Straight Versus Branched Chain Substituents in 4′-(Butoxyphenyl)-3,2′:6′,3″-terpyridines: Effects on (4,4) Coordination Network Assemblies




Abstract
:
1. Introduction
2. Materials and Methods
2.1. General
2.2. Compound rac-2
2.3. Compound 4
2.4. Compound 3a
2.5. Compound 3
2.6. Crystal Growth of [{Co(rac-2)2(NCS)2}·CHCl3]n
2.7. Crystal Growth of [Co(3)2(NCS)2]n
2.8. Crystal Growth of [{Co(4)2(NCS)2}·CHCl3]n
2.9. Crystallography
2.10. [{Co(rac-2)2(NCS)2}·CHCl3]n
2.11. [Co(3)2(NCS)2]n
2.12. [{Co(4)2(NCS)2}·CHCl3]n
3. Results and Discussion
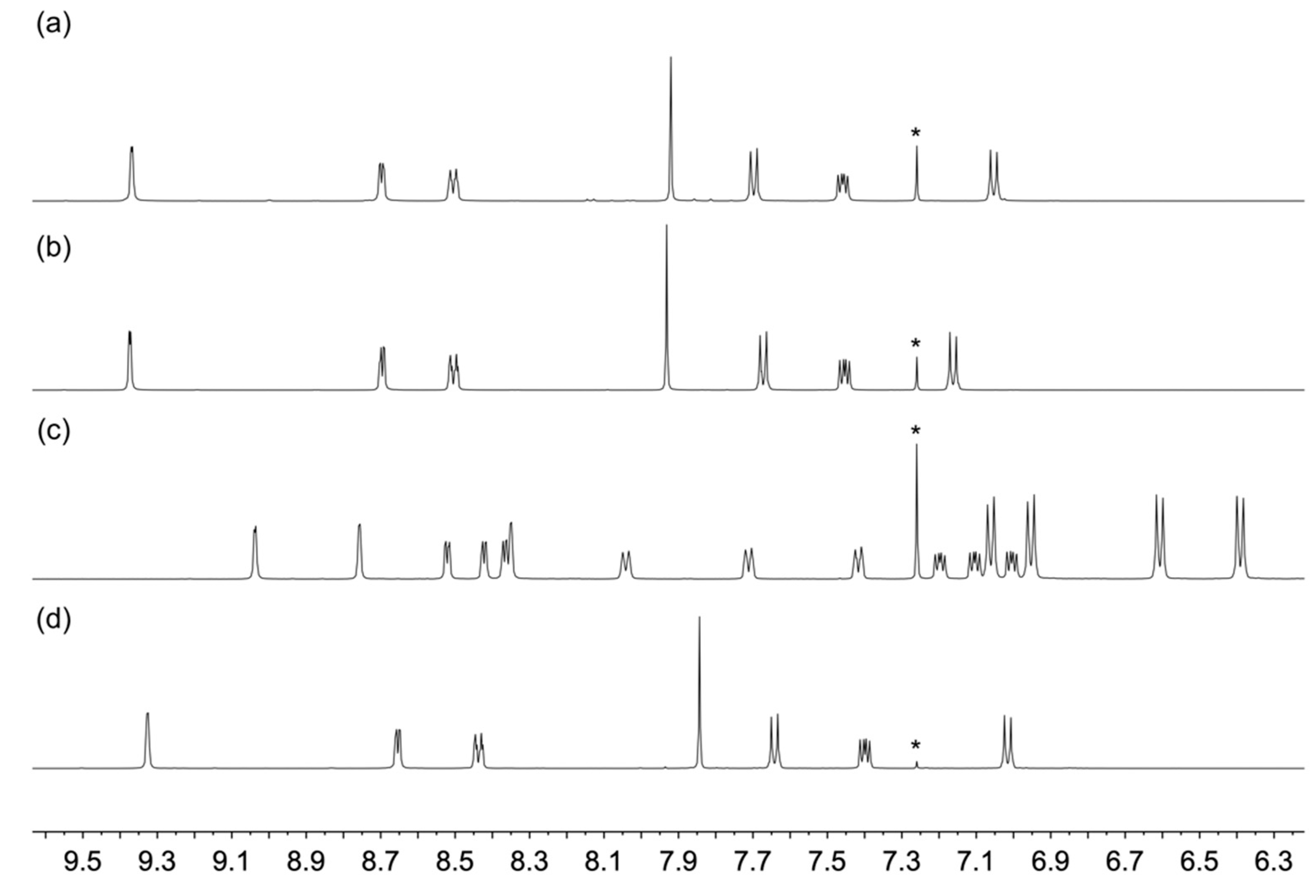
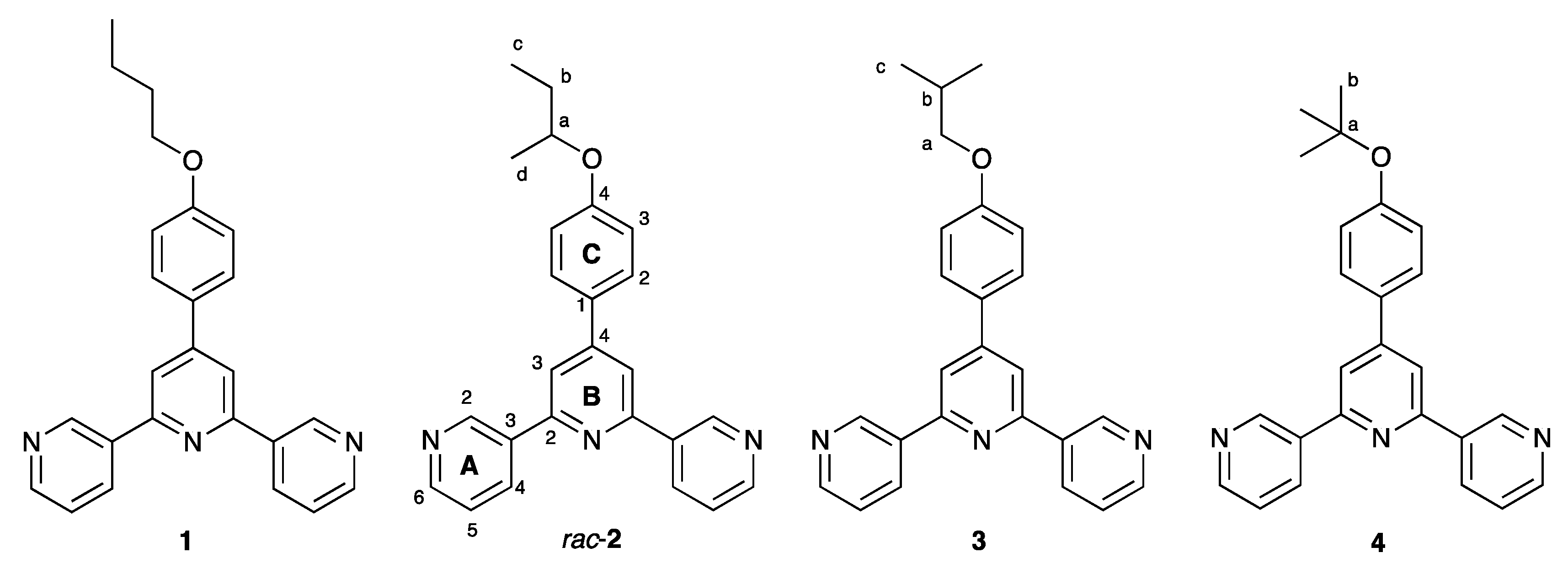
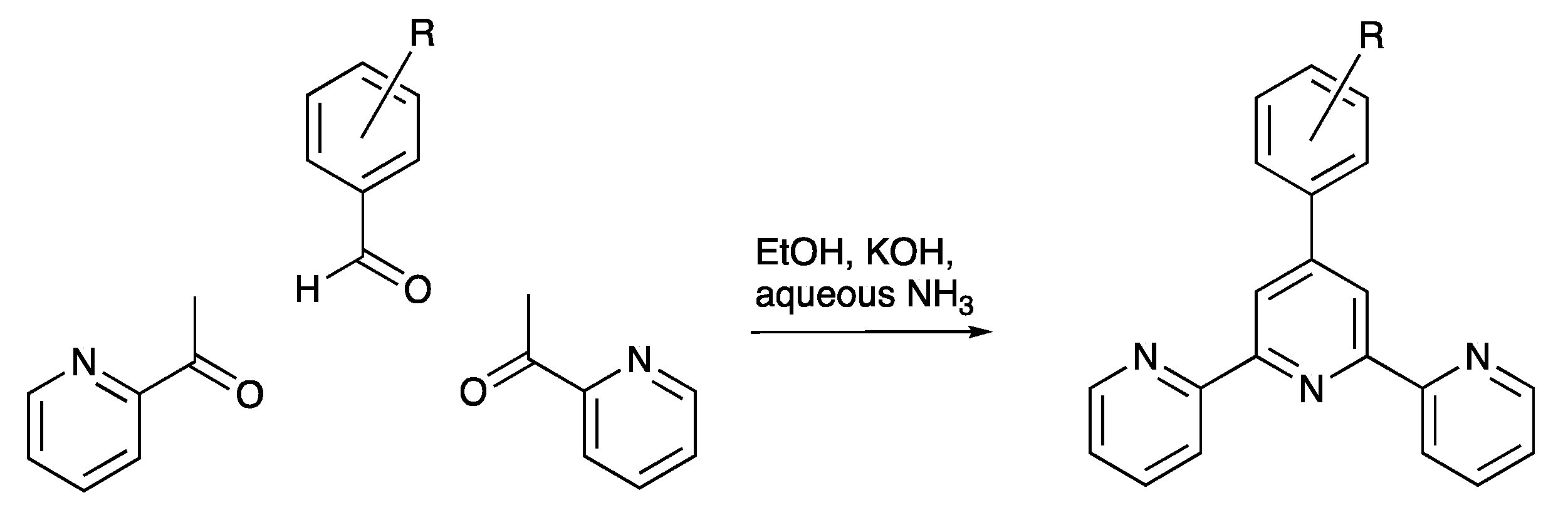
3.1. Synthesis and Characterization of Compounds rac-2, 3 and 4
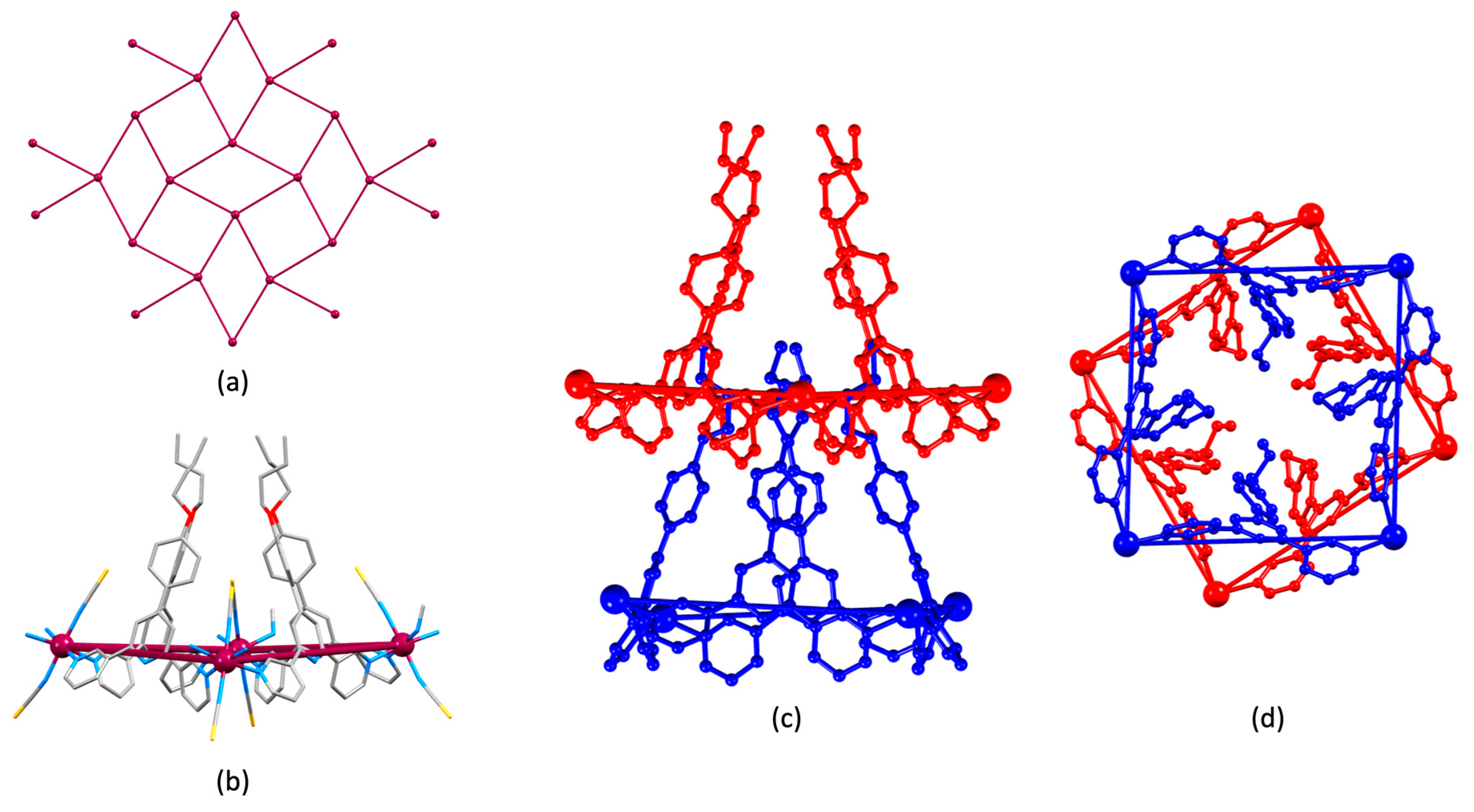
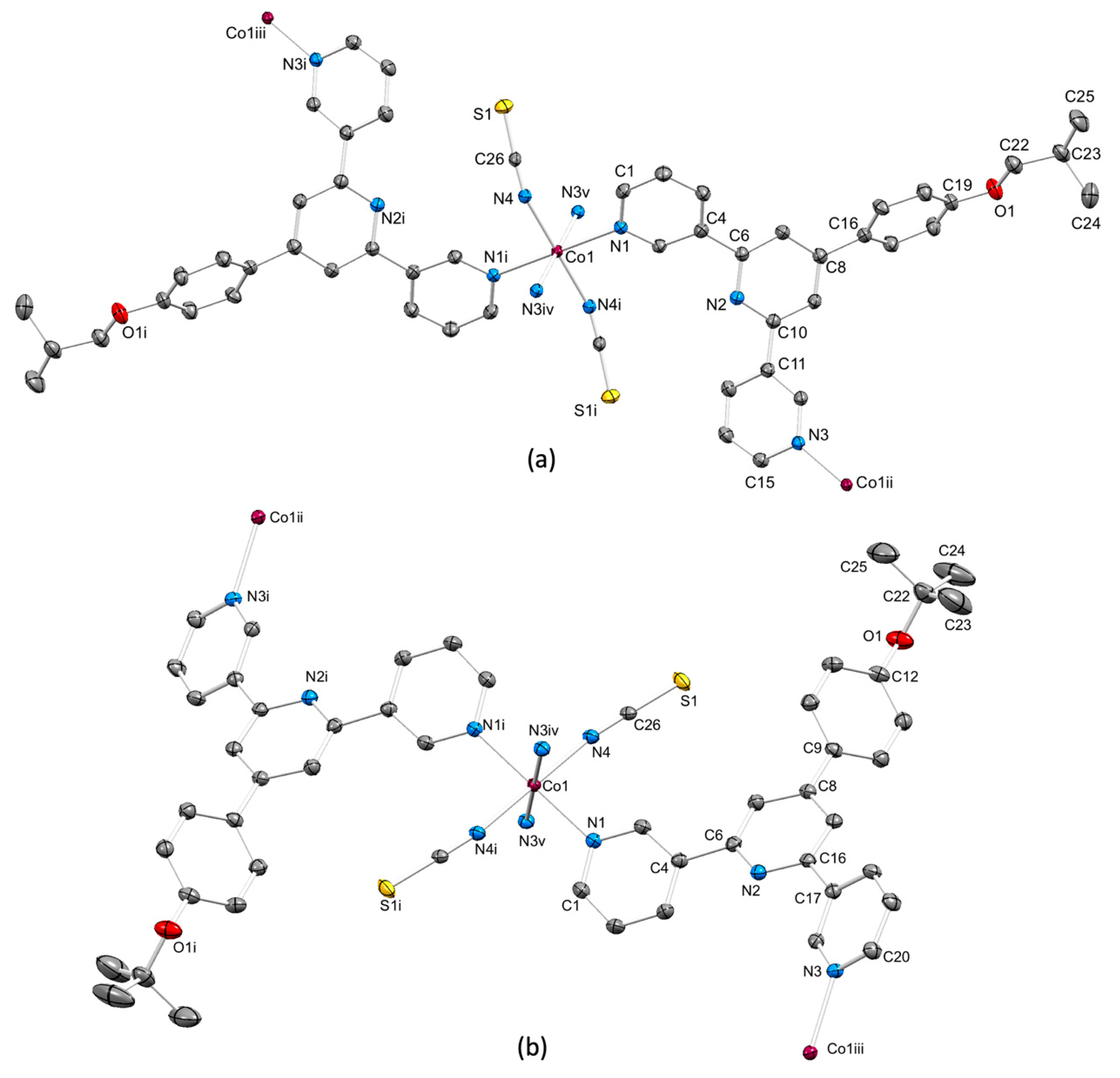
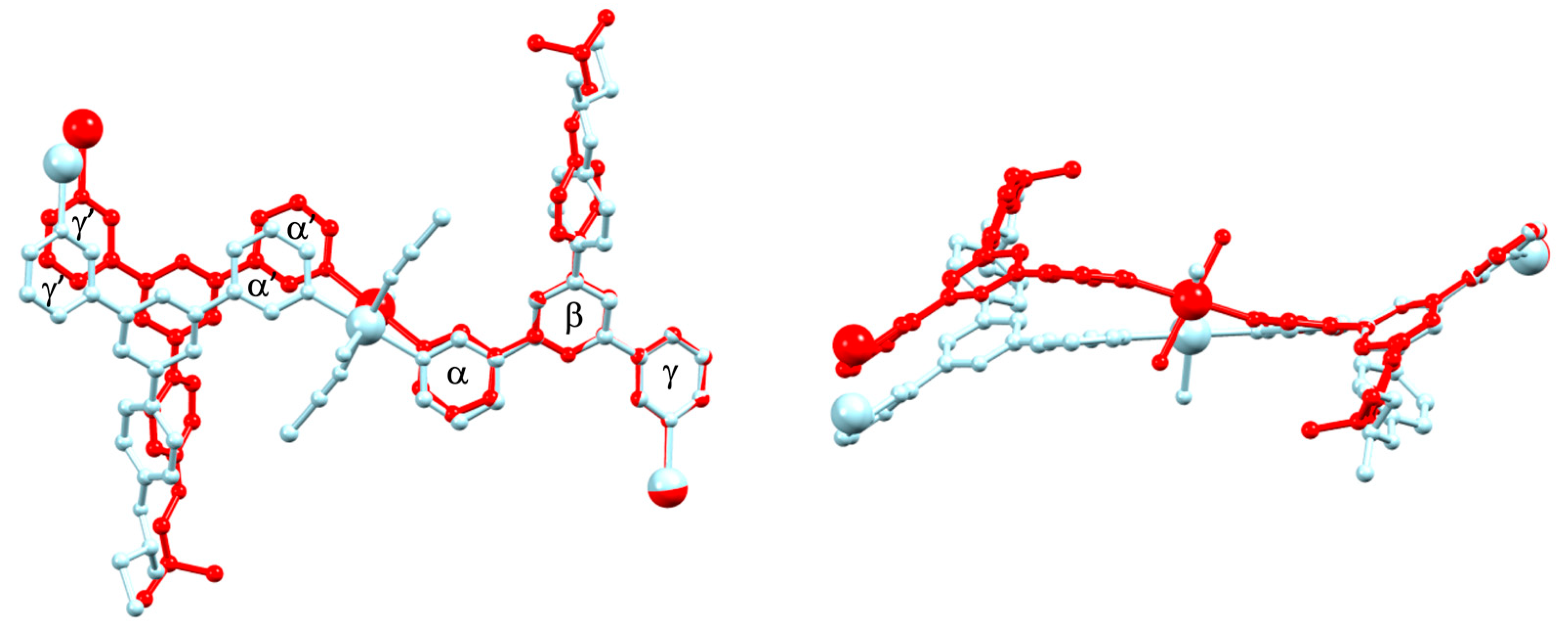
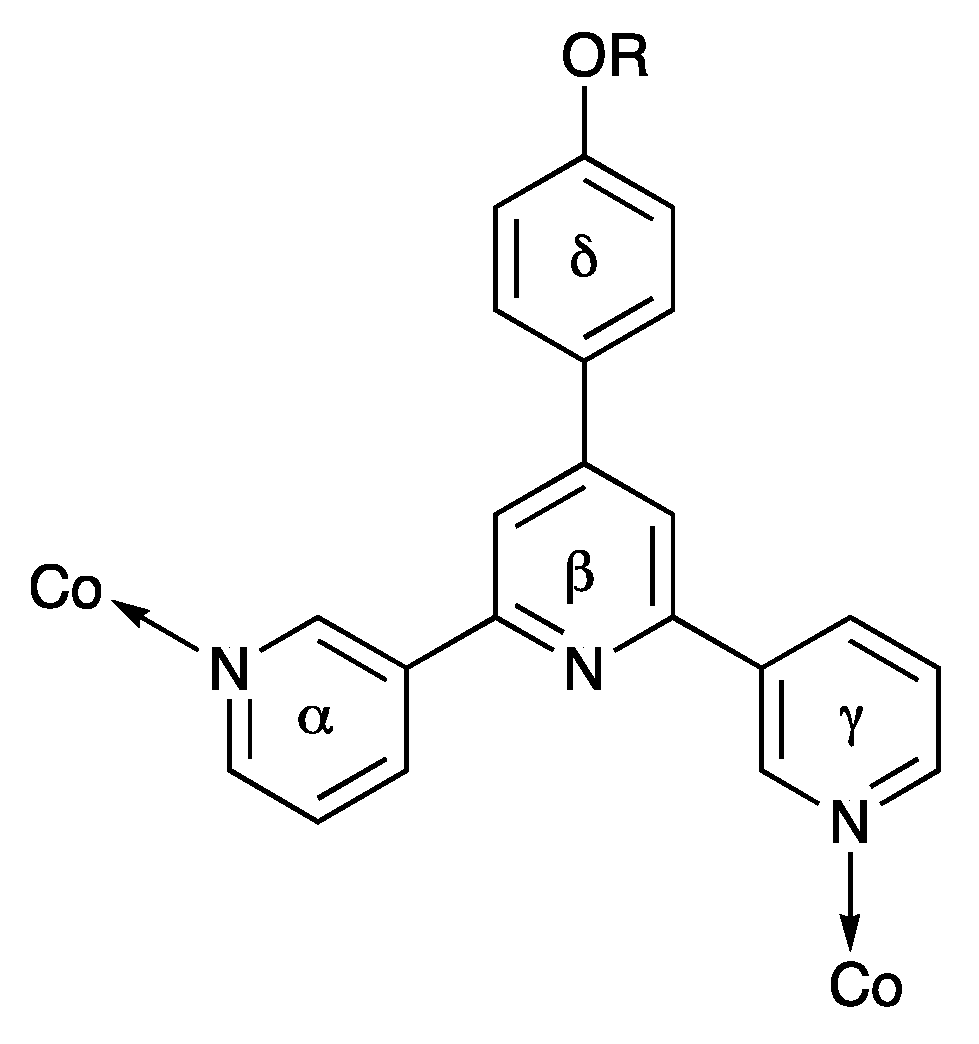
