Curing Behaviors of Alkynyl-Terminated Copolyether with Glycidyl Azide Polymer in Energetic Plasticizers


Abstract
:1. Introduction
2. Experimental
2.1. Materials and Methods
2.2. Measurements and Analysis
3. Results and Discussion
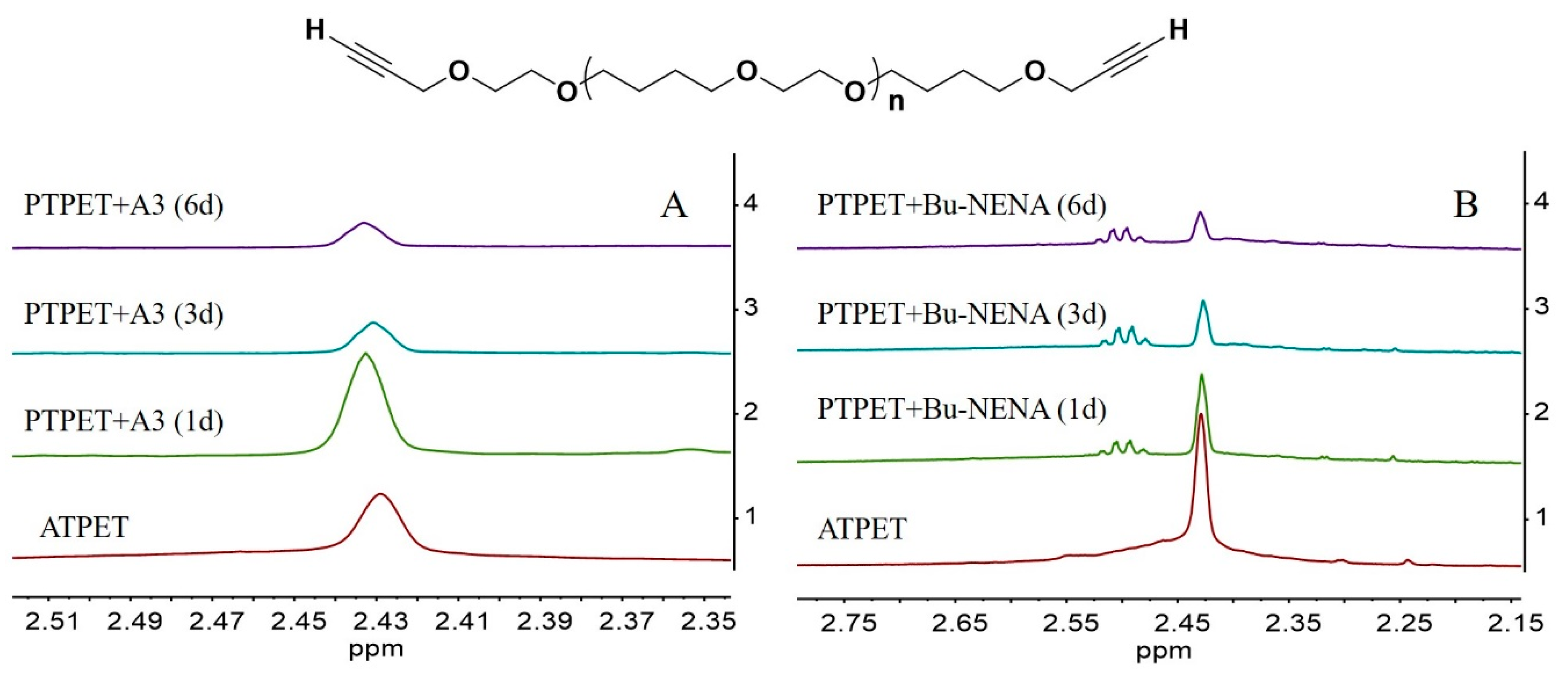
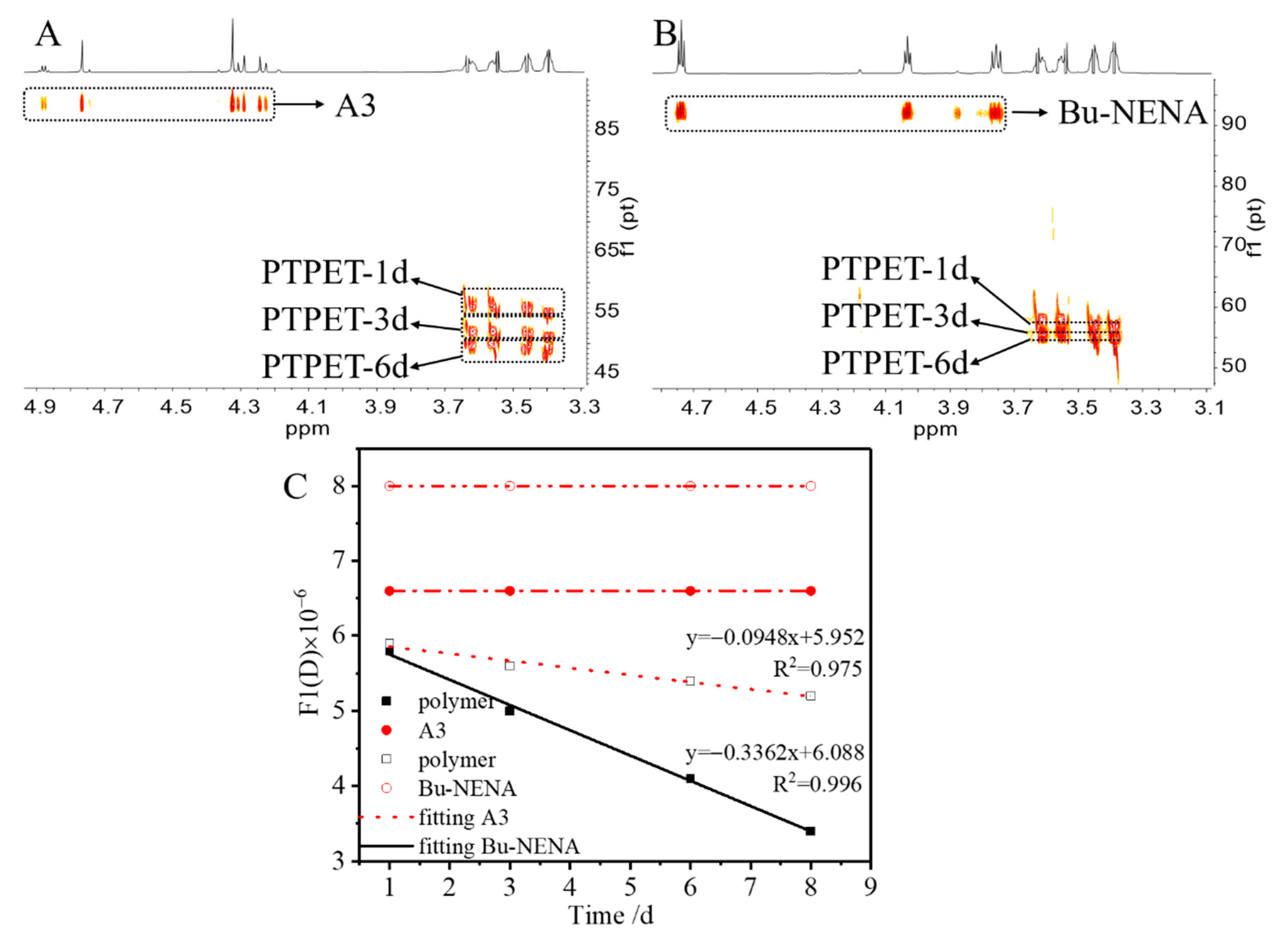
3.1. H-NMR and DOSY Analysis
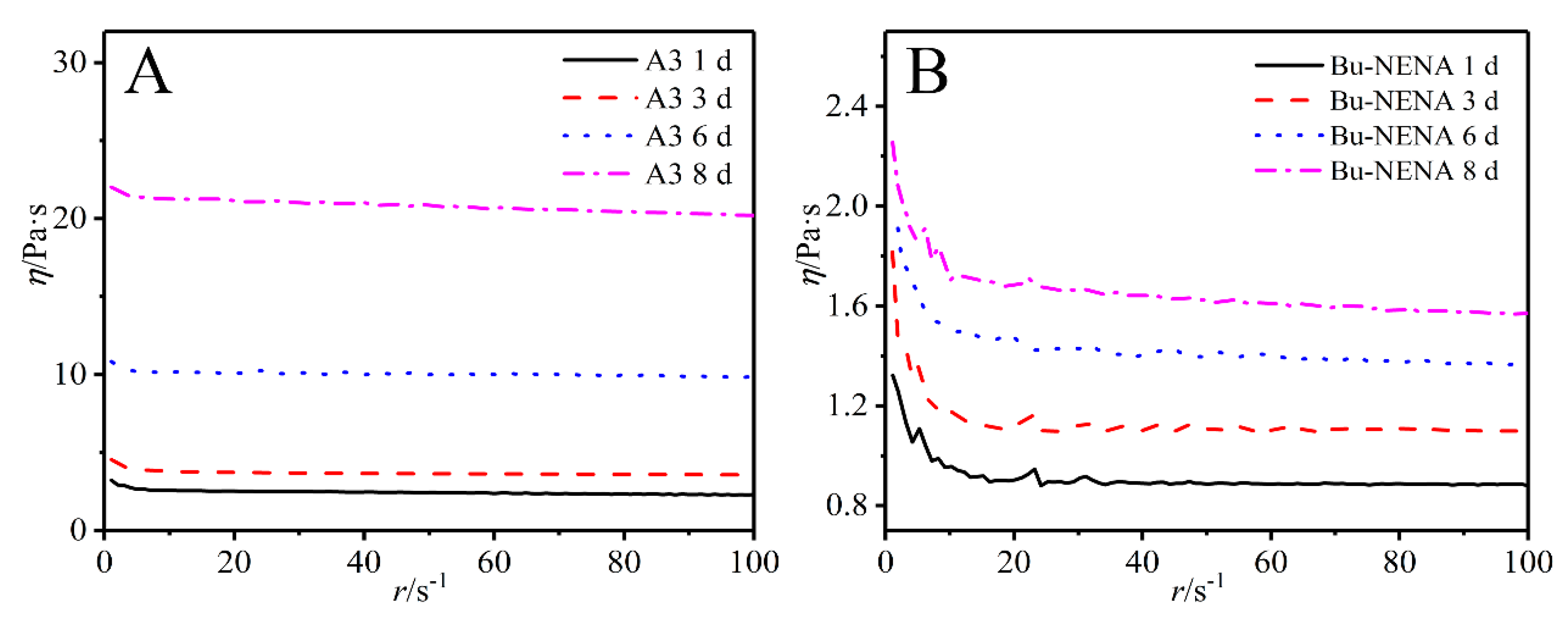
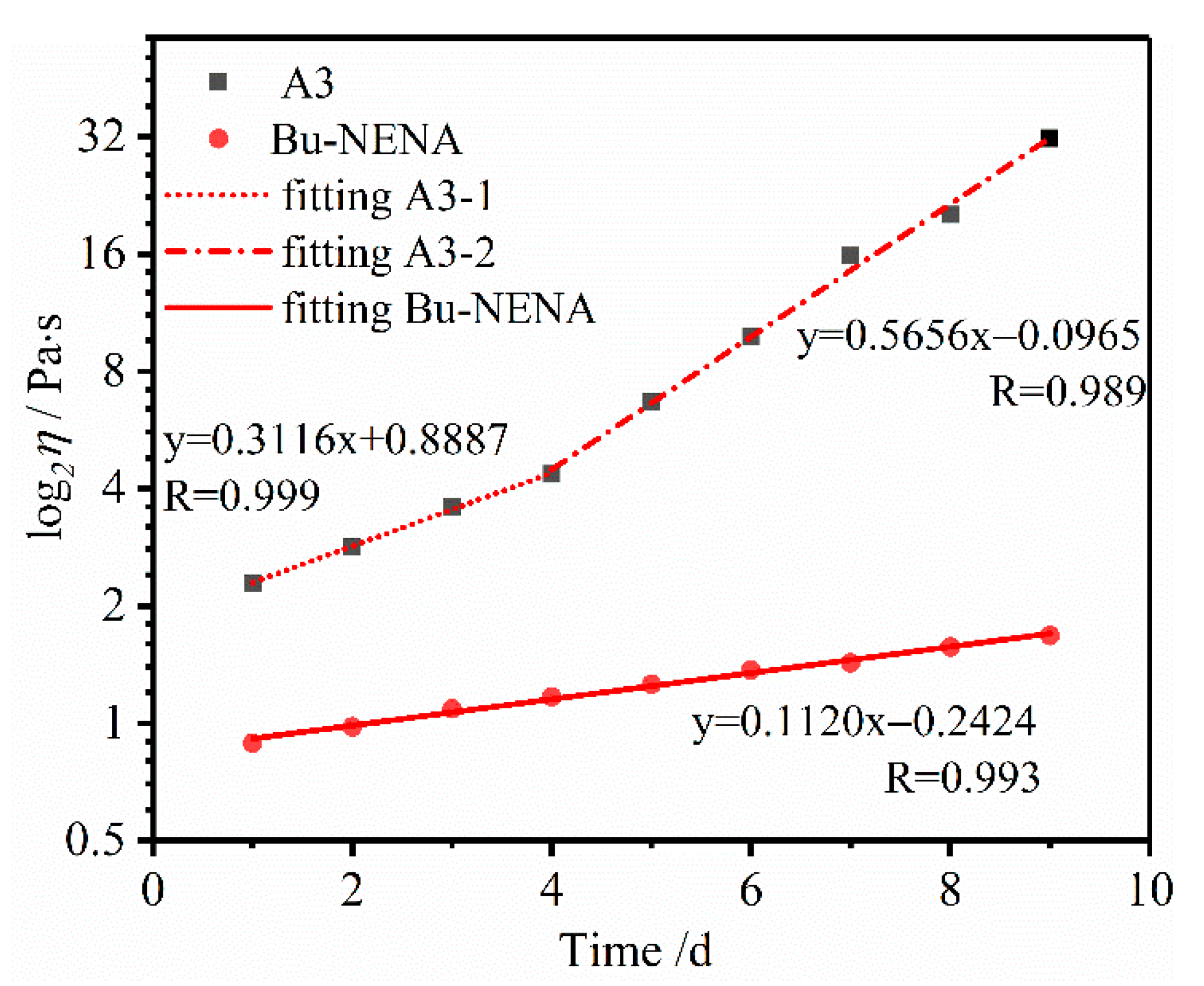
3.2. Viscosity Analysis
3.3. Gel Permeation Chromatography (GPC) Analysis
3.4. X-ray Photoelectron Spectroscopy Analysis
3.5. Solubility Parameter Analysis
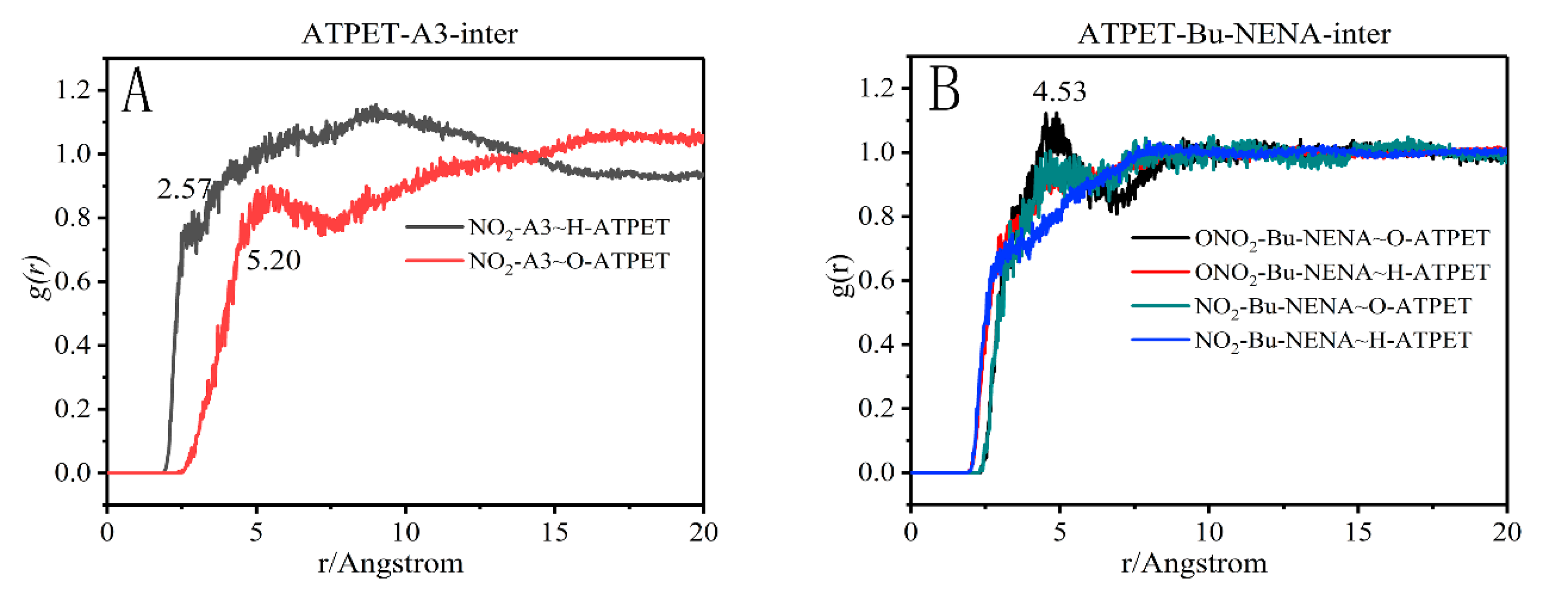
3.6. Radial Distribution Function Analysis
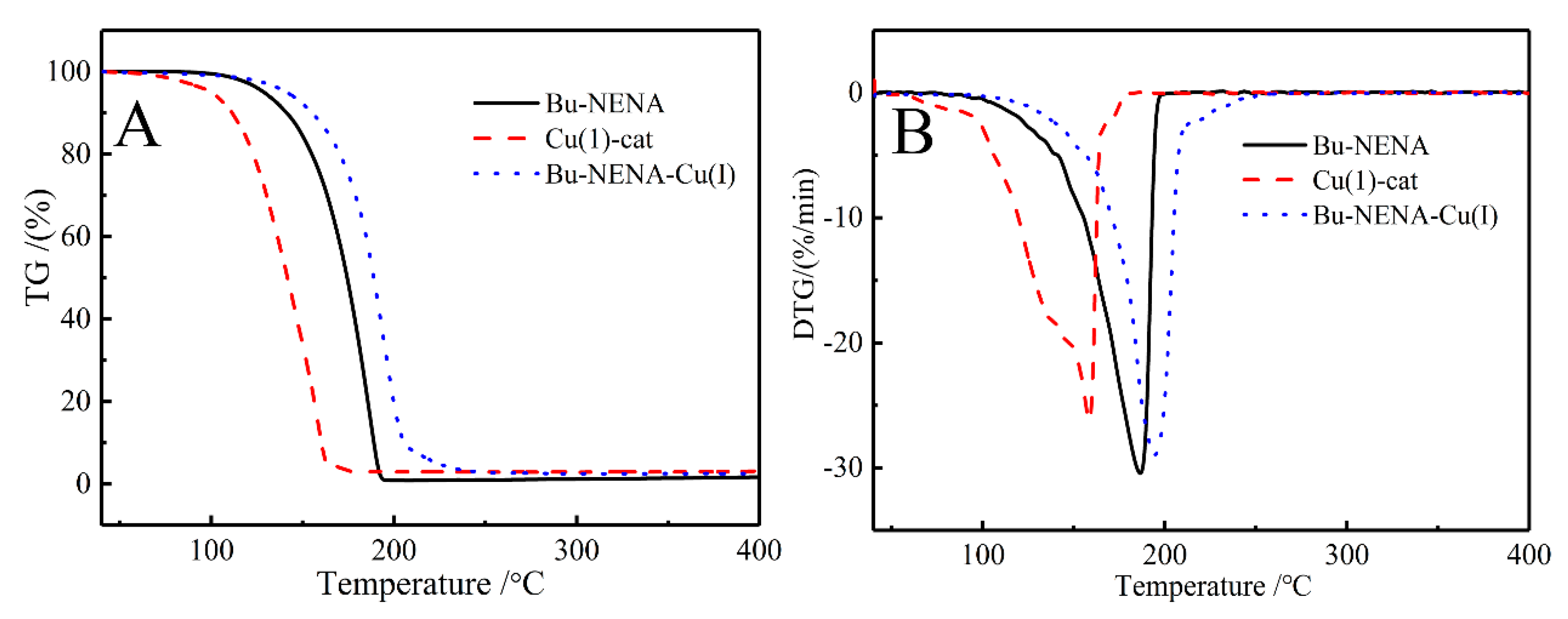
3.7. Thermogravimetric (TG) Analysis
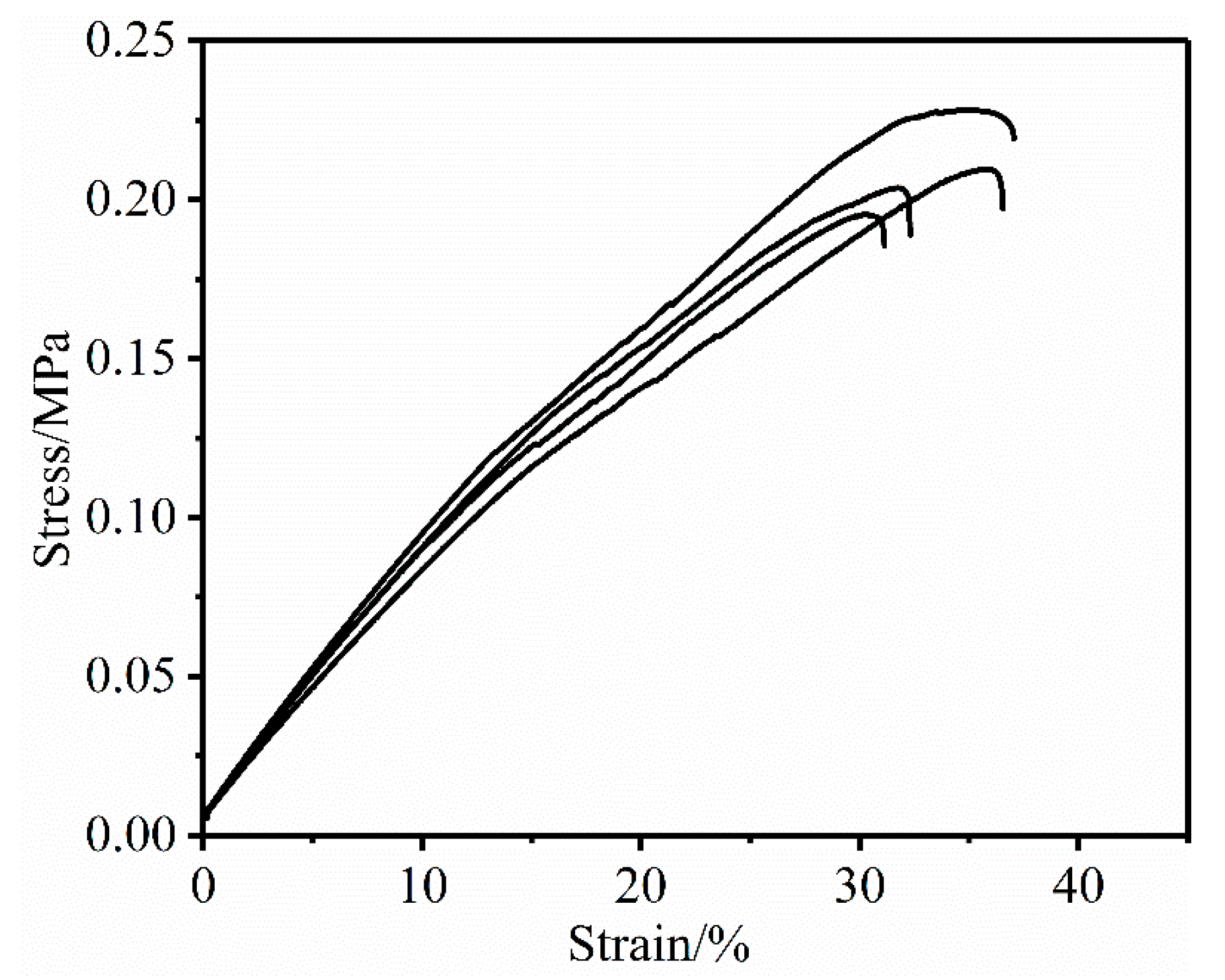
3.8. An Alternative Method
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Damse, R.S.; Singh, A. Evaluation of energetic plasticisers for solid gun propellant. Def. Sci. J. 2008, 58, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Damse, R.S.; Omprakash, B.; Tope, B.G.; Chakraborthy, T.K.; Singh, A. Study of N-n-butyl-N-(2-nitroxyethyl)nitramine in RDX based gun propellant. J. Hazard. Mater. 2009, 167, 1222–1225. [Google Scholar] [CrossRef]
