Influence of the Architecture of Soft Polymer-Functionalized Polymer Nanoparticles on Their Dynamics in Suspension


Abstract
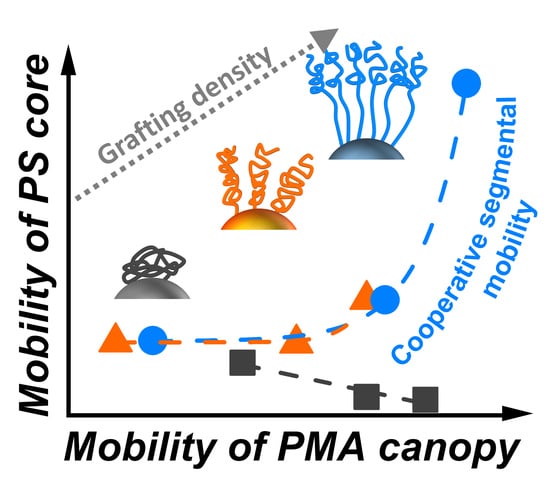
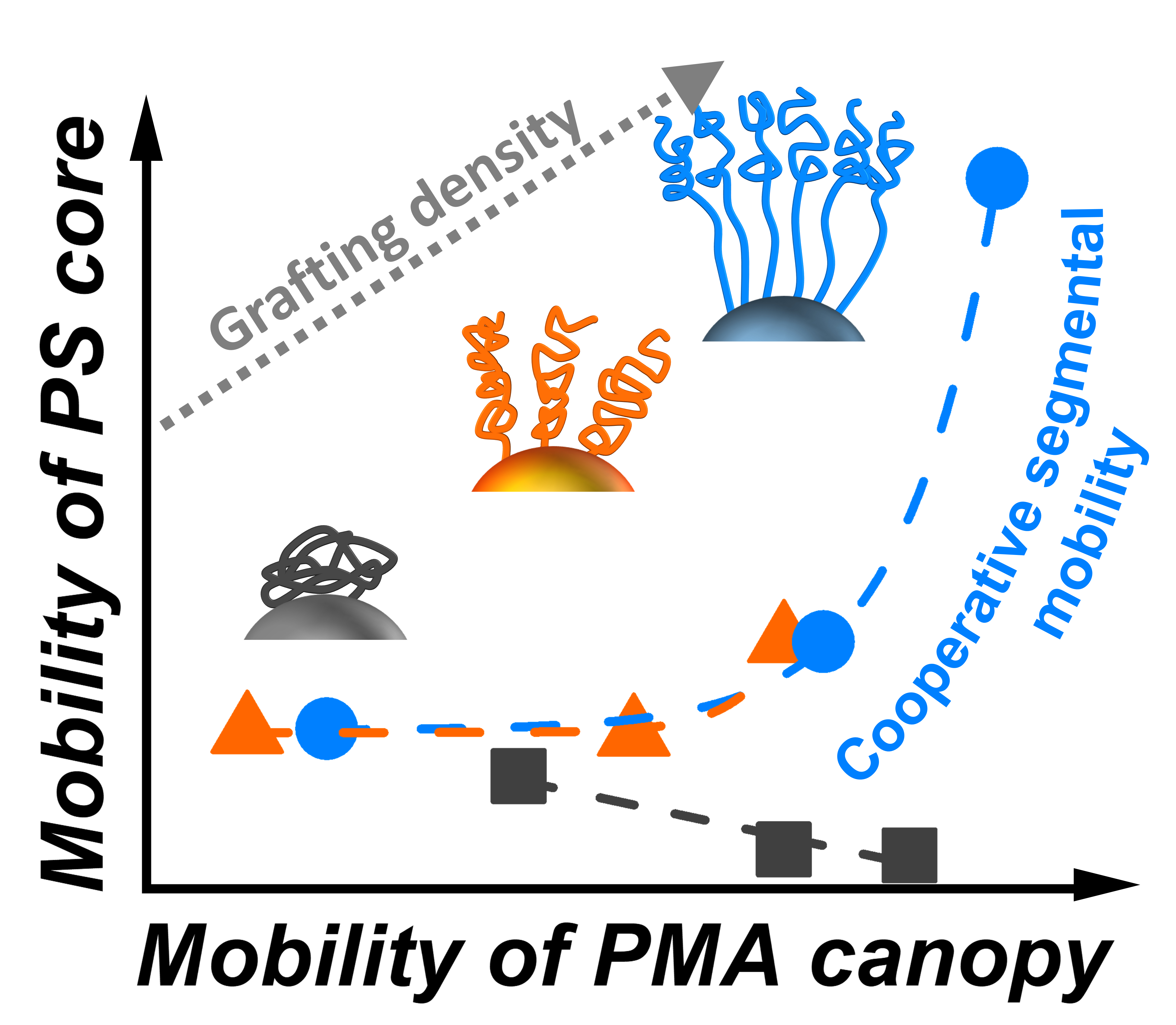
:
1. Introduction
2. Materials and Methods
2.1. Materials
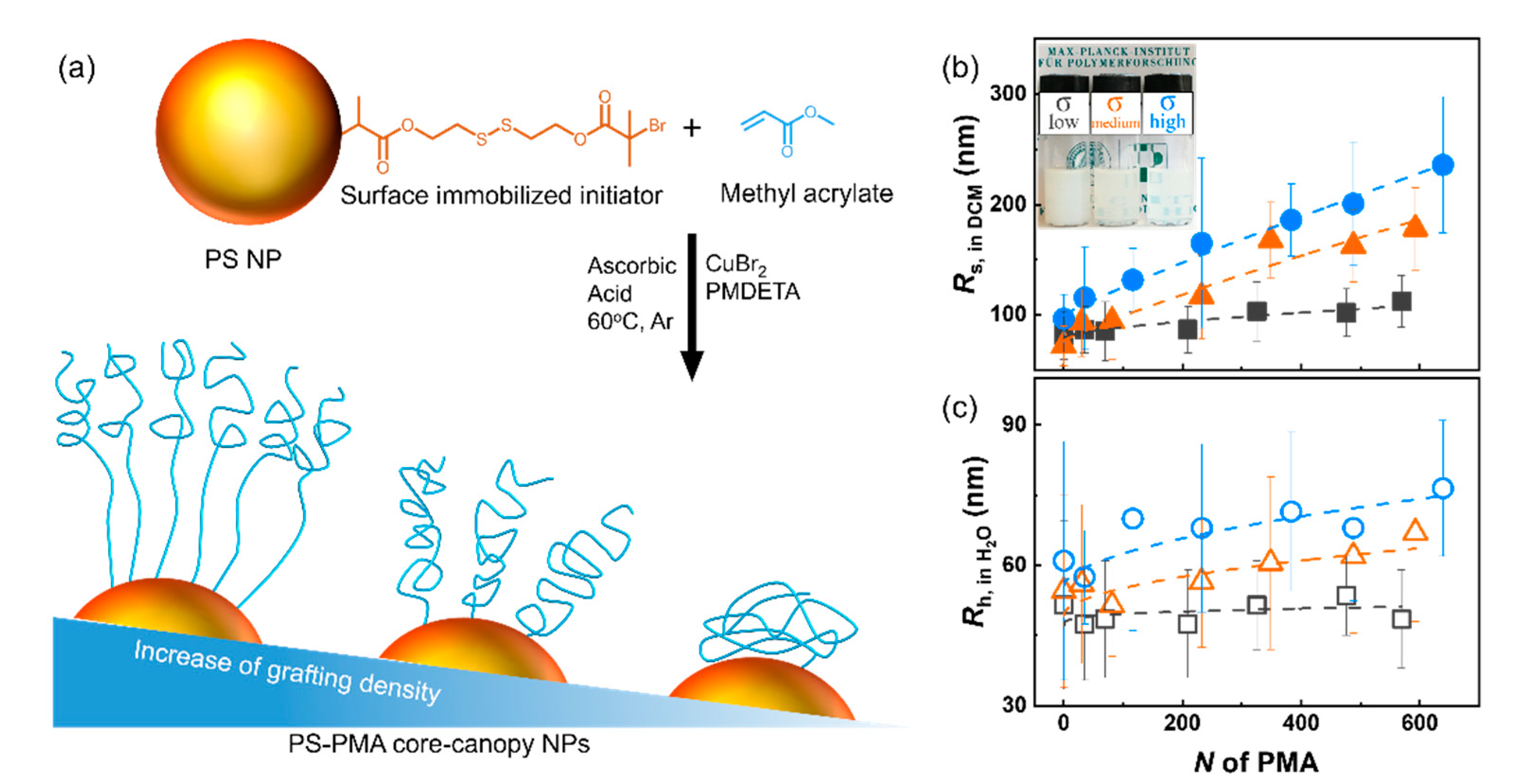
2.2. Synthesis of Nanoparticles Functionalized with ATRP Inimer (PS-Br NPs)
2.3. Synthesis of the End-Tethered Canopy of PMA on the Surface of the PS Core (PS-PMA NPs)
2.4. Characterization
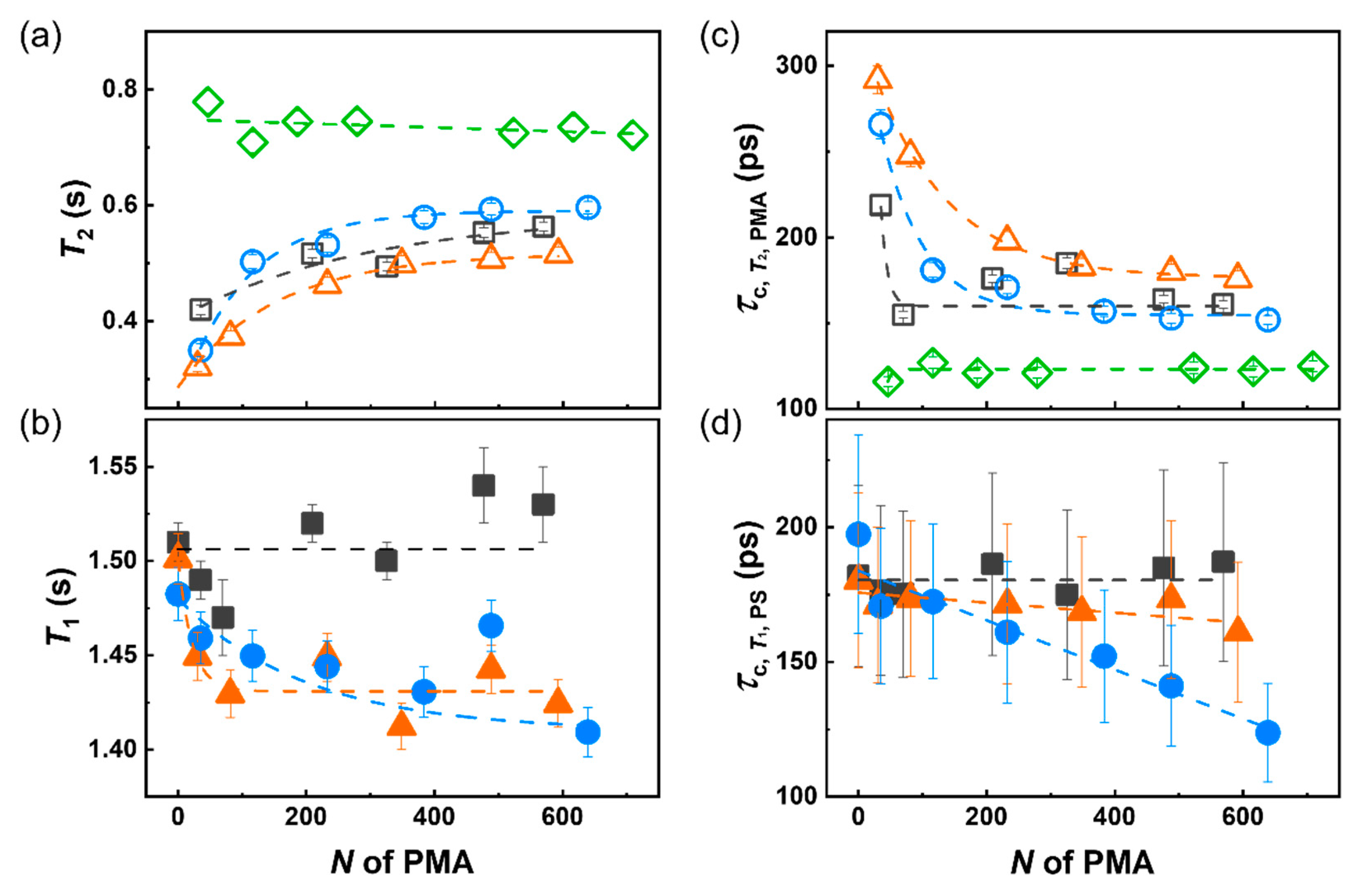
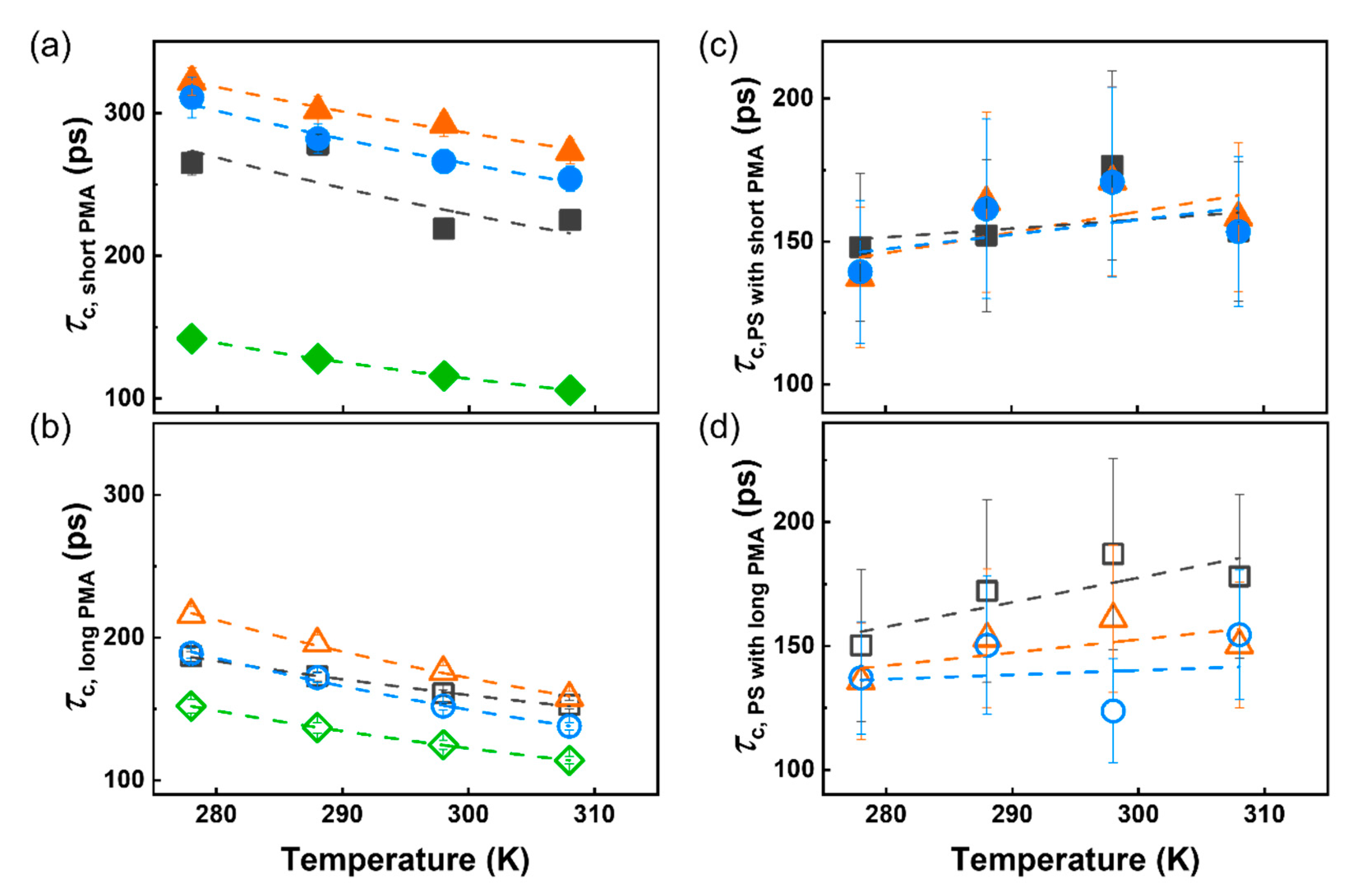
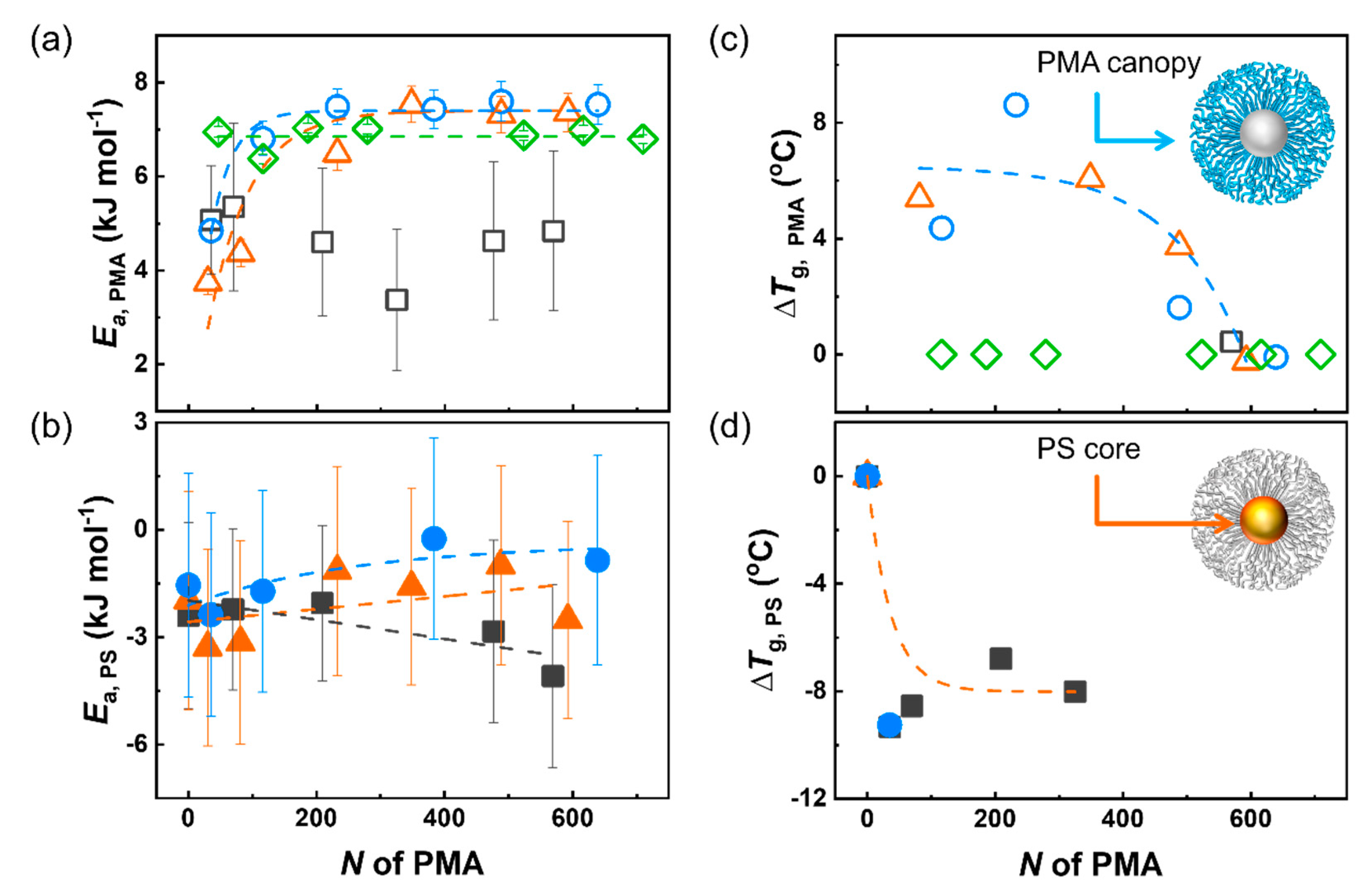
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Srivastava, S.; Agarwal, P.; Archer, L.A. Tethered nanoparticle-polymer composites: Phase stability and curvature. Langmuir 2012, 28, 6276–6281. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Xiu, K.M.; Xu, S.L.; Yang, W.T.; Xu, F.J. Functionalized layered double hydroxide nanoparticles conjugated with disulfide-linked polycation brushes for advanced gene delivery. Bioconjugate Chem. 2013, 24, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzis, G.G.; Theodorou, D.N. Structure of polymer layers grafted to nanoparticles in silica-polystyrene nanocomposites. Macromolecules 2013, 46, 4670–4683. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Ohishi, T.; Higaki, Y.; Takahara, A.; Otsuka, H. Radical crossover reactions of alkoxyamine-based dynamic covalent polymer brushes on nanoparticles and the effect on their dispersibility. Polym. J. 2016, 48, 147–155. [Google Scholar] [CrossRef]
