Lastingly Colored Polylactide Synthesized by Dye-Initiated Polymerization




Abstract
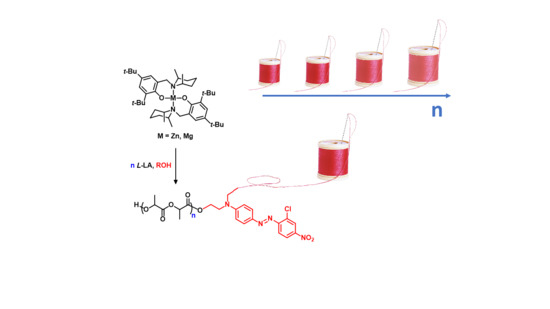
:
1. Introduction
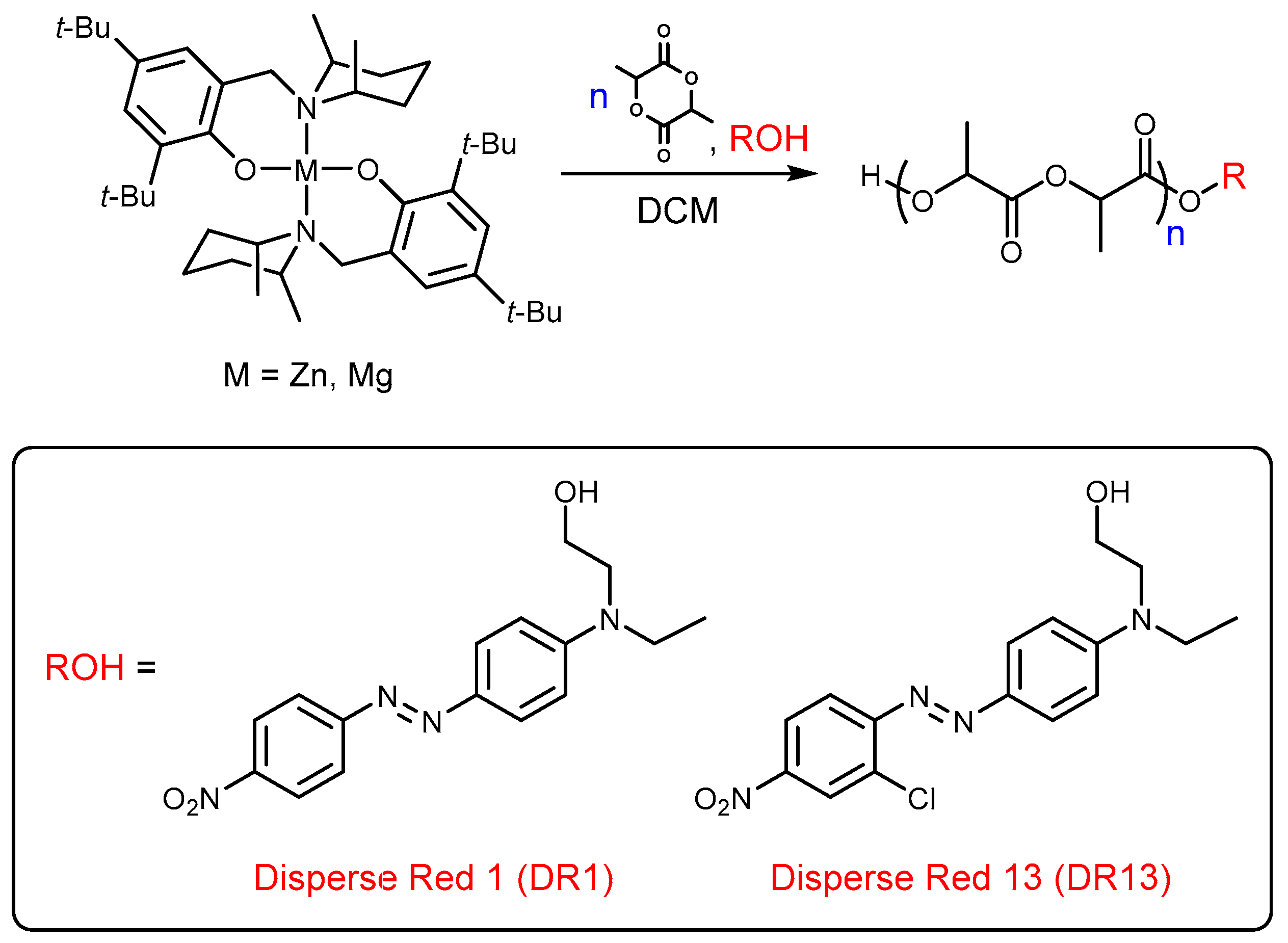
2. Materials and Methods
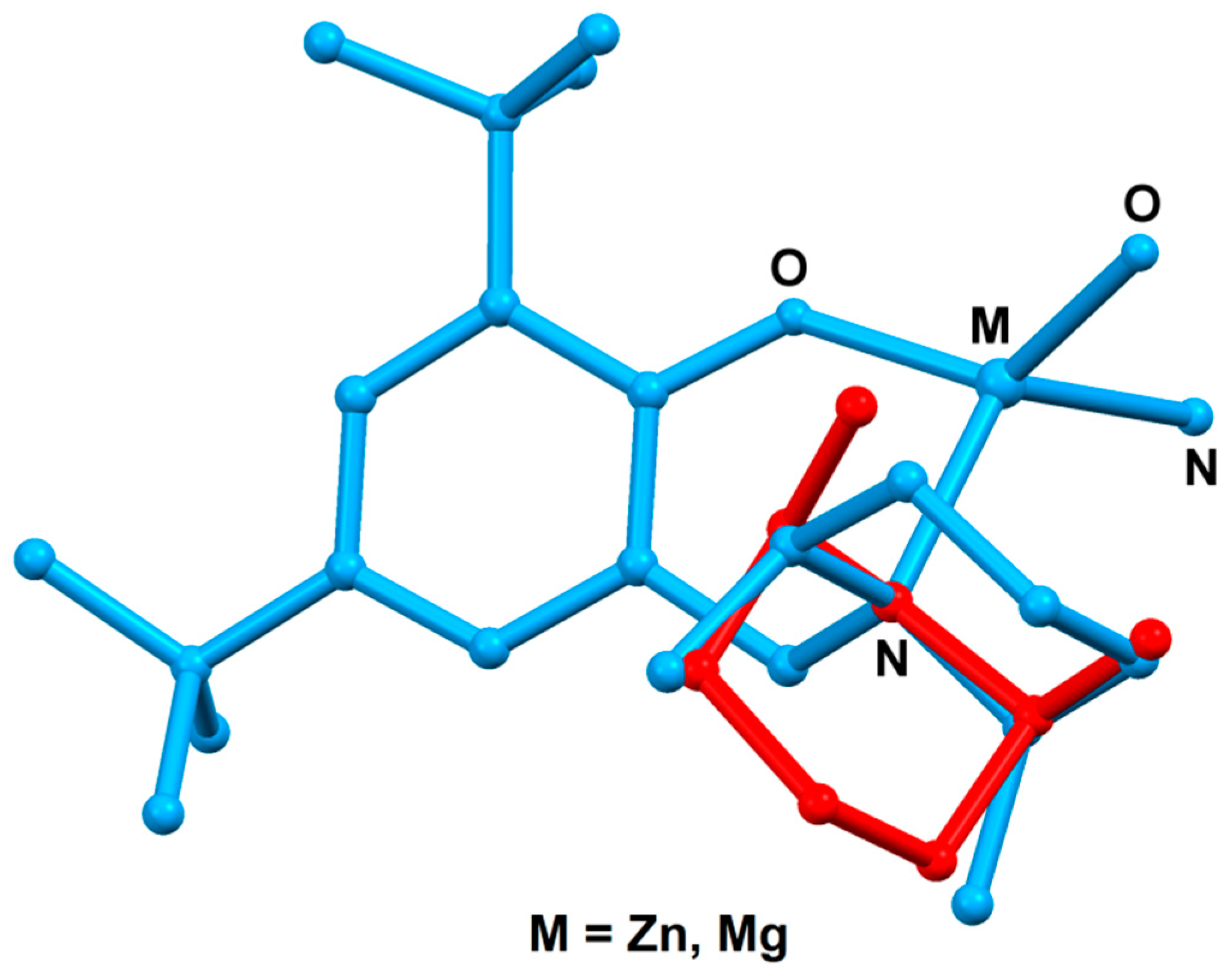
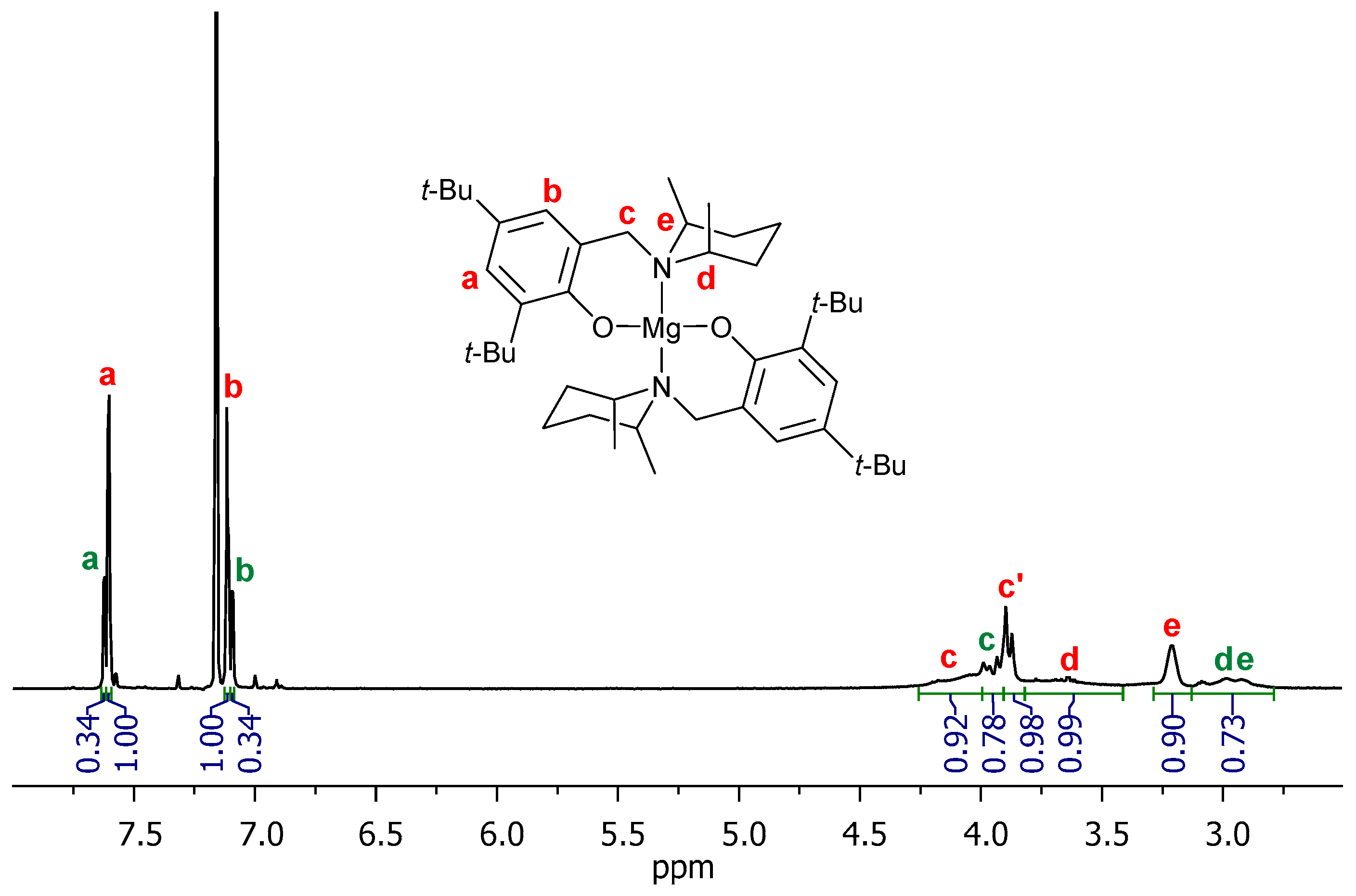
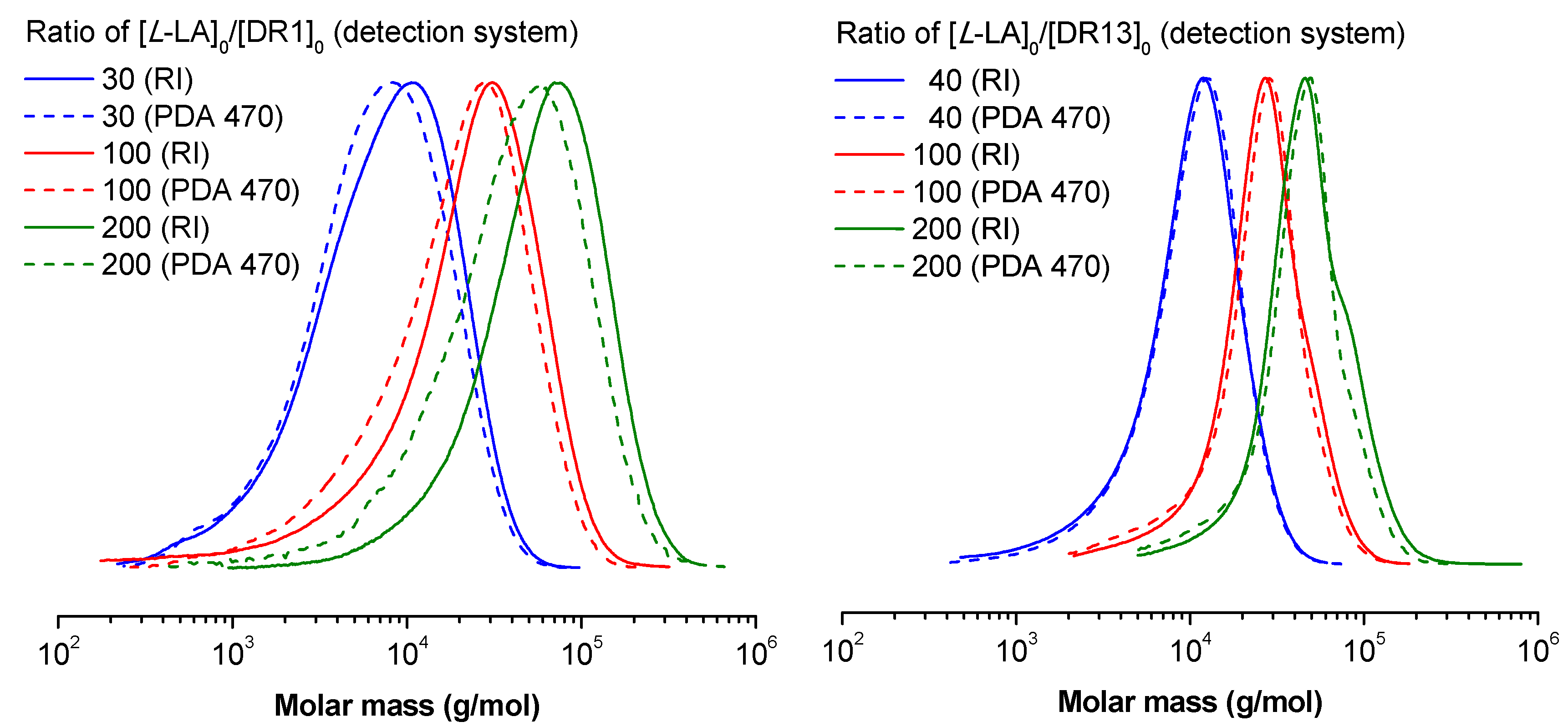
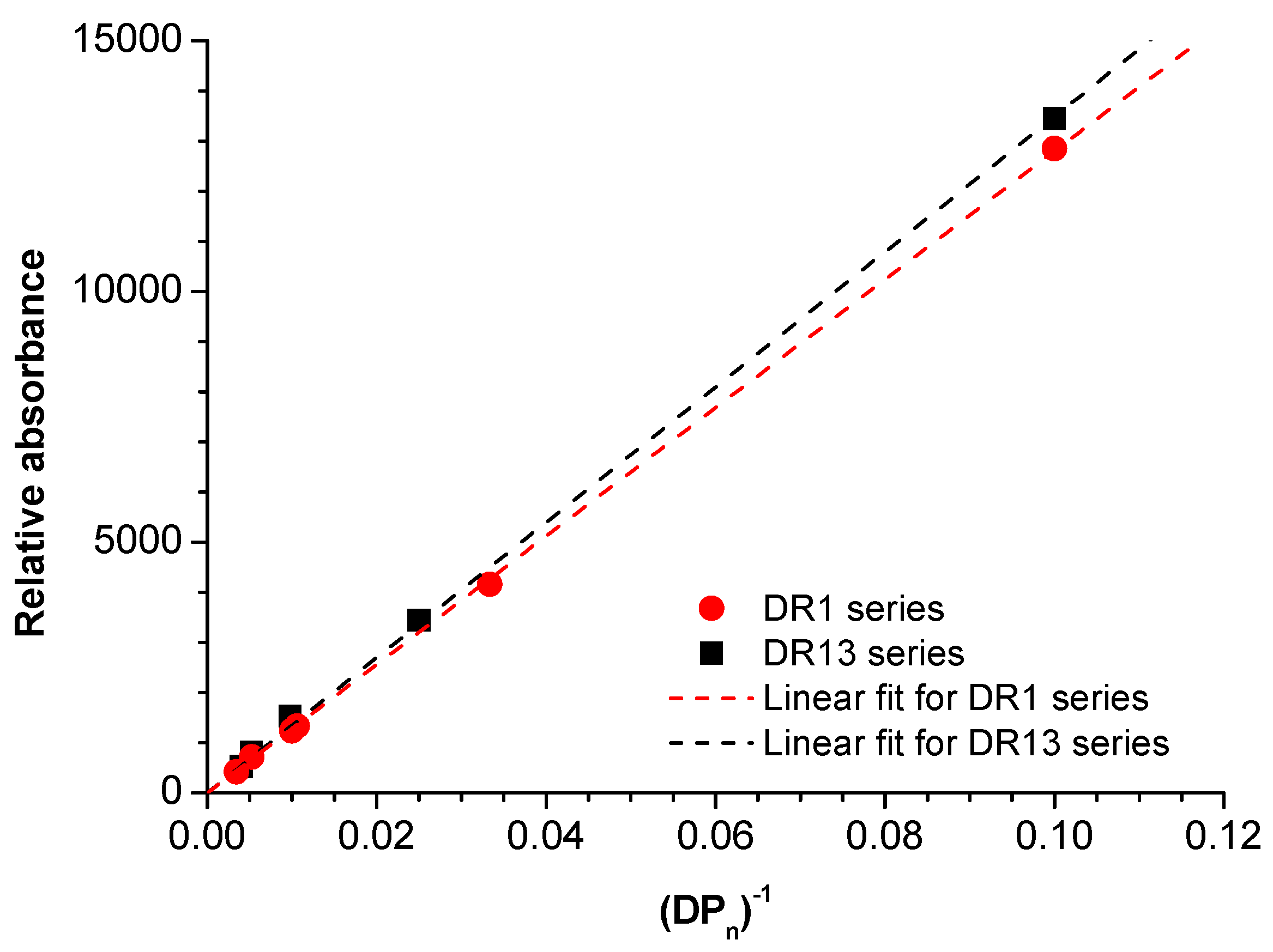
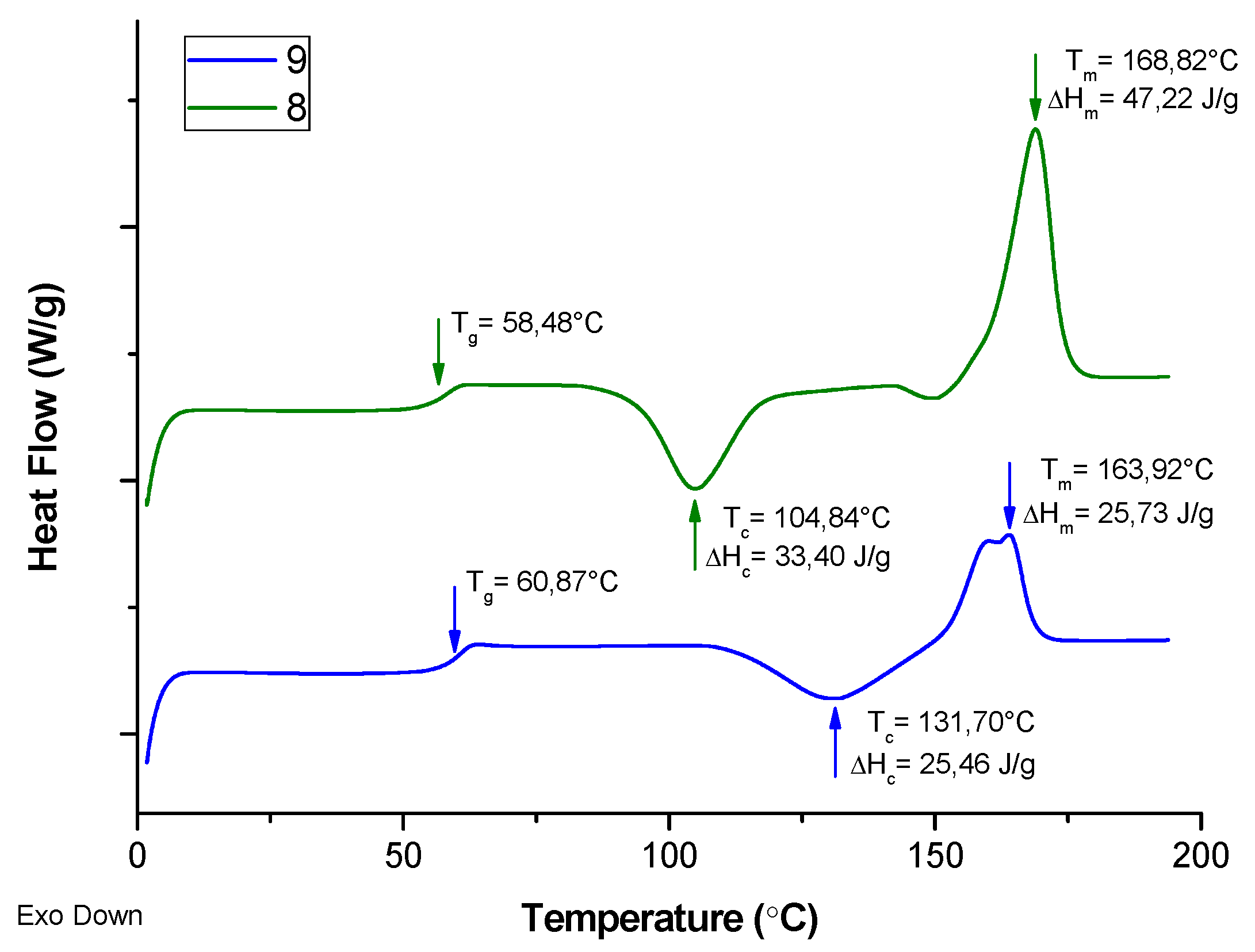
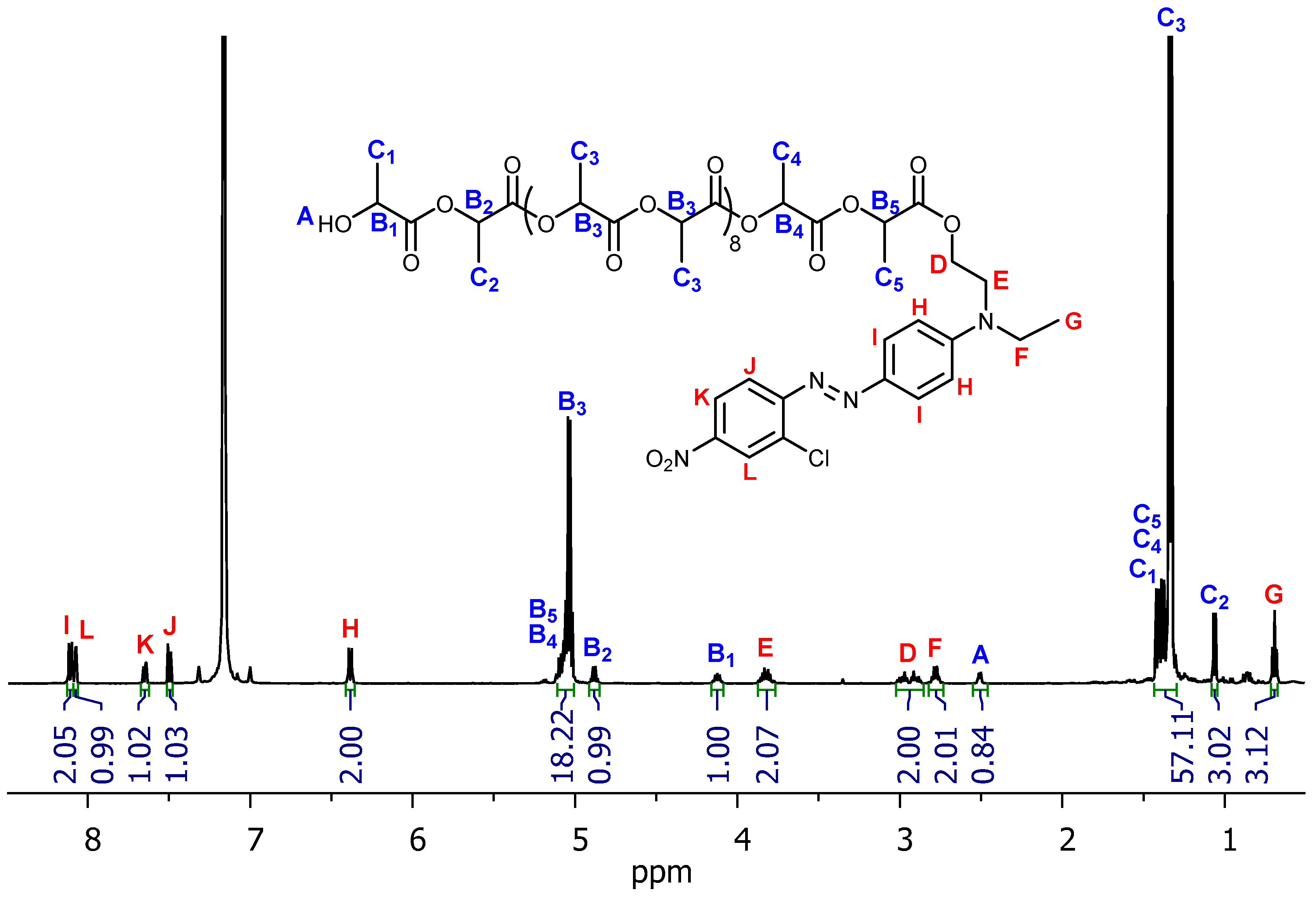
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
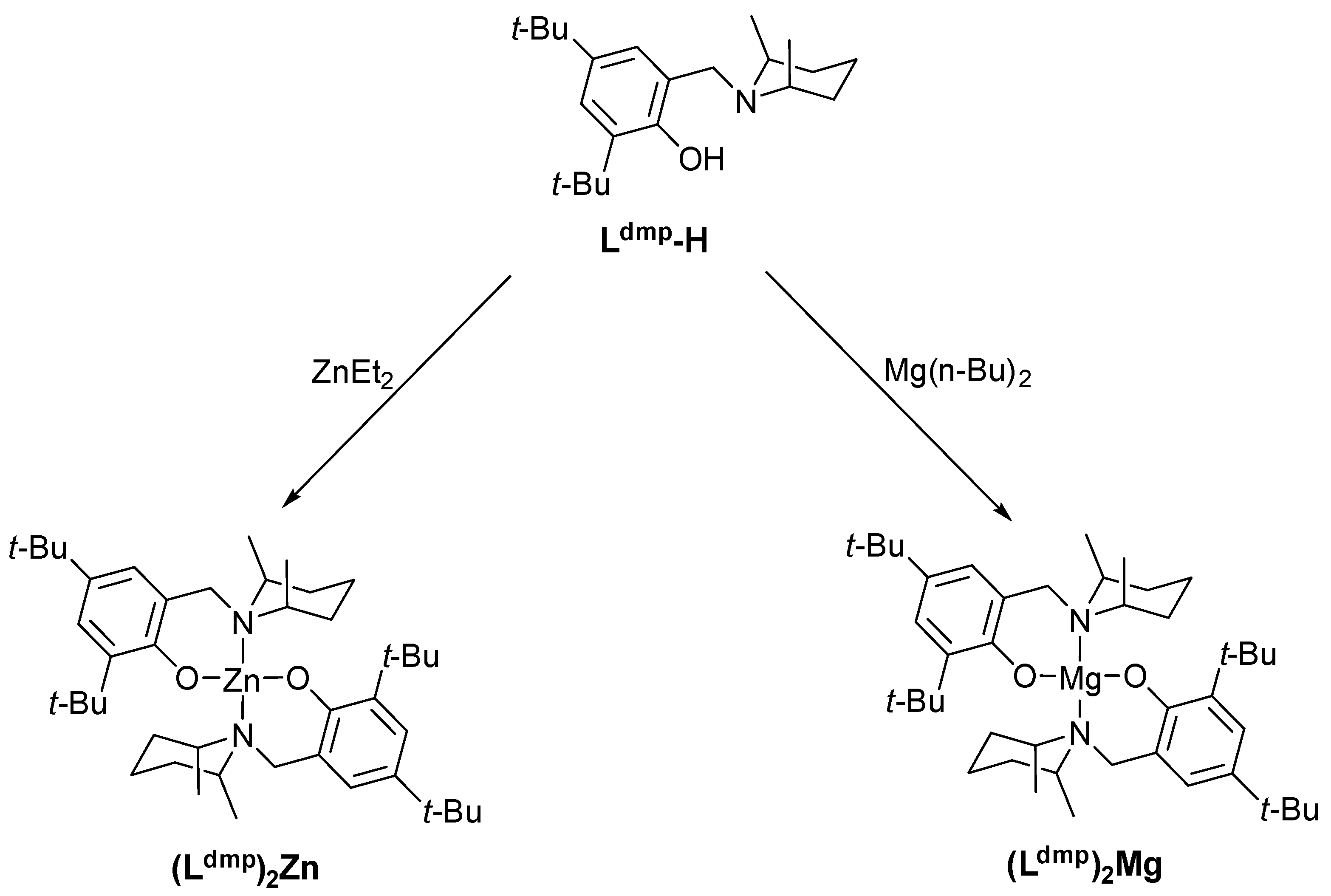
Appendix A.1. Syntheses
Appendix A.1.1. 2:4-di-tert-butyl-6-(((2R,6S)-2,6-dimethylpiperidin-1-yl)methyl)phenol, Ldmp-H
Appendix A.1.2. (Ldmp)2Zn
Appendix A.1.3. (Ldmp)2Mg
Appendix A.1.4. Representative Procedure for Solution Polymerization
Appendix A.1.5. PLA-10-DR1
Appendix A.1.6. PLA-10-DR13
References
- Schneiderman, D.K.; Hillmyer, M.A. 50th Anniversary Perspective: There Is a Great Future in Sustainable Polymers. Macromolecules 2017, 50, 3733–3749. [Google Scholar] [CrossRef]