- Wang, W.; Zhou, J. Review on Synthesis of BDNPA/F Plasticizer and its Development. Chin. J. Energ. Mater. 2007, 1, 90–93. [Google Scholar]
- Zhao, B.; Xia, M.; Huang, J.; Li, W.; Liu, Q.; Luo, Y. Characterization of Bu-NENA/NC Low Sensitive Double-base Propellant. Chin. J. Energ. Mater. 2017, 25, 794–798. [Google Scholar]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004. [Google Scholar] [CrossRef]
- Demko, Z.P.; Sharpless, K.B. A click chemistry approach to tetrazoles by Huisgen 1,3-dipolar cycloaddition: Synthesis of 5-sulfonyl tetrazoles from azides and sulfonyl cyanides. Angew. Chem. Int. Ed. 2002, 41, 2110–2113. [Google Scholar] [CrossRef]
- Diaz, D.D.; Punna, S.; Holzer, P.; McPherson, A.K.; Sharpless, K.B.; Fokin, V.V.; Finn, M.G. Click chemistry in materials synthesis. 1. Adhesive polymers from copper-catalyzed azide-alkyne cycloaddition. J. Polym. Sci. Part A-Polym. Chem. 2004, 42, 4392–4403. [Google Scholar] [CrossRef]
- Hawker, C.J.; Fokin, V.V.; Finn, M.G.; Sharpless, K.B. Bringing efficiency to materials synthesis: The philosophy of click chemistry. Aust. J. Chem. 2007, 60, 381–383. [Google Scholar] [CrossRef]
- Castro, V.; Rodriguez, H.; Albericio, F. CuAAC: An efficient click chemistry reaction on solid phase. ACS Comb. Sci. 2016, 18, 1–14. [Google Scholar] [CrossRef]
