Production of Biodegradable Palm Oil-Based Polyurethane as Potential Biomaterial for Biomedical Applications

 , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
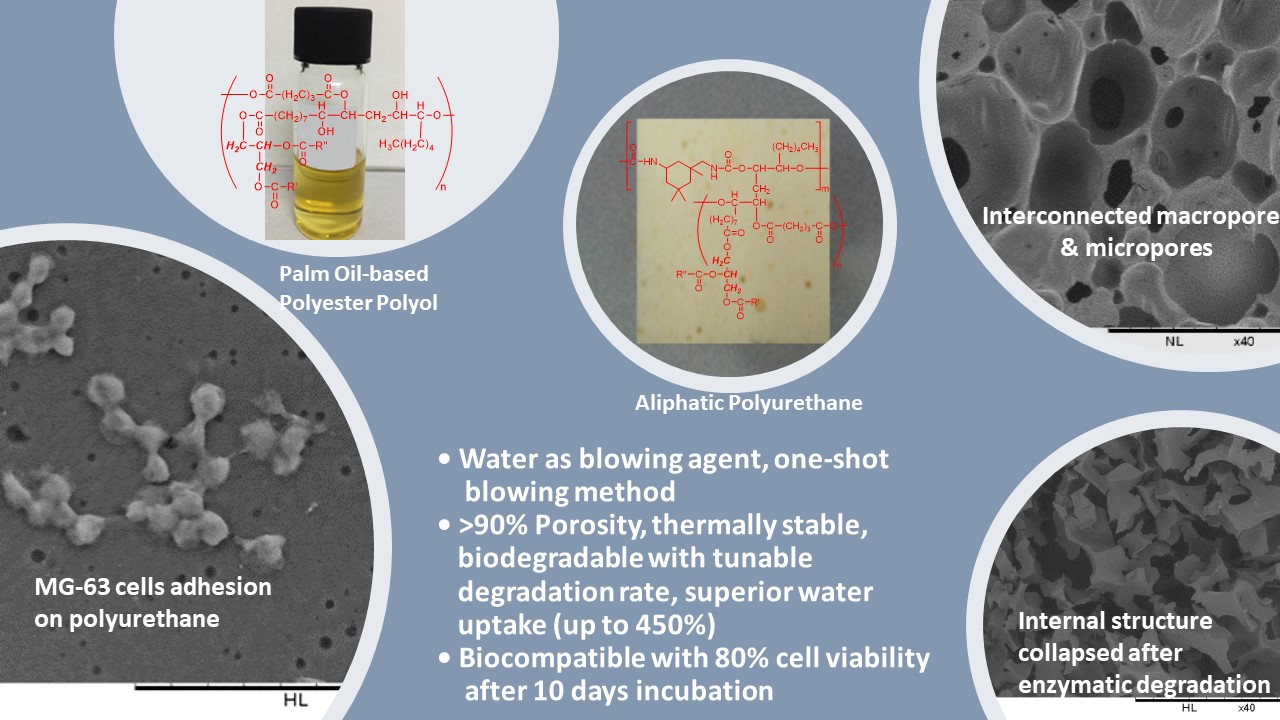
2.2. Synthesis of Palm Oil-based Polyester Polyols (PPP)
2.3. Production of Polyurethane
2.4. Characterization of Polyurethane
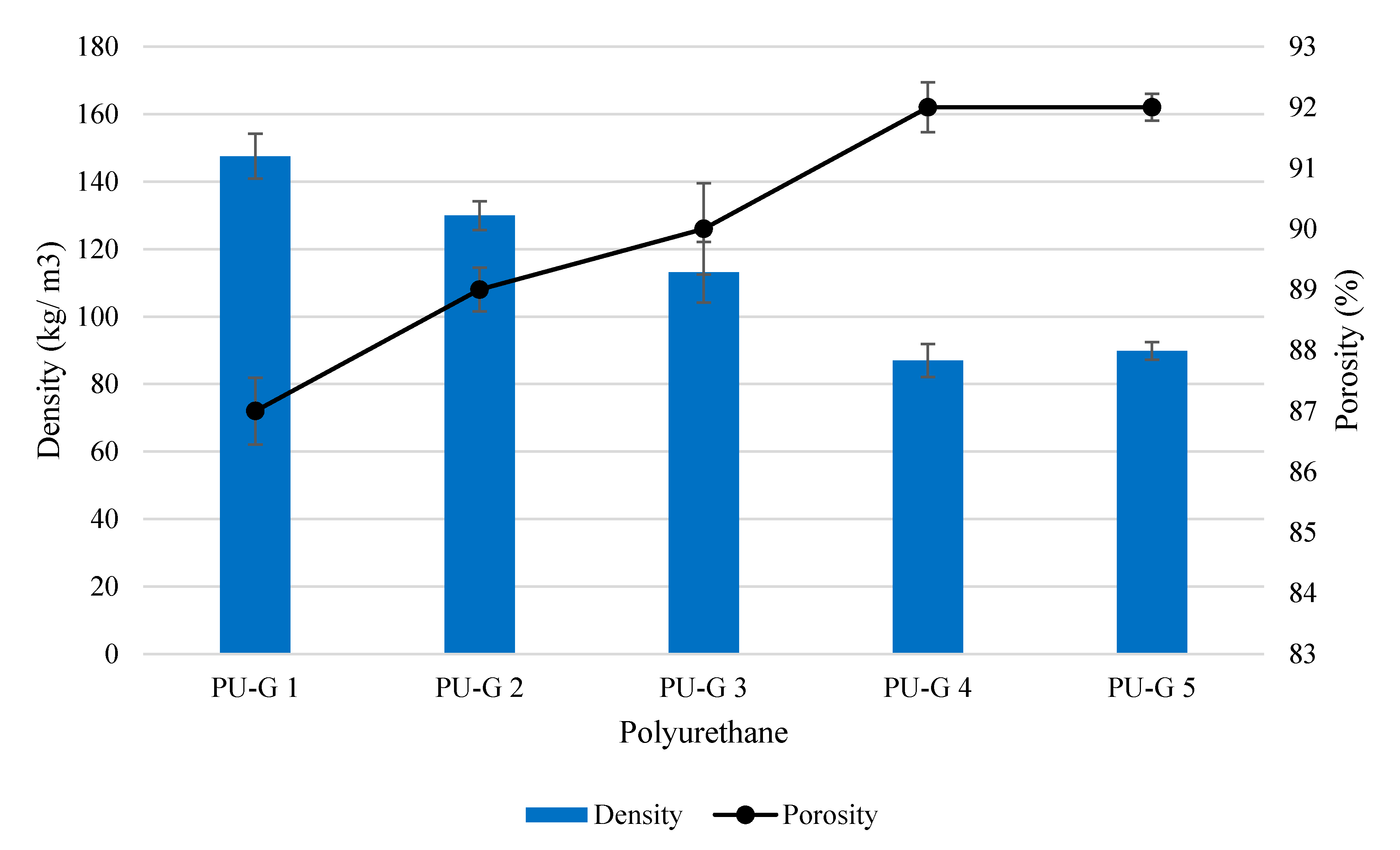
2.4.1. Morphology, Density and Porosity of Polyurethane
2.4.2. ATR-FTIR Analysis
2.4.3. Thermal Analysis
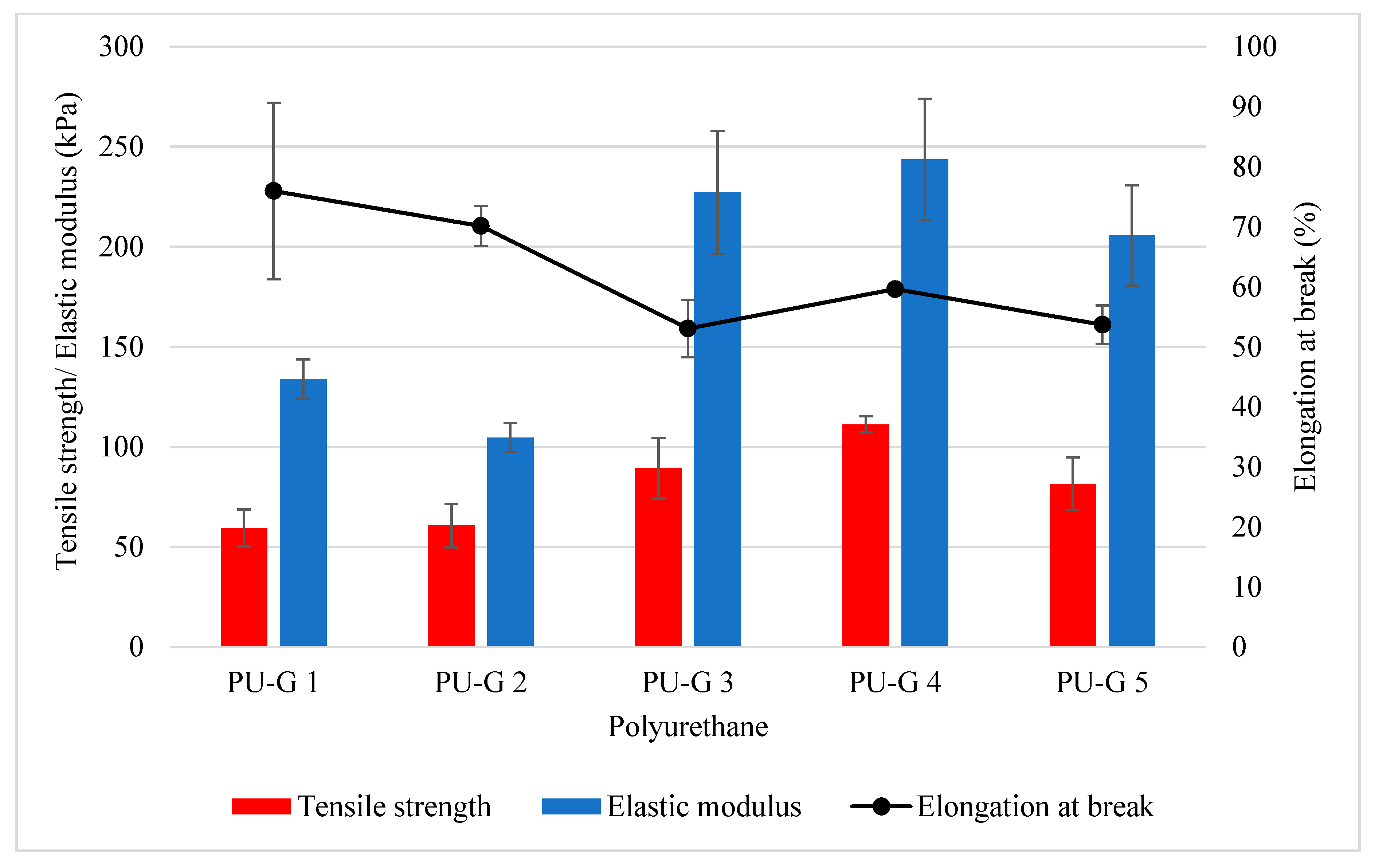
2.4.4. Mechanical Properties
2.4.5. In Vitro Enzymatic Degradation
2.4.6. Cytotoxicity Test
2.4.7. Cell Adhesion Test
3. Results and Discussion
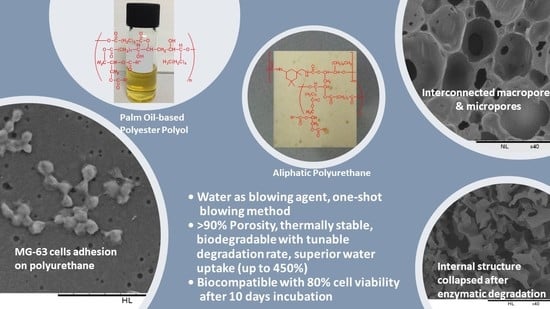
3.1. Production of Biodegradable Polyurethane
3.2. The Effect of Water Content
3.3. The Effect of Isocyanate Index
3.4. ATR-FTIR Spectroscopy
3.5. Thermal Analysis
3.6. In Vitro Enzymatic Degradation
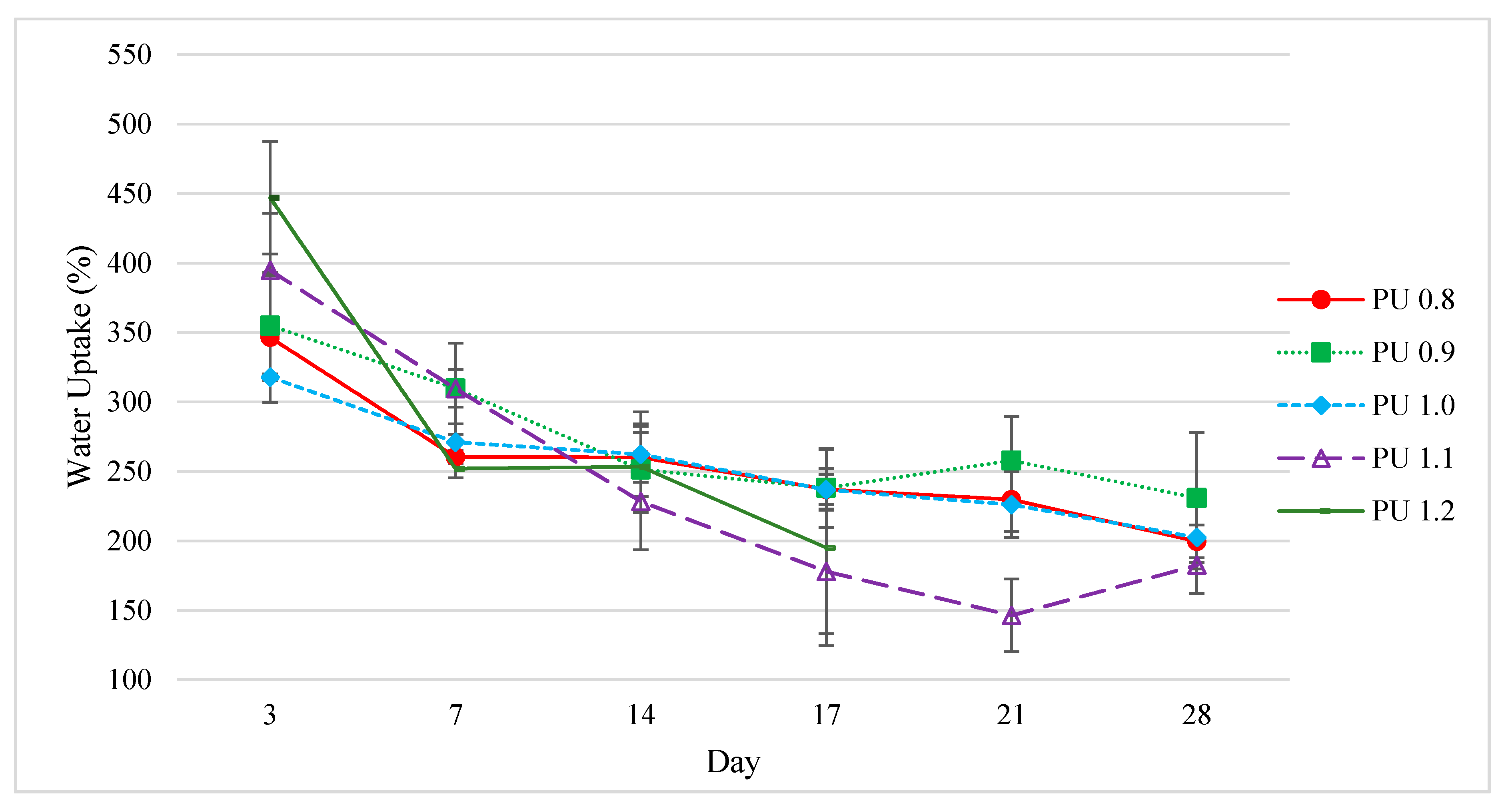
3.6.1. Water Uptake
3.6.2. Mass Loss
3.6.3. pH Measurement
3.6.4. ATR-FTIR Spectroscopy
3.6.5. Thermal Analysis
3.6.6. Physical Appearance and Morphology
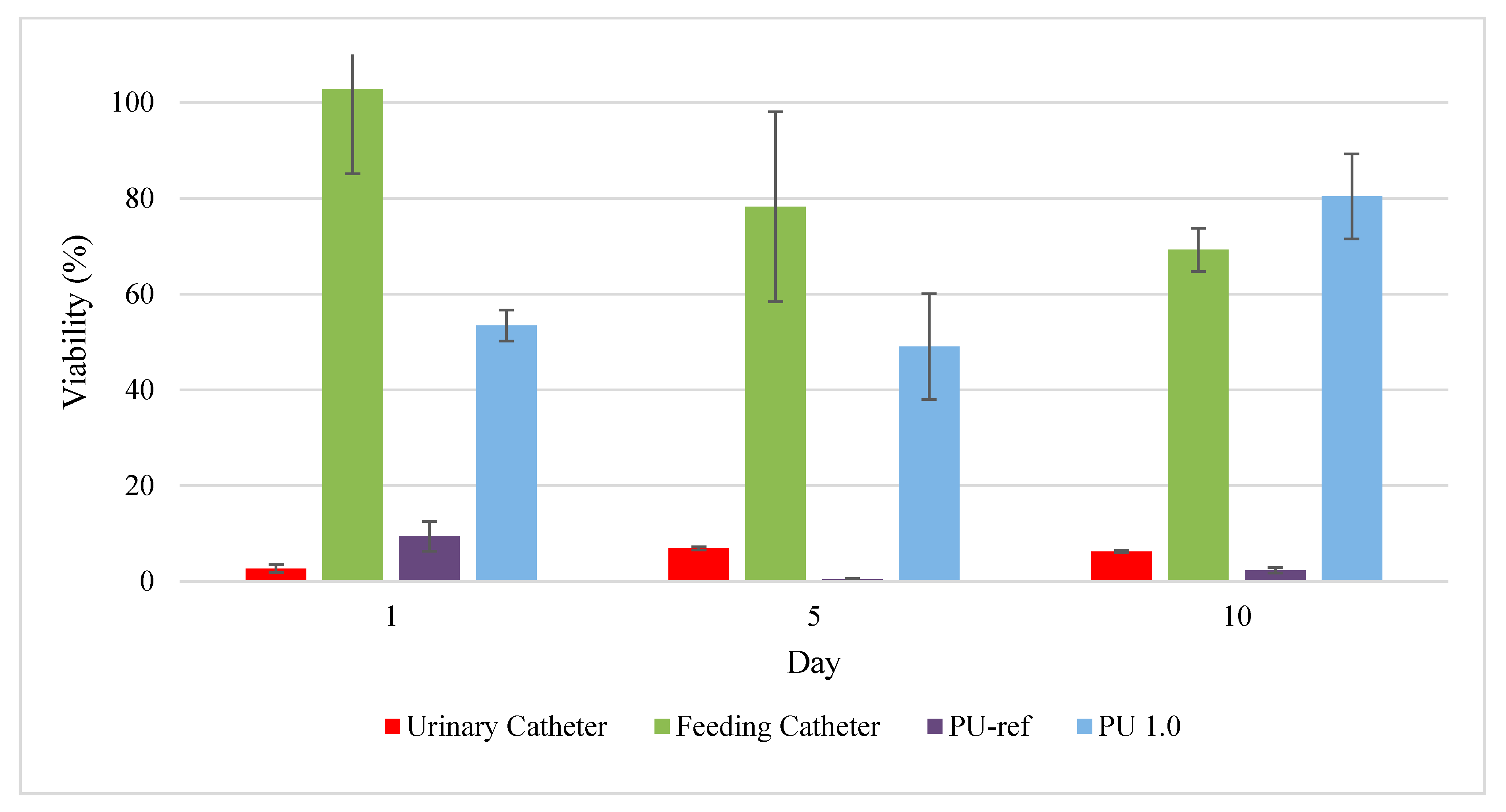
3.7. Cytotoxicity by MTT Assay
3.8. Cell Adhesion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jaganathan, S.; Mani, M.; Khudzari, A. Electrospun combination of peppermint oil and copper sulphate with conducive physico-chemical properties for wound dressing applications. Polymers 2019, 11, 586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira Rodrigues, I.C.; Tamborlin, L.; Rodrigues, A.A.; Jardini, A.L.; Ducati Luchessi, A.; Maciel Filho, R.; Najar Lopes, E.S.; Gabriel, L.P. Fibrous membranes tailored by rotary jet spinning for tissue engineering applications. J. Appl. Polym. Sci. 2020, 137, 48455–48464. [Google Scholar] [CrossRef]
- Wang, C.; Xie, J.; Xiao, X.; Chen, S.; Wang, Y. Development of nontoxic biodegradable polyurethanes based on polyhydroxyalkanoate and l-lysine diisocyanate with improved mechanical properties as new elastomers scaffolds. Polymers 2019, 11, 1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajau, R.; Rohani, R.; Wan Isahak, W.N.R.; Salleh, M.Z.; Ghazali, Z. Development of new bio-based polyol ester from palm oil for potential polymeric drug carrier. Adv. Polym. Tech. 2018, 37, 3552–3560. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Chen, J.; Shi, M.; Zhang, H.; Ma, P.; Guo, B. Electroactive anti-oxidant polyurethane elastomers with shape memory property as non-adherent wound dressing to enhance wound healing. Chem. Eng. J. 2019, 375, 121999–122012. [Google Scholar] [CrossRef]
- Peng, Z.; Zhou, P.; Zhang, F.; Peng, X. Preparation and properties of polyurethane hydrogels based on hexamethylene diisocyanate/polycaprolactone-polyethylene glycol. J. Macromol. Sci. Part B 2018, 57, 187–195. [Google Scholar] [CrossRef]