- Law, K.L.; Thompson, R.C. Microplastics in the seas. Science 2014, 345, 144–145. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.A. Sustainable Polymers: Opportunities for the Next Decade. ACS Macro Lett. 2013, 2, 550–554. [Google Scholar] [CrossRef] [Green Version]
- Vink, E.T.H.; Rábago, K.R.; Glassner, D.A.; Gruber, P.R. Applications of life cycle assessment to NatureWorksTM polylactide (PLA) production. Polym. Degrad. Stab. 2003, 80, 403–419. [Google Scholar] [CrossRef]
- Brusseau, M.L. Chapter 32-Sustainable Development and Other Solutions to Pollution and Global Change. In Environmental and Pollution Science, 3rd ed.; Brusseau, M.L., Pepper, I.L., Gerba, C.P., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 585–603. [Google Scholar] [CrossRef]
- Suzuki, S.; Ikada, Y. Chapter 27 Medical Applications. In Poly(Lactic Acid): Synthesis, Structure, Properties, Processing and Applications; Auras, R.A., Lim, L.-T., Selke, S.E.M., Tsuji, H., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp. 443–456. [Google Scholar] [CrossRef]
- Castro-Aguirre, E.; Iniguez-Franco, F.; Samsudin, H.; Fang, X.; Auras, R. Poly(lactic acid)-Mass production, processing, industrial applications, and end of life. Adv. Drug. Deliv. Rev. 2016, 107, 333–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dusselier, M.; Van Wouwe, P.; Dewaele, A.; Jacobs, P.A.; Sels, B.F. Shape-selective zeolite catalysis for bioplastics production. Science 2015, 349, 78–80. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, J. Drug-Initiated Synthesis of Polymer Prodrugs: Combining Simplicity and Efficacy in Drug Delivery. Chem. Mater. 2016, 28, 1591–1606. [Google Scholar] [CrossRef]
- Yu, W.; Foster, J.C.; Dove, A.P.; O’Reilly, R.K. Length Control of Biodegradable Fiber-Like Micelles via Tuning Solubility: A Self-Seeding Crystallization-Driven Self-Assembly of Poly(ϵ-caprolactone)-Containing Triblock Copolymers. Macromolecules 2020, 53, 1514–1521. [Google Scholar] [CrossRef]
- Christian D’Alterio, M.; De Rosa, C.; Talarico, G. Stereoselective Lactide Polymerization: The Challenge of Chiral Catalyst Recognition. ACS Catal. 2020, 10, 2221–2225. [Google Scholar] [CrossRef]
- McKeown, P.; Román-Ramírez, L.A.; Bates, S.; Wood, J.; Jones, M.D. Zinc Complexes for PLA Formation and Chemical Recycling: Towards a Circular Economy. ChemSusChem 2019, 12, 5233–5238. [Google Scholar] [CrossRef]