3.2. Coordination Networks with Co(NCS)2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Housecroft, C.E. 4,2′:6′,4″-Terpyridines: Diverging and Diverse Building Blocks in Coordination Polymers and Metallomacrocycles. Dalton Trans. 2014, 43, 6594–6604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Housecroft, C.E. Divergent 4,2′:6′,4′′- and 3,2′:6′,3′′-Terpyridines as Linkers in 2- and 3-Dimensional Architectures. CrystEngComm 2015, 17, 7461–7468. [Google Scholar] [CrossRef] [Green Version]
- Housecroft, C.E.; Constable, E.C. Ditopic and tetratopic 4,2′:6′,4″-Terpyridines as Structural Motifs in 2D- and 3D-Coordination Assemblies. Chimia 2019, 73, 462–467. [Google Scholar] [CrossRef]
- Constable, E.C.; Housecroft, C.E. Ligand and Metalloligand Design for Macrocycles, Multimetallic Arrays, Coordination Polymers and Assemblies; Elsevier Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Reedijk, J., Eds.; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar] [CrossRef]
- Burd, S.D.; Nugent, P.S.; Mohamed, M.H.; Elsaidi, S.K.; Zaworotko, M.J. Square Grid and Pillared Square Grid Coordination Polymers–Fertile Ground for Crystal Engineering of Structure and Function. Chimia 2013, 67, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Rocco, D.; Prescimone, A.; Constable, E.C.; Housecroft, C.E. Directing 2D-Coordination Networks: Combined Effects of a Conformationally Flexible 3,2′:6′,3″-Terpyridine and Chain Length Variation in 4′-(4-n-Alkyloxyphenyl) Substituents. Molecules 2020, 25, 1663. [Google Scholar] [CrossRef] [Green Version]
- Rocco, D.; Prescimone, A.; Constable, E.C.; Housecroft, C.E. Switching the conformation of 3,2′:6′,3″-tpy domains in 4′-(4-n-alkyloxyphenyl)-3,2′:6′,3″-terpyridines. Molecules 2020, 25, 3162. [Google Scholar] [CrossRef]
- Kröhnke, F. The Specific Synthesis of Pyridines and Oligopyridines. Synthesis 1976, 1, 1–24. [Google Scholar] [CrossRef]
- Wang, J.; Hanan, G.S. A facile route to sterically hindered and non-hindered 4′-aryl-2,2′:6′,2″- terpyridines. Synlett 2005, 8, 1251–1254. [Google Scholar] [CrossRef]