- Zoppe, J.O.; Ataman, N.C.; Mocny, P.; Wang, J.; Moraes, J.; Klok, H.-A. Surface-initiated controlled radical polymerization: State-of-the-art, opportunities, and challenges in surface and interface engineering with polymer brushes. Chem. Rev. 2017, 117, 1105–1318. [Google Scholar] [CrossRef] [Green Version]
- Schöttler, S.; Becker, G.; Winzen, S.; Steinbach, T.; Mohr, K.; Landfester, K.; Mailänder, V.; Wurm, F.R. Protein adsorption is required for stealth effect of poly(ethylene glycol)- and poly(phosphoester)-coated nanocarriers. Nat. Nanotechnol. 2016, 11, 372–377. [Google Scholar] [CrossRef]
- Prencipe, G.; Tabakman, S.M.; Welsher, K.; Liu, Z.; Goodwin, A.P.; Zhang, L.; Henry, J.; Dai, H. PEG branched polymer for functionalization of nanomaterials with ultralong blood circulation. J. Am. Chem. Soc. 2009, 131, 4783–4787. [Google Scholar] [CrossRef] [Green Version]
- Koerner, H.; Drummy, L.F.; Benicewicz, B.; Li, Y.; Vaia, R.A. Nonisotropic self-organization of single-component hairy nanoparticle assemblies. ACS Macro Lett. 2013, 2, 670–676. [Google Scholar] [CrossRef]
- Dahal, U.; Wang, Z.; Dormidontova, E.E. Hydration of spherical PEO-grafted gold nanoparticles: Curvature and grafting density effect. Macromolecules 2018, 51, 5950–5961. [Google Scholar] [CrossRef]
- Ethier, J.G.; Hall, L.M. Structure and entanglement network of model polymer-grafted nanoparticle monolayers. Macromolecules 2018, 51, 9878–9889. [Google Scholar] [CrossRef]
- Sakib, N.; Koh, Y.P.; Huang, Y.; Mongcopa, K.I.S.; Le, A.N.; Benicewicz, B.C.; Krishnamoorti, R.; Simon, S.L. Thermal and rheological analysis of polystyrene-grafted silica nanocomposites. Macromolecules 2020. [Google Scholar] [CrossRef]