- Suesat, J.; Suwanruji, P. Dyeing and Fastness Properties of Disperse Dyes on Poly(Lactic Acid) Fiber. In Textile Dyeing; Hauser, P., Ed.; InTech: Rijeka, Croatia, 2011; pp. 351–372. [Google Scholar] [CrossRef]
- Farah, S.; Anderson, D.G.; Langer, R. Physical and mechanical properties of PLA, and their functions in widespread applications-A comprehensive review. Adv. Drug. Deliv. Rev. 2016, 107, 367–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasprilla, A.J.R.; Martinez, G.A.R.; Lunelli, B.H.; Jardini, A.L.; Maciel, R. Poly-lactic acid synthesis for application in biomedical devices-A review. Biotechnol. Adv. 2012, 30, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Standau, T.; Zhao, C.J.; Castellon, S.M.; Bonten, C.; Altstadt, V. Chemical Modification and Foam Processing of Polylactide (PLA). Polymers 2019, 11, 306. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.G.; Cui, W.J.; Bei, J.Z. Bulk and surface modifications of polylactide. Anal. Bioanal. Chem. 2005, 381, 547–556. [Google Scholar] [CrossRef]
- Saulnier, B.; Ponsart, S.; Coudane, J.; Garreau, H.; Vert, M. Lactic acid-based functionalized polymers via copolymerization and chemical modification. Macromol. Biosci. 2004, 4, 232–237. [Google Scholar] [CrossRef]
- Rasal, R.M.; Janorkar, A.V.; Hirt, D.E. Poly(lactic acid) modifications. Prog. Polym. Sci. 2010, 35, 338–356. [Google Scholar] [CrossRef]
- Jamshidian, M.; Tehrany, E.A.; Imran, M.; Jacquot, M.; Desobry, S. Poly-Lactic Acid: Production, Applications, Nanocomposites, and Release Studies. Compr. Rev. Food Sci. Food Saf. 2010, 9, 552–571. [Google Scholar] [CrossRef]
- Kim, H.-S.; Park, Y.-K.; Jo, A.-R.; Lee, J.-J. Dispersant-free Dyeing of Poly(lactic acid) Knitted Fabric with Temporarily Solubilized Azo Disperse Dyes. Fibers Polym. 2017, 18, 1263–1268. [Google Scholar] [CrossRef]
- Phillips, D.; Suesat, J.; Taylor, J.A.; Wilding, M.; Farrington, D.; Bone, J.; Dervan, S. Thermal migration of selected disperse dyes on poly(ethylene terephthalate) and poly(lactic acid) (Ingeo) fibres. Color. Technol. 2004, 120, 260–264. [Google Scholar] [CrossRef]