- Shahrousvand, M.; Mir Mohamad Sadeghi, G.; Salimi, A. Artificial extracellular matrix for biomedical applications: Biocompatible and biodegradable poly(tetramethylene ether) glycol/poly(ε-caprolactone diol)-based polyurethanes. J. Biomater. Sci. Polym. Ed. 2016, 27, 1712–1728. [Google Scholar] [CrossRef]
- Park, J.S.; Kang, H.J.; Lee, B.-T.; Choi, J.S.; Yim, J.-H. Mechanically and electrically enhanced polyurethane-poly(3,4-ethylenedioxythiophene) conductive foams with aligned pore structures promote MC3T3-E1 cell growth and proliferation. ACS Appl. Polym. Mater. 2020, 2, 1482–1490. [Google Scholar] [CrossRef]
- Yeganeh, H.; Hojati-Talemi, P. Preparation and properties of novel biodegradable polyurethane networks based on castor oil and poly(ethylene glycol). Polym. Degrad. Stab. 2007, 92, 480–489. [Google Scholar] [CrossRef]
- Miao, S.; Sun, L.; Wang, P.; Liu, R.; Su, Z.; Zhang, S. Soybean oil-based polyurethane networks as candidate biomaterials: Synthesis and biocompatibility. Eur. J. Lipid Sci. Technol. 2012, 114, 1165–1174. [Google Scholar] [CrossRef]
- Luo, X.; Yu, Z.; Cai, Y.; Wu, Q.; Zeng, J. Facile fabrication of environmentally-friendly hydroxyl-functionalized multiwalled carbon nanotubes/soy oil-based polyurethane nanocomposite bioplastics with enhanced mechanical, thermal, and electrical conductivity properties. Polymers 2019, 11, 763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zieleniewska, M.; Auguścik, M.; Prociak, A.; Rojek, P.; Ryszkowska, J. Polyurethane-urea substrates from rapeseed oil-based polyol for bone tissue cultures intended for application in tissue engineering. Polym. Degrad. Stab. 2014, 108, 241–249. [Google Scholar] [CrossRef]
- Zieleniewska, M.; Leszczyński, M.; Kurańska, M.; Prociak, A.; Szczepkowski, L.; Krzyżowska, M.; Ryszkowska, J. Preparation and characterisation of rigid polyurethane foams using a rapeseed oil-based polyol. Ind. Crop. Prod. 2015, 74, 887–897. [Google Scholar] [CrossRef]
- Kong, X.; Liu, G.; Curtis, J.M. Characterization of canola oil based polyurethane wood adhesives. Int. J. Adhes. Adhes. 2011, 31, 559–564. [Google Scholar] [CrossRef]
- Pillai, P.K.S.; Li, S.; Bouzidi, L.; Narine, S.S. Polyurethane foams from chlorinated and non-chlorinated metathesis modified canola oil polyols. J. Appl. Polym. Sci. 2018, 135, 46616–46628. [Google Scholar] [CrossRef]
- Ang, K.P.; Lee, C.S.; Cheng, S.F.; Chuah, C.H. Synthesis of palm oil-based polyester polyol for polyurethane adhesive production. J. Appl. Polym. Sci. 2014, 131, 39967–39974. [Google Scholar] [CrossRef]
- Chuayjuljit, S.; Sangpakdee, T.; Saravari, O. Processing and properties of palm oil-based rigid polyurethane foam. J. Met. Mater. Miner. 2007, 17, 17–23. [Google Scholar]
- Pawlik, H.; Prociak, A. Influence of palm oil-based polyol on the properties of flexible polyurethane foams. J. Polym. Environ. 2012, 20, 438–445. [Google Scholar] [CrossRef]