- Kim, Y.-G.; Wagner, M.; Thérien-Aubin, H. Dynamics of soft and hairy polymer nanoparticles in a suspension by NMR relaxation. Macromolecules 2020, 53, 844–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daoud, M.; Cotton, J.P. Star shaped polymers: A model for the conformation and its concentration dependence. J. Phys. 1982, 43, 531–538. [Google Scholar] [CrossRef]
- Ohno, K.; Morinaga, T.; Takeno, S.; Tsujii, Y.; Fukuda, T. Suspensions of silica particles grafted with concentrated polymer brush: Effects of graft chain length on brush layer thickness and colloidal crystallization. Macromolecules 2007, 40, 9143. [Google Scholar] [CrossRef]
- Dukes, D.; Li, Y.; Lewis, S.; Benicewicz, B.; Schadler, L.; Kumar, S.K. Conformational transitions of spherical polymer brushes: Synthesis, characterization, and theory. Macromolecules 2010, 43, 1564–1570. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.A.; Mangal, R.; Archer, L.A. Relaxation dynamics of nanoparticle-tethered polymer chains. Macromolecules 2015, 48, 6280. [Google Scholar] [CrossRef]
- Akcora, P.; Liu, H.; Kumar, S.K.; Moll, J.; Li, Y.; Benicewicz, B.C.; Schadler, L.S.; Acehan, D.; Panagiotopoulos, A.Z.; Pryamitsyn, V.; et al. Anisotropic self-assembly of spherical polymer-grafted nanoparticles. Nat. Mater. 2009, 8, 354–359. [Google Scholar] [CrossRef]
- Wichaita, W.; Kim, Y.-G.; Tangboriboonrat, P.; Thérien-Aubin, H. Polymer-functionalized polymer nanoparticles and their behaviour in suspensions. Polym. Chem. 2020, 11, 2119–2128. [Google Scholar] [CrossRef]