- Meldal, M. Polymer “Clicking” by CuAAC reactions. Macromol. Rapid Commun. 2008, 29, 1016–1051. [Google Scholar] [CrossRef]
- Moses, J.E.; Moorhouse, A.D. The growing applications of click chemistry. Chem. Soc. Rev. 2007, 36, 1249–1262. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Zhai, J.; Yang, R. Comparison between properties of polyether polytriazole elastomers and polyether polyurethane elastomers. Polym. Adv. Technol. 2014, 25, 314–321. [Google Scholar] [CrossRef]
- Hu, J.; Li, Y.; Xiao, F.; Zhang, Y.; He, J.; Yang, R. Thermal degradation and aging behavior of polytriazole polyethylene oxide-tetrahydrofuran elastomer based on click-chemistry. J. Appl. Polym. Sci. 2020, 48974. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Burnham, A.K.; Criado, J.M.; Pérez-Maqueda, L.A.; Sbirrazzuoli, N. ICTAC Kinetics Commitee Recommendations for Performing Kinetic Computations on Thermal Analysis Data. Thermochim. Acta 2011, 520, 1–19. [Google Scholar] [CrossRef]
- Jouyandeh, M.; Paran, S.M.R.; Jannesari, A.; Puglia, D.; Saeb, M.R. Protocol for nonisothermal cure analysis of thermoset composites. Prog. Org. Coat. 2019, 131, 333–339. [Google Scholar] [CrossRef]