- Ng, W.S.; Lee, C.S.; Chuah, C.H.; Cheng, S.F. Preparation and modification of water-blown porous biodegradable polyurethane foams with palm oil-based polyester polyol. Ind. Crop. Prod. 2017, 97, 65–78. [Google Scholar] [CrossRef]
- Somarathna, H.; Raman, S.; Badri, K.; Mutalib, A.; Mohotti, D.; Ravana, S. Quasi-static behavior of palm-based elastomeric polyurethane: For strengthening application of structures under impulsive loadings. Polymers 2016, 8, 202. [Google Scholar] [CrossRef] [Green Version]
- Badri, K.H.; Othman, Z.; Ahmad, S.H. Rigid polyurethane foams from oil palm resources. J. Mater. Sci. 2004, 39, 5541–5542. [Google Scholar] [CrossRef]
- Liu, J.; Yang, Y.; Gao, B.; Li, Y.C.; Xie, J. Bio-based elastic polyurethane for controlled-release urea fertilizer: Fabrication, properties, swelling and nitrogen release characteristics. J. Clean. Prod. 2019, 209, 528–537. [Google Scholar] [CrossRef]
- Yaakob, Z.; Min, A.M.; Hilmi, M.M.; Zaman, H.M.D.K.; Kamarudin, S.K.K. Effect of compactabilization of polymer on the properties of polyurethane-palm fiber composites. J. Polym. Eng. 2009, 29, 503–520. [Google Scholar] [CrossRef]
- Hassouna, Y.M.; Somayeh, Z.K.; Kafienahb, W.; Husam, M.Y. Synthesis, characterization & cytocompatibility of poly(diol-co-tricarballylate) based thermally crosslinked elastomers for drug delivery & tissue engineering applications. Mater. Sci. Eng. C 2018, 93, 254–264. [Google Scholar]
- Yang, J.; Antonio, R.W.; Samuel, J.P.; Hageman, G.; Guillermo, A.A. Synthesis and evaluation of poly(diol citrate) biodegradable elastomers. Biomaterials 2006, 27, 1889–1898. [Google Scholar] [CrossRef]
- Mani, M.P.; Jaganathan, S.K.; Prabhakaran, P.; Nageswaran, G.; Krishnasamy, N.P. Electrospun polyurethane patch in combination with cedarwood and cobalt nitrate for cardiac applications. J. Appl. Polym. Sci. 2019, 136, 48226–48234. [Google Scholar] [CrossRef]
- Molina, G.A.; Elizalde-Mata, A.; Hernández-Martínez, Á.R.; Fonseca, G.; Cruz Soto, M.; Rodríguez-Morales, Á.L.; Estevez, M. Synthesis and characterization of inulin-based responsive polyurethanes for breast cancer applications. Polymers 2020, 12, 865. [Google Scholar] [CrossRef] [Green Version]
- Jaganathan, S.K.; Mani, M.P. Single-stage synthesis of electrospun polyurethane scaffold impregnated with zinc nitrate nanofibers for wound healing applications. J. Appl. Polym. Sci. 2019, 136, 46942–46950. [Google Scholar] [CrossRef]
- Xue, L.; Greisler, H.P. Biomaterials in the development and future of vascular grafts. J. Vasc. Surg. 2003, 37, 472–480. [Google Scholar] [CrossRef] [Green Version]
- Pfister, D.P.; Xia, Y.; Larock, R.C. Recent advances in vegetable oil-based polyurethanes. ChemSusChem 2011, 4, 703–717. [Google Scholar] [CrossRef]