- Wei, Y.; Xu, Y.; Faraone, A.; Hore, M.J.A. Local structure and relaxation dynamics in the brush of polymer-grafted silica nanoparticles. ACS Macro Lett. 2018, 7, 699–704. [Google Scholar] [CrossRef]
- Savin, D.A.; Pyun, J.; Patterson, G.D.; Kowalewski, T.; Matyjaszewski, K. Synthesis and characterization of silica-graft-polystyrene hybrid nanoparticles: Effect of constraint on the glass-transition temperature of spherical polymer brushes. J. Polym. Sci. Part B Polym. Phys. 2002, 40, 2667–2676. [Google Scholar] [CrossRef]
- Dang, A.; Hui, C.M.; Ferebee, R.; Kubiak, J.; Li, T.; Matyjaszewski, K.; Bockstaller, M.R. Thermal properties of particle brush materials: Effect of polymer graft architecture on the glass transition temperature in polymer-grafted colloidal systems. Macromol. Symp. 2013, 331–332, 9–16. [Google Scholar] [CrossRef]
- Askar, S.; Li, L.; Torkelson, J.M. Polystyrene-grafted silica nanoparticles: Investigating the molecular weight dependence of glass transition and fragility behavior. Macromolecules 2017, 50, 1589–1598. [Google Scholar] [CrossRef]
- Holley, D.W.; Ruppel, M.; Mays, J.W.; Urban, V.S.; Baskaran, D. Polystyrene nanoparticles with tunable interfaces and softness. Polymer 2014, 55, 58–65. [Google Scholar] [CrossRef]
- Arbe, A.; Pomposo, J.A.; Moreno, A.J.; LoVerso, F.; González-Burgos, M.; Asenjo-Sanz, I.; Iturrospe, A.; Radulescu, A.; Ivanova, O.; Colmenero, J. Structure and dynamics of single-chain nano-particles in solution. Polymer 2016, 105, 532–544. [Google Scholar] [CrossRef]