- Avinc, O.; Phillips, D.; Wilding, M. Influence of different finishing conditions on the wet fastness of selected disperse dyes on polylactic acid fabrics. Color. Technol. 2009, 125, 288–295. [Google Scholar] [CrossRef]
- Thongkham, S.; Monot, J.; Martin-Vaca, B.; Bourissou, D. Simple in based dual catalyst enables siginificant progress in decalactone ring-opening (co)polymerization. Macromolecules 2019, 52, 8102–8113. [Google Scholar] [CrossRef]
- Tanzi, M.C.; Verderio, P.; Lampugnani, M.G.; Resnati, M.; Dejana, E.; Sturani, E. Cytotoxicity of some catalysts commonly used in the synthesis of copolymers for biomedical. J. Mat. Sci. Mat. Med. 1994, 5, 393–396. [Google Scholar] [CrossRef]
- Ghosh, S.; Glöcker, E.; Wölper, C.; Tjaberings, A.; Gröschel, A.H.; Schultz, S. Heteroleptic β-Ketoiminate magnesium catalysts for the Ring-Opening Polymerization of Lactide. Organometallics 2020. [Google Scholar] [CrossRef]
- De Groot, A.P.; Feron, V.J.; Til, H.P. Short-term toxicity studies on some salts and oxides of tin in rats. Food Cosmet. Toxicol. 1973, 11, 19–30. [Google Scholar] [CrossRef]