- Yeoh, F.H.; Lee, C.S.; Kang, Y.B.; Wong, S.F.; Cheng, S.F. One-pot synthesis of palm oil-based polyester polyol for production of biodegradable and biocompatible polyurethane. J. Appl. Polym. Sci. 2018, 135, 46861. [Google Scholar] [CrossRef]
- Skrobot, J.; Ignaczak, W.; el Fray, M. Hydrolytic and enzymatic degradation of flexible polymer networks comprising fatty acid derivatives. Polym. Degrad. Stab. 2015, 120, 368–376. [Google Scholar] [CrossRef]
- Song, N.; Jiang, X.; Li, J.; Pang, Y.; Li, J.; Tan, H.; Fu, Q. The degradation and biocompatibility of waterborne biodegradable polyurethanes for tissue engineering. Chin. J. Polym. Sci. 2013, 31, 1451–1462. [Google Scholar] [CrossRef]
- Rottmar, M.; Richter, M.; Mäder, X.; Grieder, K.; Nuss, K.; Karol, A.; Rechenberg, B.; Zimmermann, E.; Buser, S.; Dobmann, A.; et al. In vitro investigations of a novel wound dressing concept based on biodegradable polyurethane. Sci. Tech. Adv. Mater. 2015, 16, 034606–034615. [Google Scholar] [CrossRef] [Green Version]
- Barrioni, B.R.; de Carvalho, S.M.; Oréfice, R.L.; de Oliveira, A.A.R.; Pereira, M.D.M. Synthesis and characterization of biodegradable polyurethane films based on hdi with hydrolyzable crosslinked bonds and a homogeneous structure for biomedical applications. Mater. Sci. Eng C 2015, 52, 22–30. [Google Scholar] [CrossRef]
- Aastha, S.D. Polyurethane foam chemistry. In Recycling of Polyurethane Foams; Thomas, S., Rane, A.V., Kanny, K., Abitha, V.K., Thomas, M.G., Eds.; William Andrew: New York, NY, USA, 2018; pp. 17–27. [Google Scholar]
- Pinchuk, L. A review of the biostability and carcinogenicity of polyurethanes in medicine and the new generation of “biostable” polyurethanes. J. Biomater. Sci., Polym. Ed. 1995, 6, 225–267. [Google Scholar] [CrossRef]
- Gabriel, L.; Zavaglia, C.; Jardini, A.; Dias, C.; Maciel Filho, R. Isocyanates as precursors to biomedical polyurethanes. Chem. Eng. Trans. 2014, 38, 253–258. [Google Scholar]
- Zhang, X.D.; Macosko, C.W.; Davis, H.T.; Nikolov, A.D.; Wasan, D.T. Role of silicone surfactant in flexible polyurethane foam. J. Colloid Interface Sci. 1999, 215, 270–279. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, B.K.; Lim, H. Effect of isocyanate index on the properties of rigid polyurethane foams blown by HFC 365mfc. Macromol. Res. 2008, 16, 467–472. [Google Scholar] [CrossRef]
- Dworakowska, S.; Bogdal, D.; Prociak, A. Microwave-assisted synthesis of polyols from rapeseed oil and properties of flexible polyurethane foams. Polymers 2012, 4, 1462–1477. [Google Scholar] [CrossRef] [Green Version]
- Gogolewski, S.; Gorna, K.; Turner, A.S. Regeneration of bicortical defects in the iliac crest of estrogen-deficient sheep, using new biodegradable polyurethane bone graft substitutes. J. Biomed. Mater. Res. Part A 2006, 77, 802–810. [Google Scholar] [CrossRef] [PubMed]