- Klonos, P.A.; Patelis, N.; Glynos, E.; Sakellariou, G.; Kyritsis, A. Molecular dynamics in polystyrene single-chain nanoparticles. Macromolecules 2019, 52, 9334–9340. [Google Scholar] [CrossRef]
- Richter, D.; Kruteva, M. Polymer dynamics under confinement. Soft Matter 2019, 15, 7316–7349. [Google Scholar] [CrossRef]
- Munkhbat, O.; Canakci, M.; Zheng, S.; Hu, W.; Osborne, B.; Bogdanov, A.A.; Thayumanavan, S. 19F MRI of polymer nanogels aided by improved segmental mobility of embedded fluorine moieties. Biomacromolecules 2019, 20, 790–800. [Google Scholar] [CrossRef]
- Heatley, F. Nuclear magnetic-relaxation of synthetic-polymers in dilute-solution. Prog. Nucl. Magn. Reson. Spectrosc. 1979, 13, 47–85. [Google Scholar] [CrossRef]
- McCall, D.W. Nuclear magnetic resonance studies of molecular relaxation mechanisms in polymers. Acc. Chem. Res. 1971, 4, 223–232. [Google Scholar] [CrossRef]
- Graff, R.W.; Wang, X.; Gao, H. Exploring self-condensing vinyl polymerization of inimers in microemulsion to regulate the structures of hyperbranched polymers. Macromolecules 2015, 48, 2118–2126. [Google Scholar] [CrossRef]
- Hore, M.J.A. Polymers on nanoparticles: Structure & dynamics. Soft Matter 2019, 15, 1120–1134. [Google Scholar] [CrossRef] [PubMed]
- Birshtein, T.M.; Zhulina, E.B. Conformations of star-branched macromolecules. Polymer 1984, 25, 1453–1461. [Google Scholar] [CrossRef]
- Grest, G.S.; Murat, M. Structure of grafted polymeric brushes in solvents of varying quality: A molecular dynamics study. Macromolecules 1993, 26, 3108–3117. [Google Scholar] [CrossRef]