- European Food Safety Authority. Opinion of the Scientific Panel on Contaminants in the Food Chain on a request from the Commission to assess the health risks to consumers associated with exposure to organotin in foodstuff. EFSA J. 2004, 102, 1–119. [Google Scholar]
- Mori, T.; Nishida, H.; Shirai, Y.; Endo, T. Effect of chain end structures on pyrolysis of poly(l-lactic acid) containing tin atoms. Polym. Degrad. Stab. 2004, 84, 243–251. [Google Scholar] [CrossRef]
- Cam, D.; Marucci, M. Influence of residual monomers and metals on poly(L-lactide) thermal stability. Polymer 1997, 38, 1879–1884. [Google Scholar] [CrossRef]
- Wojtczak, E.; Kubisa, P.; Bednarek, M. Thermal stability of polylactide with different and groups depending on the catalyst used for the polymerization. Polym. Degrad. Stab. 2018, 151, 100–104. [Google Scholar] [CrossRef]
- Kopinke, F.-D.; Remmler, M.; Mackenzie, K.; Moder, M.; Wachsen, O. Thermal decomposition of biodegradable polyesrers-II. Poly (lactic acid). Polym. Derrad. Stab. 1996, 53, 329–342. [Google Scholar] [CrossRef]
- Dos Santos Vieira, I.; Herres-Pawlis, S. Lactide Polymerisation with Complexes of Neutral N-Donors–New Strategies for Robust Catalysts. Eur. J. Inorg. Chem. 2012, 765–774. [Google Scholar] [CrossRef]