- Polo-Corrales, L.; Latorre-Esteves, M.; Ramirez-Vick, J.E. Scaffold design for bone regeneration. J. Nanosci. Nanotechnol. 2014, 14, 15–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucinska-Lipka, J.; Marzec, M.; Gubanska, I.; Janik, H. Porosity and swelling properties of novel polyurethane–ascorbic acid scaffolds prepared by different procedures for potential use in bone tissue engineering. J. Elastom. Plast. 2017, 49, 440–456. [Google Scholar] [CrossRef]
- Spaans, C.; Belgraver, V.; Rienstra Ode Groot, J.; Veth, R.; Pennings, A. Solvent-free fabrication of micro-porous polyurethane amide and polyurethane-urea scaffolds for repair and replacement of the knee-joint meniscus. Biomaterials 2000, 21, 2453–2460. [Google Scholar] [CrossRef]
- Kucińska-Lipka, J.; Gubanska, I.; Skwarska, A. Microporous polyurethane thin layer as a promising scaffold for tissue engineering. Polymers 2017, 9, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafeman, A.; Yoshii, B.; Zienkiewicz, T.; Davidson, K.J.; Guelcher, S. Injectable biodegradable polyurethane scaffolds with release of platelet-derived growth factor for tissue repair and regeneration. Pharm. Res. 2008, 25, 2387–2399. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Li, Y.; Zou, Q. Degradation and biocompatibility of porous nano hydroxyapatite/polyurethane composite scaffold for bone tissue engineering. Appl. Surf. Sci. 2009, 255, 6087–6091. [Google Scholar] [CrossRef]
- Lu, Y.; Larock, R.C. Soybean-oil-based waterborne polyurethane dispersions: Effects of polyol functionality and hard segment content on properties. Biomacromolecules 2008, 9, 3332–4330. [Google Scholar] [CrossRef]
- Asensio, M.; Costa, V.; Nohales, A.; Bianchi, O.; Gómez, C.M. Tunable structure and properties of segmented thermoplastic polyurethanes as a function of flexible segment. Polymers 2019, 11, 1910. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Peng, Y.; Tong, R.; Wang, Y.; Wu, Z. The effects of isocyanate index on the properties of aliphatic waterborne polyurethaneureas. J. Appl. Polym. Sci. 2010, 118, 920–927. [Google Scholar] [CrossRef]
- Kattiyaboot, T.; Thongpin, C. Effect of natural oil based polyols on the properties of flexible polyurethane foams blown by distilled water. Energy Procedia 2016, 89, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Prociak, A.; Malewska, E.; Bąk, S. Influence of isocyanate index on selected properties of flexible polyurethane foams modified with various bio-components. J. Renew. Mater. 2016, 4, 78–85. [Google Scholar] [CrossRef]
- Fan, H.; Tekeei, A.; Suppes, G.J.; Hsieh, F.H. Physical properties of soy-phosphate polyol-based rigid polyurethane foams. Int. J. Polym. Sci. 2012, 2012, 907049. [Google Scholar] [CrossRef] [Green Version]
- Bil, M.; Ryszkowska, J.; Kurzydłowski, K.J. Effect of polyurethane composition and the fabrication process on scaffold properties. J. Mater. Sci. 2009, 44, 1469–1476. [Google Scholar] [CrossRef]