- Dimitrov, D.I.; Milchev, A.; Binder, K. Polymer brushes in solvents of variable quality: Molecular dynamics simulations using explicit solvent. J. Chem. Phys. 2007, 127, 084905. [Google Scholar] [CrossRef] [PubMed]
- Bloembergen, N.; Purcell, E.M.; Pound, R.V. Relaxation effects in nuclear magnetic resonance absorption. Phys. Rev. 1948, 73, 679–712. [Google Scholar] [CrossRef]
- Holt, A.P.; Bocharova, V.; Cheng, S.; Kisliuk, A.M.; White, B.T.; Saito, T.; Uhrig, D.; Mahalik, J.P.; Kumar, R.; Imel, A.E.; et al. Controlling interfacial dynamics: Covalent bonding versus physical adsorption in polymer nanocomposites. ACS Nano 2016, 10, 6843–6852. [Google Scholar] [CrossRef]
- Alegria, A.; Lund, R.; Barroso-Bujans, F.; Arbe, A.; Colmenero, J. Component dynamics in nanostructured PI-PDMS diblock copolymers with PI segregated in lamellas, cylinders, and spheres. Colloid Polym. Sci. 2014, 292, 1863–1876. [Google Scholar] [CrossRef]
- Lund, R.; Willner, L.; Alegría, A.; Colmenero, J.; Richter, D. Self-concentration and interfacial fluctuation effects on the local segmental dynamics of nanostructured diblock copolymer melts. Macromolecules 2008, 41, 511–514. [Google Scholar] [CrossRef]
- Helfand, E. Dynamics of conformational transitions in polymers. Science 1984, 226, 647–650. [Google Scholar] [CrossRef]
- Ngai, K.L.; Roland, C.M. Chemical structure and intermolecular cooperativity: Dielectric relaxation results. Macromolecules 1993, 26, 6824–6830. [Google Scholar] [CrossRef] [Green Version]
- Pethrick, R.A. Molecular-motion of polymers in solution. Sci. Prog. 1975, 62, 599–631. [Google Scholar]