- Avram, L.; Cohen, Y. Diffusion NMR of molecular cages and capsules. Chem. Soc. Rev. 2015, 44, 586–602. [Google Scholar] [CrossRef] [PubMed]
- Bellachioma, G.; Ciancaleoni, G.; Zuccaccia, C.; Zuccaccia, D.; Macchioni, A. NMR investigation of non-covalent aggregation of coordination compounds ranging from dimers and ion pairs up to nano-aggregates. Coord. Chem. Rev. 2008, 252, 2224–2238. [Google Scholar] [CrossRef]
- Zaccaria, F.; Zuccaccia, C.; Cipullo, R.; Macchioni, A. Extraction of Reliable Molecular Information from Diffusion NMR Spectroscopy: Hydrodynamic Volume or Molecular Mass? Chem. -A Eur. J. 2019, 25, 9930–9937. [Google Scholar] [CrossRef]
- Pages, G.; Gilard, V.; Martino, R.; Malet-Martino, M. Pulsed-field gradient nuclear magnetic resonance measurements (PFG NMR) for diffusion ordered spectroscopy (DOSY) mapping. Analyst 2017, 142, 3771–3796. [Google Scholar] [CrossRef]
- Pregosin, P.S. Applications of NMR diffusion methods with emphasis on ion pairing in inorganic chemistry: A mini-review. Magn. Reson. Chem. 2017, 55, 405–413. [Google Scholar] [CrossRef]