- Modesti, M.; Lorenzetti, A. An experimental method for evaluating isocyanate conversion and trimer formation in polyisocyanate–polyurethane foams. Eur. Polym. J. 2001, 37, 949–954. [Google Scholar] [CrossRef]
- Rojek, P.; Prociak, A. Effect of different rapeseed-oil-based polyols on mechanical properties of flexible polyurethane foams. J. Appl. Polym. Sci. 2012, 125, 2936. [Google Scholar] [CrossRef]
- Gorna, K.; Gogolewski, S. Preparation, degradation, and calcification of biodegradable polyurethane foams for bone graft substitutes. J. Biomed. Mater. Res. 2003, 67, 813. [Google Scholar] [CrossRef]
- Das, B.; Konwar, U.; Mandal, M.; Karak, N. Sunflower oil based biodegradable hyperbranched polyurethane as a thin film material. Ind. Crop. Prod. 2013, 44, 396–404. [Google Scholar] [CrossRef]
- Ryszkowska, J.L.; Auguścik, M.; Sheikh, A.; Boccaccini, A.R. Biodegradable polyurethane composite scaffolds containing Bioglass® for bone tissue engineering. Compos. Sci. Technol. 2010, 70, 1894–1908. [Google Scholar] [CrossRef] [Green Version]
- Cangemi, J.M.; Claro Neto, S.; Chierice, G.O.; Santos, A.M.D. Study of the biodegradation of a polymer derived from castor oil by scanning electron microscopy, thermogravimetry and infrared spectroscopy. Polímeros 2006, 16, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Javni, I.; Petrovi, Z.S.; Guo, A.; Fuller, R. Thermal stability of polyurethanes based on vegetable oils. J. Appl. Polym. Sci. 2000, 77, 1723–1734. [Google Scholar] [CrossRef]
- Gómez, E.F.; Luo, X.; Li, C.; Michel, F.C.; Li, Y. Biodegradability of crude glycerol-based polyurethane foams during composting, anaerobic digestion and soil incubation. Polym. Degrad. Stab. 2014, 102, 195–203. [Google Scholar] [CrossRef]
- Wang, J.; Zheng, Z.; Wang, Q.; Du, P.; Shi, J.; Wang, X. Synthesis and characterization of biodegradable polyurethanes based on L-cystine/cysteine and poly(ϵ-caprolactone). J. Appl. Polym. Sci. 2013, 128, 4047–4057. [Google Scholar] [CrossRef]
- Prociak, A.; Rojek, P.; Pawlik, H. Flexible polyurethane foams modified with natural oil based polyols. J. Cell. Plast. 2012, 48, 489–499. [Google Scholar] [CrossRef]
- Norouz, F.; Halabian, R.; Salimi, A.; Ghollasi, M. A new nanocomposite scaffold based on polyurethane and clay nanoplates for osteogenic differentiation of human mesenchymal stem cells in vitro. Mater. Sci. Eng. C 2019, 103, 109857–109869. [Google Scholar] [CrossRef]
- Tawagi, E.; Ganesh, T.; Cheng, H.L.M.; Santerre, J.P. Synthesis of degradable-polar-hydrophobic-ionic co-polymeric microspheres by membrane emulsion photopolymerization: In vitro and in vivo studies. Acta Biomater. 2019, 89, 279–288. [Google Scholar] [CrossRef]