- Erman, B.; Baysal, B.M. Temperature dependence of swelling of polystyrene networks. Macromolecules 1985, 18, 1696–1700. [Google Scholar] [CrossRef]
- Fox, T.G.; Flory, P.J. Second-order transition temperatures and related properties of polystyrene. I. Influence of molecular weight. J. Appl. Phys. 1950, 21, 581–591. [Google Scholar] [CrossRef]
- Fox, T.G.; Flory, P.J. The glass temperature and related properties of polystyrene. Influence of molecular weight. J. Polym. Sci. 1954, 14, 315–319. [Google Scholar] [CrossRef]
- Zuo, B.A.; Zhang, S.S.; Niu, C.; Zhou, H.; Sun, S.Z.; Wang, X.P. Grafting density dominant glass transition of dry polystyrene brushes. Soft Matter 2017, 13, 2426–2436. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Grafting Density (Chains nm⁻2) | N | Mn, NMR (kDa) | Mn, SEC (kDa) | Ð | Rs in DCM (nm) |
|---|---|---|---|---|---|---|
| PS-σlow | 0.07 ± 0.01 | 0 | 81 ± 20 | |||
| PS-PMA3k-σlow | 35 | 3 | 4 | 1.8 | 86 ± 20 | |
| PS-PMA6k-σlow | 70 | 6 | 8 | 2.1 | 85 ± 30 | |
| PS-PMA18k-σlow | 209 | 18 | 19 | 2.3 | 90 ± 20 | |
| PS-PMA28k-σlow | 325 | 28 | 15 | 2.2 | 100 ± 30 | |
| PS-PMA41k-σlow | 476 | 41 | 18 | 2.7 | 100 ± 20 | |
| PS-PMA49k-σlow | 569 | 49 | 34 | 2.2 | 110 ± 20 | |
| PS-σmedium | 0.46 ± 0.02 | 0 | 70 ± 20 | |||
| PS-PMA3k-σmedium | 30 | 3 | 4 | 1.9 | 90 ± 30 | |
| PS-PMA7k-σmedium | 81 | 7 | 9 | 2.6 | 90 ± 30 | |
| PS-PMA20k-σmedium | 232 | 20 | 20 | 2.2 | 120 ± 40 | |
| PS-PMA30k-σmedium | 348 | 30 | 27 | 2.1 | 170 ± 30 | |
| PS-PMA42k-σmedium | 488 | 42 | 29 | 2.1 | 160 ± 30 | |
| PS-PMA51k-σmedium | 592 | 51 | 35 | 2.4 | 180 ± 40 | |
| PS-σhigh | 1.00 ± 0.12 | 0 | 90 ± 20 | |||