- Constable, E.C.; Housecroft, C.E. Tetratopic bis(4,2′:6′,4″-terpyridine) and bis(3,2′:6′,3″-terpyridine) ligands as 4-connecting nodes in 2D-coordination networks and 3D-frameworks. J. Inorg. Organomet. Polym. Mater. 2018, 28, 414–427. [Google Scholar] [CrossRef] [Green Version]
- Klein, Y.M.; Prescimone, A.; Constable, E.C.; Housecroft, C.E. 2-Dimensional networks assembled using 4′-functionalized 4,2′:6′,4″-terpyridines and Co(NCS)2. Polyhedron 2016, 103, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Rocco, D.; Prescimone, A.; Klein, Y.M.; Gawryluk, D.J.; Constable, E.C.; Housecroft, C.E. Competition in coordination assemblies: 1D-coordination polymer or 2D-nets based on Co(NCS)2 and 4′-(4-methoxyphenyl)-3,2′:6′,3″-terpyridine. Polymers 2019, 11, 1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, Y.M.; Prescimone, A.; Constable, E.C.; Housecroft, C.E. 4,2′:6′,4″- and 3,2′:6′,3″-terpyridines: The conflict between well-defined vectorial properties and serendipity in the assembly of 1D-, 2D- and 3D-architectures. Materials 2017, 10, 