- Cohen, Y.; Avram, L.; Frish, L. Diffusion NMR spectroscopy in supramolecular and combinatorial chemistry: An old parameter—New insights. Angew. Chem. Int. Ed. 2005, 44, 520–554. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, G.; Li, J.; Luo, Y. Preparation and properties of semi-interpenetrating networks combined by thermoplastic polyurethane and a thermosetting elastomer. New J. Chem. 2018, 42, 3087–3096. [Google Scholar] [CrossRef]
- Zou, Y.; Yang, R.; Zhai, J. Polytriazole polyether elastomers with widely tunable mechanical properties: The role of network structure and crystallization behavior. J. Appl. Polym. Sci. 2017, 134. [Google Scholar] [CrossRef]
- Lan, Y.; Li, D.; Zhai, J.; Yang, R. Molecular Dynamics Simulation on the Binder of Ethylene Oxide-Tetrahydrofuran Copolyether Cross-Linked with N100. Ind. Eng. Chem. Res. 2015, 54, 3563–3569. [Google Scholar] [CrossRef]
- Le Gresley, A.; Broadberry, G.; Robertson, C.; Peron, J.-M.R.; Robinson, J.; O’Leary, S. Application of pure shift and diffusion NMR for the characterisation of crude and processed pyrolysis oil. J. Anal. Appl. Pyrolysis 2019, 140, 281–289. [Google Scholar] [CrossRef] [Green Version]
- Dudas, E.F.; Bodor, A. Quantitative, Diffusion NMR Based Analytical Tool To Distinguish Folded, Disordered, and Denatured Biomolecules. Anal. Chem. 2019, 91, 4929–4933. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Laryea, E.; Wilhelm, M.; Luy, B.; Nirschl, H.; Guthausen, G. Diffusion in Polymer Solutions: Molecular Weight Distribution by PFG-NMR and Relation to SEC. Macromol. Chem. Phys. 2017, 218. [Google Scholar] [CrossRef]
- Berry, G.C. Translational frictional constant of comb-branched polymers. J. Polym. Sci. Part A-2 Polym. Phys. 1968, 6, 1551. [Google Scholar] [CrossRef]
- Ajroldi, G.; Marchionni, G.; Pezzin, G. The viscosity-molecular weight relationships for diolic perfluoropolyethers. Polymer 1999, 40, 4163–4164. [Google Scholar] [CrossRef]
- Wanger, C.D.; Riggs, W.M.; Davis, L.E.; Moulder, G.F. Handbook of X-ray Photoelectron Spectroscopy; Heyden & Son Ltd.: London, UK, 1979. [Google Scholar] [CrossRef]
- Sperling, L.H. Introduction to Physical Polymer Science, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2005. [Google Scholar] [CrossRef]
- Steiner, T.; Desiraju, G.R. Distinction between the weak hydrogen bond and the van der Waals interaction. Chem. Commun. 1998, 8, 891–892. [Google Scholar] [CrossRef]
- Fu, X.; Fan, X.; Ju, X.; Qi, X.; Li, J.; Yu, H. Molecular dynamic simulations on the interaction between an HTPE polymer and energetic plasticizers in a solid propellant. RSC Adv. 2015, 5, 52844–52851. [Google Scholar] [CrossRef]