| PS-PMA3k-σhigh | 35 | 3 | 4 | 1.7 | 110 ± 50 | |
| PS-PMA10k-σhigh | 116 | 10 | 12 | 2.1 | 130 ± 30 | |
| PS-PMA20k-σhigh | 232 | 20 | 21 | 2.1 | 160 ± 70 | |
| PS-PMA33k-σhigh | 383 | 33 | 27 | 2.4 | 190 ± 30 | |
| PS-PMA42k-σhigh | 488 | 42 | 40 | 2.5 | 200 ± 60 | |
| PS-PMA55k-σhigh | 639 | 55 | 44 | 2.0 | 240 ± 60 |
| Grafting Density (σ) | Grafting Density in H2O (Chains nm⁻2) | Grafting Density in DCM (Chains nm⁻2) | Scaling Exponent in H2O | Scaling Exponent in DCM |
|---|---|---|---|---|
| Low | 0.17 | 0.07 | 0.27 | 0.63 |
| Medium | 0.80 | 0.46 | 0.50 | 0.88 |
| High | 2.50 | 1.00 | 0.55 | 0.91 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.-G.; Wichaita, W.; Thérien-Aubin, H. Influence of the Architecture of Soft Polymer-Functionalized Polymer Nanoparticles on Their Dynamics in Suspension. Polymers 2020, 12, 1844. https://doi.org/10.3390/polym12081844
Kim Y-G, Wichaita W, Thérien-Aubin H. Influence of the Architecture of Soft Polymer-Functionalized Polymer Nanoparticles on Their Dynamics in Suspension. Polymers. 2020; 12(8):1844. https://doi.org/10.3390/polym12081844
Chicago/Turabian StyleKim, Young-Gon, Waraporn Wichaita, and Héloïse Thérien-Aubin. 2020. "Influence of the Architecture of Soft Polymer-Functionalized Polymer Nanoparticles on Their Dynamics in Suspension" Polymers 12, no. 8: 1844. https://doi.org/10.3390/polym12081844
APA StyleKim, Y. -G., Wichaita, W., & Thérien-Aubin, H. (2020). Influence of the Architecture of Soft Polymer-Functionalized Polymer Nanoparticles on Their Dynamics in Suspension. Polymers, 12(8), 1844. https://doi.org/10.3390/polym12081844