728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, A.K.; Khatua, S.; Tomar, K.; Konar, S. Field-Induced Single-Ion-Magnetic Behavior of Octahedral CoII in a Two-Dimensional Coordination Polymer. Eur. J. Inorg. Chem. 2016, 22, 3545–3552. [Google Scholar] [CrossRef]
- Constable, E.C.; Housecroft, C.E.; Neuburger, M.; Vujovic, S.; Zampese, J.A.; Zhang, G. Cobalt(II) coordination polymers with 4′-substituted 4,2′:6′,4″- and 3,2′:6′,3″-terpyridines: Engineering a switch from planar to undulating chains and sheets. CrystEngComm. 2012, 14, 3554–3563. [Google Scholar] [CrossRef] [Green Version]
- Klein, Y.M.; Prescimone, A.; Pitak, M.B.; Coles, S.J.; Constable, E.C.; Housecroft, C.E. Constructing chiral MOFs by functionalizing 4,2′:6′,4″-terpyridine with long-chain alkoxy domains: Rare examples of neb nets. CrystEngComm 2016, 18, 4704–4707. [Google Scholar] [CrossRef] [Green Version]
- Constable, E.C.; Housecroft, C.E.; Vujovic, S.; Zampese, J.A. 2D→2D Parallel interpenetration of (4,4) sheets constructed from a ditopic bis(4,2′:6′,4″-terpyridine). CrystEngComm 2014, 16, 3494–3497. [Google Scholar] [CrossRef] [Green Version]
- Vujovic, S.; Constable, E.C.; Housecroft, C.E.; Morris, C.D.; Neuburger, M.; Prescimone, A. Engineering 2D→2D parallel interpenetration using long alkoxy-chain substituents. Polyhedron 2015, 92, 77–83. [Google Scholar] [CrossRef] [Green Version]
- Klein, Y.M.; Prescimone, A.; Karpacheva, M.; Constable, E.C.; Housecroft, C.E. Sometimes the same, sometimes different: Understanding self-assembly algorithms in coordination networks. Polymers 2018, 10, 1369. [Google Scholar] [CrossRef] [Green Version]
- Klein, Y.M.; Constable, E.C.; Housecroft, C.E.; Zampese, J.A.; Crochet, A. Greasy tails switch 1D-coordination [Zn2(OAc)4(4′-(4-ROC6H4)-4,2′:6′,4″-tpy)]n polymers to discrete [Zn2(OAc)4(4′-(4-ROC6H4)-4,2′:6′,4″-tpy)2] complexes. CrystEngComm 2014, 16, 9915–9929. [Google Scholar] [CrossRef] [Green Version]
- Software for the Integration of CCD Detector System Bruker Analytical X-ray Systems; Bruker axs: Madison, WI, USA, 2013.