- Tran, T.V.; Couture, G.; Do, L.H. Evaluation of dicopper azacryptand complexes in aqueous CuAAC reactions and their tolerance toward biological thiols. Dalton Trans. 2019. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Tang, W.; Li, Y.; He, J.; Guo, X.; Yang, R. The Effect of Glycidyl Azide Polymer Grafted Tetrafunctional Isocyanate on Polytriazole Polyethylene Oxide-Tetrahydrofuran Elastomer and its Propellant Properties. Polymers 2020, 12, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y. Research on the Mechanical Properties of Azide Polyuretgane Elastomers and Its Propellant. Ph.D. Thesis, Nanjing University of Science and Technology, Nanjing, China, 2018. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A3 Sample | Mn (g/mol) | Mw (g/mol) | PDI | Bu-NENA Sample | Mn (g/mol) | Mw (g/mol) | PDI |
|---|---|---|---|---|---|---|---|
| 1 d | 5341 | 10,159 | 1.66 | 1 d | 5277 | 9957 | 1.68 |
| 3 d | 5498 | 10,379 | 1.68 | 3 d | 5343 | 10,748 | 1.69 |
| 4 d | 5915 | 11,535 | 1.77 | 4 d | 5376 | 10,793 | 1.72 |
| 6 d | 6236 | 12,128 | 1.95 | 6 d | 5897 | 11,494 | 1.81 |
| 8 d | 6343 | 12,191 | 2.38 | 8 d | 5947 | 11,546 | 1.83 |
| Material | m (Bu-NENA:ATPET) = 1:1 | m (A3/ATPET) = 1:1 | ATPET |