- Shen, Z.; Lu, D.; Li, Q.; Zhang, Z.; Zhu, Y. Synthesis and characterization of biodegradable polyurethane for hypopharyngeal tissue engineering. BioMed Res. Int. 2015, 2015, 871202. [Google Scholar] [CrossRef]
- Pan, Z.; Ding, J. Poly(lactide-co-glycolide) Porous scaffolds for tissue engineering and regenerative medicine. Interface Focus. 2012, 2, 366–377. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.H.; Hibbs, D.E.; Howard, N.T. Conformations, energies, and intramolecular hydrogen bonds in dicarboxylic acids: Implications for the design of synthetic dicarboxylic acid receptors. J. Comput. Chem. 2005, 26, 1233–1241. [Google Scholar] [CrossRef]
- Wang, Z.; Yu, L.; Ding, M.; Tan, H.; Li, J.; Fu, Q. Preparation and rapid degradation of nontoxic biodegradable polyurethanes based on poly(lactic acid)-poly(ethylene glycol)-poly(lactic acid) and l-lysine diisocyanate. Polym. Chem. 2011, 2, 601–607. [Google Scholar] [CrossRef]
- Guelcher, S.; Srinivasan, A.; Hafeman, A.; Gallagher, K.; Doctor, J.; Khetan, S.; McBride, S.; Hollinger, J. Synthesis, in vitro degradation, and mechanical properties of two-component poly(ester urethane)urea scaffolds: Effects of water and polyol composition. Tissue Eng. 2007, 13, 2321–2333. [Google Scholar] [CrossRef] [PubMed]
- Bugajny, M.; le Bras, M.; Bourbigot, S. Thermoplastic polyurethanes as carbonization agents in intumescent blends. Part 2: Thermal behavior of polypropylene/thermoplastic polyurethane/ammonium polyphosphate blends. J. Fire Sci. 2000, 18, 7–27. [Google Scholar] [CrossRef]
- Choi, H.; Lee, J.; Lee, J.; Sung, H.; Shin, J.; Shin, J.; Wu, Y.; Kim, J. MG-63 cells proliferation following various types of mechanical stimulation on cells by auxetic hybrid scaffolds. Biomater. Res. 2016, 20, 32–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Encalada-Diaz, I.; Cole, B.; MacGillivray, J.; Ruiz-Suarez, M.; Kercher, J.; Friel, N.; Valero-Gonzalez, F. Rotator cuff repair augmentation using a novel polycarbonate polyurethane patch: Preliminary results at 12 months’ follow-up. J. Shoulder Elb. Surg. 2011, 20, 788–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Li, Y.; Zuo, Y.; Zhang, L.; Zou, Q.; Cheng, L.; Jiang, H. Porous bioactive scaffold of aliphatic polyurethane and hydroxyapatite for tissue regeneration. Biomed. Mater. 2009, 4, 025003–025009. [Google Scholar] [CrossRef] [PubMed]
- Lönnroth, E.C. Toxicity of medical glove materials: A pilot study. Int. J. Occup. Saf. Ergon. 2005, 11, 131–139. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physico-Chemical Properties | Value | |
|---|---|---|
| Acid Value (mg KOH/g sample) | 1.95 | |
| Oxirane oxygen content (%) | 0.35 | |
| Hydroxyl value (mg KOH/g sample) | 84.50 | |
| Mn | 6698 | |
| Viscosity (Pa.s) | 25 °C: | 24.55 |
| 40 °C: | 8.68 | |
| Pour Point (°C) | 12 | |
| Cloud Point (°C) | 12 | |
| Physical Properties | Liquid | |
| Components | Composition (g) | |