- Sheldrick, G.M. ShelXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with ShelXL. Acta Crystallogr. C 2015, 27, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Palatinus, L.; Chapuis, G. Superflip-A Computer Program for the Solution of Crystal Structures by Charge Flipping in Arbitrary Dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. [Google Scholar] [CrossRef] [Green Version]
- Palatinus, L.; Prathapa, S.J.; van Smaalen, S. EDMA: A Computer Program for Topological Analysis of Discrete Electron Densities. J. Appl. Crystallogr. 2012, 45, 575–580. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spek, A.L. PLATON SQUEEZE: A Tool for the Calculation of the Disordered Solvent Contribution to the Calculated Structure Factors. Acta Crystallogr. C 2015, 71, 9–18. [Google Scholar] [CrossRef] [Green Version]
- LeBail, A.; Duroy, H.; Fourquet, J.L. Ab-initio structure determination of LiSbWO6 by X-ray powder diffraction. Mat. Res. Bull. 1988, 23, 447–452. [Google Scholar] [CrossRef]
- Pawley, G.S. Unit-cell refinement from powder diffraction scans. J. Appl. Crystallogr. 1981, 14, 357–361. [Google Scholar] [CrossRef]
- Rodríguez-Carvajal, J. Recent Advances in Magnetic Structure Determination by Neutron Powder Diffraction. Physica B 1993, 192, 55–69. [Google Scholar] [CrossRef]
- Roisnel, T.; Rodríguez-Carvajal, J. WinPLOTR: A Windows tool for powder diffraction patterns analysis Materials Science Forum. In Proceedings of the Seventh European Powder Diffraction Conference (EPDIC 7), Barcelona, Spain, 20–23 May 2000; Volume 378, pp. 118–123. [Google Scholar]
- Rocco, D.; Housecroft, C.E.; Constable, E.C. Synthesis of Terpyridines: Simple Reactions–What Could Possibly Go Wrong? Molecules 2019, 24, 1799. [Google Scholar] [CrossRef] [Green Version]
- Rocco, D.; Manfroni, G.; Prescimone, A.; Klein, Y.M.; Gawryluk, D.J.; Constable, E.C.; Housecroft, C.E. Single and double-stranded 1D-coordination polymers with 4′-(4-alkyloxyphenyl)-3,2′:6′,3″-terpyridines and {Cu2(μ-OAc)4} or {Cu4(μ3-OH)2(μ-OAc)2(μ3-OAc)2(AcO-κO)2} motifs. Polymers 2020, 12, 318. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhang, Y.Z.; Yang, C.; Liu, E.; Golen, J.A.; Zhang, G. One-dimensional copper(II) coordination polymers built on 4′-substituted 4,2′:6′,4″- and 3,2′:6′,3″-terpyridines: Syntheses, structures and catalytic properties. Polyhedron 2016, 105, 115–122. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