|---|---|---|---|
| CED/(J cm−3) | 426.2 | 423.9 | 338.9 |
| CEDv/(J cm−3) | 335.3 | 297.6 | 296.3 |
| CEDe/(J cm−3) | 74.66 | 110.5 | 263.5 |
| δ/(J cm−3)0.5 | 20.65 | 20.59 | 18.40 |
| δv/(J cm−3)0.5 | 18.31 | 17.25 | 17.21 |
| δe/(J cm−3)0.5 | 8.64 | 10.51 | 5.13 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, J.; Liu, Y.; Cong, K.; He, J.; Yang, R. Curing Behaviors of Alkynyl-Terminated Copolyether with Glycidyl Azide Polymer in Energetic Plasticizers. Polymers 2020, 12, 1199. https://doi.org/10.3390/polym12051199
Hu J, Liu Y, Cong K, He J, Yang R. Curing Behaviors of Alkynyl-Terminated Copolyether with Glycidyl Azide Polymer in Energetic Plasticizers. Polymers. 2020; 12(5):1199. https://doi.org/10.3390/polym12051199
Chicago/Turabian StyleHu, Jinghui, Yina Liu, Kun Cong, Jiyu He, and Rongjie Yang. 2020. "Curing Behaviors of Alkynyl-Terminated Copolyether with Glycidyl Azide Polymer in Energetic Plasticizers" Polymers 12, no. 5: 1199. https://doi.org/10.3390/polym12051199
APA StyleHu, J., Liu, Y., Cong, K., He, J., & Yang, R. (2020). Curing Behaviors of Alkynyl-Terminated Copolyether with Glycidyl Azide Polymer in Energetic Plasticizers. Polymers, 12(5), 1199. https://doi.org/10.3390/polym12051199