. An overview of doxorubicin formulations in cancer therapy. J. Cancer Res. Ther. 2014, 10, 853. [Google Scholar] [CrossRef]
- Li, M.; Song, W.; Tang, Z.; Lv, S.; Lin, L.; Sun, H.; Li, Q.; Yang, Y.; Hong, H.; Chen, X. Nanoscaled poly(L-glutamic acid)/doxorubicin-amphiphile complex as pH-responsive drug delivery system for effective treatment of nonsmall cell lung cancer. ACS Appl. Mater. Interfaces 2013, 5, 1781–1792. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, L.; Liu, M.; Le, Y.; Lv, S.; Wang, J.; Chen, J.-F. Dual-responsive star-shaped polypeptides for drug delivery. RSC Adv. 2016, 6, 6368–6377. [Google Scholar] [CrossRef]
- Du, Y.; Chen, W.; Zheng, M.; Meng, F.; Zhong, Z. pH-Sensitive degradable chimaeric polymersomes for the intracellular release of doxorubicin hydrochloride. Biomaterials 2012, 33, 7291–7299. [Google Scholar] [CrossRef] [PubMed]
- Nukolova, N.V.; Oberoi, H.S.; Cohen, S.M.; Kabanov, A.V.; Bronich, T.K. Folate-decorated nanogels for targeted therapy of ovarian cancer. Biomaterials 2011, 32, 5417–5426. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.-C.; Chiang, W.-H.; Huang, Y.-F.; Lin, S.-C.; Shih, Z.-F.; Chern, C.-S.; Chiang, C.-S.; Chiu, H.-C. Nano-scaled pH-responsive polymeric vesicles for intracellular release of doxorubicin. J. Drug Target. 2011, 19, 944–953. [Google Scholar] [CrossRef]
- Manocha, B.; Margaritis, A. Controlled release of doxorubicin from doxorubicin/γ-polyglutamic acid ionic complex. J. Nanomater. 2010, 2010, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Bromberg, L.; Lin, S.N.; Hatton, T.A.; Tam, K.C. Complexation and release of doxorubicin from its complexes with pluronic P85-b-poly(acrylic acid) block copolymers. J. Control. Release 2007, 121, 137–145. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polyester a | Yield (%) | Composition b [Gl]/[PDL] (mol/mol) | Mnc (g·mol−1) | |
|---|---|---|---|---|
| Feed | Copolymer | |||
| PGl | 70 | 100/0 | 100/0 | 9700 |
| P(Gl82-r-PDL18) | 80 | 90/10 | 82/18 | 10,500 |
| P(Gl73-r-PDL27) | 75 | 70/30 | 73/27 | 9450 |
| P(Gl48-r-PDL52) | 88 | 50/50 | 48/52 | 11,900 |
| P(Gl33-r-PDL67) | 80 | 30/70 | 33/67 | 10,650 |
| P(Gl13-r-PDL87) | 90 | 10/90 | 13/87 | 11,760 |
| PPDL | 90 | 0/100 | 0/100 | 12,300 |
| Copolyester | TGA a | DSC b | |||||||
|---|---|---|---|---|---|---|---|---|---|
| First Heating | Cooling | Second Heating | |||||||
| Tda (°C) | maxTd (°C) | Rw (%) | Tm (°C) | ΔHm (J·g−1) | Tc (°C) | ΔHc (J·g−1) | Tm (°C) | ΔHm (J·g−1) | |
| PGl | 349 | 419, 466 | 4 | 42 | 48 | 17 | −35 | 27 | 43 |
| P(Gl82-r-PDL18) | 376 | 417, 466 | 2 | 54 | 60 | 38 | −40 | 53 | 55 |
| P(Gl73-r-PDL27) | 387 | 420, 469 | 1 | 65 | 89 | 50 | −46 | 66 | 66 |
| P(Gl48-r-PDL52) | 378 | 423, 470 | 1 | 74 | 120 | 57 | −76 | 74 | 91 |
| P(Gl33-r-PDL67) | 374 | 431, 469 | 1 | 81 | 124 | 65 | −93 | 80 | 97 |
| P(Gl13-r-PDL87) | 381 | 426, 470 | 1 | 90 | 137 | 74 | −101 | 90 | 107 |
| PPDL | 396 | 427, 472 | 1 | 95 | 161 | 81 | −130 | 95 | 136 |
| Copolymer a | Gl-BAET b (%) | Copolymer c Gl/PDL/LGlu | Side Chain Length d (LGlu Units) | Yield e (%) | Mnf (g·mol−1) |
|---|---|---|---|---|---|
| P[(Gl13-r-PDL87)–g–(BLG)10] | 90 | 6/38/56 | 10 | 93 | 27,700 |
| P[(Gl48-r-PDL52)–g–(BLG)2] | 50 | 32/36/32 | 2 | 85 | 16,700 |
| P[(Gl13-r-PDL87)–g–(LGA)10] | 90 | 6/38/56 | 10 | 70 | 21,400 |
| P[(Gl48-r-PDL52)–g–(LGA)2] | 50 | 32/36/32 | 2 | 74 | 15,100 |
| Copolymer | TGA a | DSC b | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| First Heating | Cooling | Second Heating | |||||||||
| Td °C | maxTd °C | Rw % | Tm °C | ΔH J·g−1 | TLCc °C | ΔH J·g−1 | Tc °C | ΔH J·g−1 | Tm °C | ΔH J·g−1 | |
| P[(Gl13-r-PDL87)–g–(BLG)10] | 235 | 230–280 | 18 | 52,76 | 4 | 115–120 | 2 | 34 | −6 | 75 | 6 |
| P[(Gl48-r-PDL52)–g–(BLG)2] | 220 | 290–320 | 12 | 68 | 21 | - | - | 43 | −13 | 68 | 16 |
| P[(Gl13-r-PDL87)–g–(LGA)10] | 285 | 310–450 | 42 | 80–90 | 9 | - | - | 58 | −0.5 | 83 | 1 |
| P[(Gl48-r-PDL52)–g–(LGA)2] | 215 | 270–400 | 29 | 72 | 18 | - | - | 48 | −9 | 67 | 11 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tinajero-Díaz, E.; Martínez de Ilarduya, A.; Muñoz-Guerra, S. Copolymacrolactones Grafted with l-Glutamic Acid: Synthesis, Structure, and Nanocarrier Properties. Polymers 2020, 12, 995. https://doi.org/10.3390/polym12040995
Tinajero-Díaz E, Martínez de Ilarduya A, Muñoz-Guerra S. Copolymacrolactones Grafted with l-Glutamic Acid: Synthesis, Structure, and Nanocarrier Properties. Polymers. 2020; 12(4):995. https://doi.org/10.3390/polym12040995
Chicago/Turabian StyleTinajero-Díaz, Ernesto, Antxon Martínez de Ilarduya, and Sebastián Muñoz-Guerra. 2020. "Copolymacrolactones Grafted with l-Glutamic Acid: Synthesis, Structure, and Nanocarrier Properties" Polymers 12, no. 4: 995. https://doi.org/10.3390/polym12040995
APA StyleTinajero-Díaz, E., Martínez de Ilarduya, A., & Muñoz-Guerra, S. (2020). Copolymacrolactones Grafted with l-Glutamic Acid: Synthesis, Structure, and Nanocarrier Properties. Polymers, 12(4), 995. https://doi.org/10.3390/polym12040995