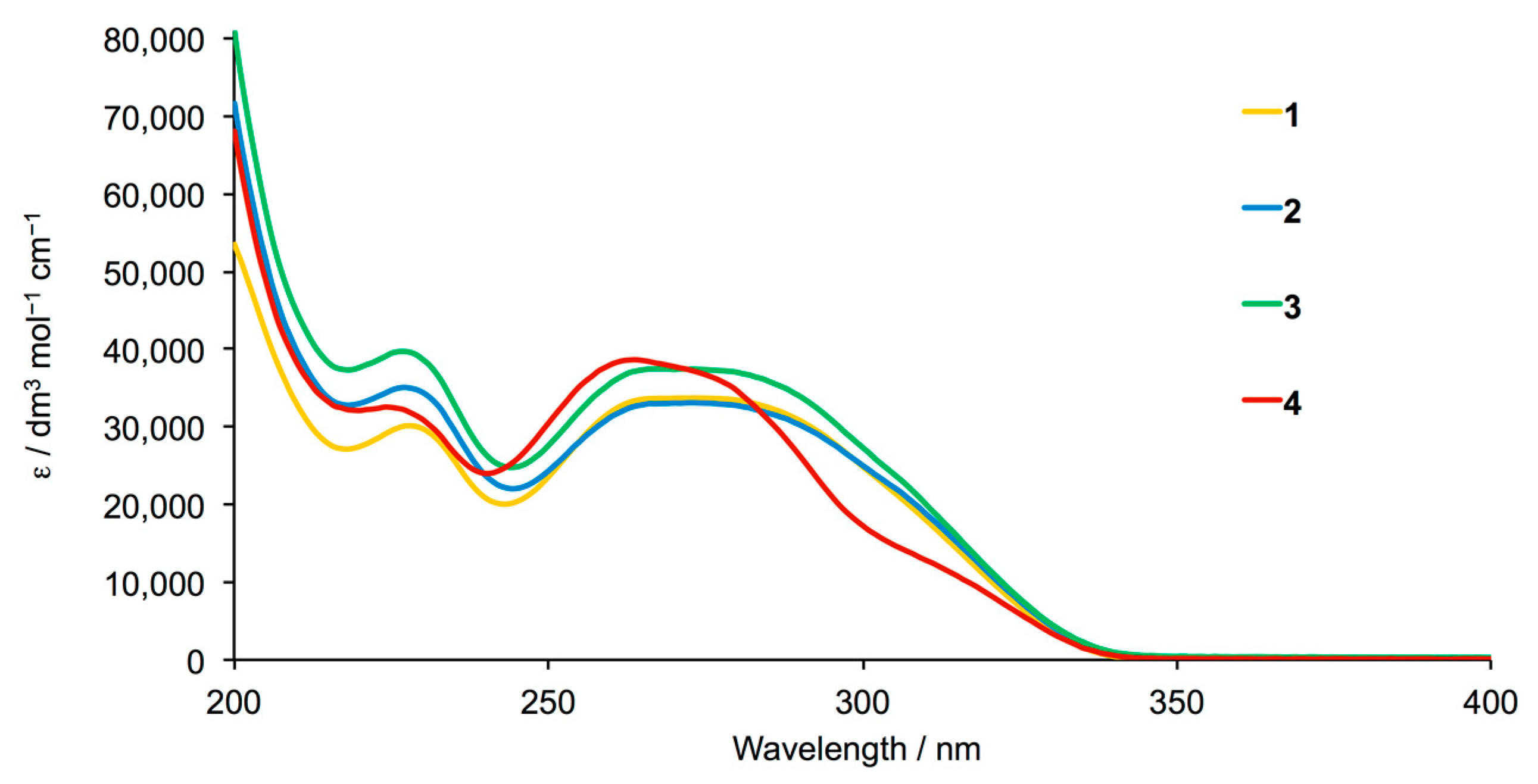
| Compound | λ/nm (ε/dm3 mol–1 cm–1) |
|---|---|
| rac-2 | 227 (35,100), 273 (33,100) |
| 3 | 227 (39,700), 269 (37,450) |
| 4 | 224 (32,550), 264 (38,600), 315 sh (10,800) |
| Compound | Co–Ntpy/Å | Co–NNCS/Å | Ntpy–Co–Ntpy1/deg | NNCS–Co–NNCS /deg |
|---|---|---|---|---|
| [{Co(rac-2)2(NCS)2}·CHCl3]n | 2.185(3), 2.211(3) | 2.079(3) | 180, 180 | 180 |
| [Co(3)2(NCS)2]n | 2.217(3), 2.219(3) | 2.072(3) | 180, 180 | 180 |
| [{Co(4)2(NCS)2}·CHCl3]n | 2.205(3), 2.230(3) | 2.066(3) | 180, 180 | 180 |
| Compound | Ring α/Ring β | Ring β/Ring γ | Ring β/Ring δ |
|---|---|---|---|
| [{Co(rac-2)2(NCS)2}·CHCl3]n | 30.6 | 32.7 | 55.7 |
| [Co(3)2(NCS)2]n | 37.7 | 27.9 | 48.1 |
| [{Co(4)2(NCS)2}·CHCl3]n | 28.0 | 28.9 | 48.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rocco, D.; Prescimone, A.; Constable, E.C.; Housecroft, C.E. Straight Versus Branched Chain Substituents in 4′-(Butoxyphenyl)-3,2′:6′,3″-terpyridines: Effects on (4,4) Coordination Network Assemblies. Polymers 2020, 12, 1823. https://doi.org/10.3390/polym12081823
Rocco D, Prescimone A, Constable EC, Housecroft CE. Straight Versus Branched Chain Substituents in 4′-(Butoxyphenyl)-3,2′:6′,3″-terpyridines: Effects on (4,4) Coordination Network Assemblies. Polymers. 2020; 12(8):1823. https://doi.org/10.3390/polym12081823
Chicago/Turabian StyleRocco, Dalila, Alessandro Prescimone, Edwin C. Constable, and Catherine E. Housecroft. 2020. "Straight Versus Branched Chain Substituents in 4′-(Butoxyphenyl)-3,2′:6′,3″-terpyridines: Effects on (4,4) Coordination Network Assemblies" Polymers 12, no. 8: 1823. https://doi.org/10.3390/polym12081823
APA StyleRocco, D., Prescimone, A., Constable, E. C., & Housecroft, C. E. (2020). Straight Versus Branched Chain Substituents in 4′-(Butoxyphenyl)-3,2′:6′,3″-terpyridines: Effects on (4,4) Coordination Network Assemblies. Polymers, 12(8), 1823. https://doi.org/10.3390/polym12081823