|---|---|---|
| PU | PU-Ref | |
| PEG 6000 | - | 40.9 |
| PCL diol 2000 | - | 9.1 |
| PPP | 50 | - |
| A33 | 2 | 2 |
| L580 | 2 | 2 |
| Glycerol | 3 | 3 |
| DBTDL | 0.4 | 0.2 |
| Water | Ranged from 0.6–1.4 | 0.5 |
| Polyurethane | Physical Properties | Tensile Properties | Compression Stress (kPa) | ||||
|---|---|---|---|---|---|---|---|
| Density (kg/m3) | Porosity (%) | Pore Size (μm) | Tensile Strength (kPa) | Elongation at Break (%) | Elastic Modulus (kPa) | ||
| PU 0.8 | 96.30 | 92.07 | 37–1322 | 48.32 ± 8.2 | 76.62 ± 8.3 | 79.83 ± 3.4 | 15.10 ± 0.4 |
| PU 0.9 | 68.00 | 94.44 | 43–1464 | 53.77 ± 8.6 | 66.83 ± 5.6 | 100.36 ± 53.2 | 16.92 ± 3.3 |
| PU 1.0 | 87.00 | 92.85 | 43–1013 | 111.25 ± 4.1 | 59.63 ± 0.7 | 243.69 ± 30.2 | 64.08 ± 12.3 |
| PU 1.1 | 71.60 | 94.13 | 49–1655 | 85.27 ± 8.5 | 45.86 ± 3.6 | 289.67 ± 58.6 | 49.19 ± 7.6 |
| PU 1.2 | 71.20 | 94.17 | 57–1700 | 70.85 ± 13.4 | 32.12 ± 1.6 | 482.69 ± 70.3 | 115.09 ± 18.2 |
| Polyurethane | Before Degradation | After Degradation | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T5% (°C) | T10% (°C) | T50% (°C) | T90% (°C) | Residue at 800 °C (%) | Tmax1 (°C) | Tmax2 (°C) | Tmax3 (°C) | T5% (°C) | T10% (°C) | T50% (°C) | T90% (°C) | Residue at 800 °C (%) | Tmax2 (°C) | Tmax3 (°C) | |
| PU 0.8 | 248 | 280 | 379 | 430 | 0 | 287 | 306 | 398 | 256 | 279 | 373 | 424 | 0.23 | 307 | 395 |
| PU 0.9 | 250 | 280 | 373 | 433 | 0 | 290 | 311 | 390 | 258 | 280 | 373 | 422 | 0.23 | 308 | 391 |
| PU 1.0 | 254 | 279 | 373 | 426 | 0 | 286 | 308 | 394 | 256 | 278 | 367 | 432 | 0.07 | 309 | 391 |
| PU 1.1 | 258 | 280 | 373 | 428 | 0 | 285 | 307 | 395 | 267 | 285 | 369 | 429 | 0.34 | 312 | 395 |
| PU 1.2 | 256 | 276 | 372 | 429 | 0 | 281 | 309 | 394 | 260 | 282 | 377 | 719 | 8.06 | 341 | 403 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeoh, F.H.; Lee, C.S.; Kang, Y.B.; Wong, S.F.; Cheng, S.F.; Ng, W.S. Production of Biodegradable Palm Oil-Based Polyurethane as Potential Biomaterial for Biomedical Applications. Polymers 2020, 12, 1842. https://doi.org/10.3390/polym12081842
Yeoh FH, Lee CS, Kang YB, Wong SF, Cheng SF, Ng WS. Production of Biodegradable Palm Oil-Based Polyurethane as Potential Biomaterial for Biomedical Applications. Polymers. 2020; 12(8):1842. https://doi.org/10.3390/polym12081842
Chicago/Turabian StyleYeoh, Fang Hoong, Choy Sin Lee, Yew Beng Kang, Shew Fung Wong, Sit Foon Cheng, and Wei Seng Ng. 2020. "Production of Biodegradable Palm Oil-Based Polyurethane as Potential Biomaterial for Biomedical Applications" Polymers 12, no. 8: 1842. https://doi.org/10.3390/polym12081842
APA StyleYeoh, F. H., Lee, C. S., Kang, Y. B., Wong, S. F., Cheng, S. F., & Ng, W. S. (2020). Production of Biodegradable Palm Oil-Based Polyurethane as Potential Biomaterial for Biomedical Applications. Polymers, 12(8), 1842. https://doi.org/10.3390/polym12081842