- Sarazin, Y.; Carpentier, J.-F. Discrete Cationic Complexes for Ring-Opening Polymerization Catalysis of Cyclic Esters and Epoxides. Chem. Rev. 2015, 115, 3564–3614. [Google Scholar] [CrossRef]
- Li, H.; Shakaroun, R.M.; Guillaume, S.M.; Carpentier, J.-F. Recent Advances in Metal-Mediated Stereoselective Ring-Opening Polymerization of Functional Cyclic Esters towards Well-Defined Poly(hydroxy acid)s: From Stereoselectivity to Sequence-Control. Chem. Eur. J. 2020, 26, 128–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jędrzkiewicz, D.; Czeluśniak, I.; Wierzejewska, M.; Szafert, S.; Ejfler, J. Well-Controlled, Zinc-Catalyzed Synthesis of Low Molecular Weight Oligolactides by Ring Opening Reaction. J. Mol. Catal. A Chem. 2015, 396, 155–163. [Google Scholar] [CrossRef]
- Naumann, S.; Scholten, P.B.V.; Wilson, J.A.; Dove, A.P. Dual Catalysis for Selective Ring-Opening Polymerization of Lactones: Evolution toward Simplicity. J. Am. Chem. Soc. 2015, 137, 14439–14445. [Google Scholar] [CrossRef] [PubMed]
- Jędrzkiewicz, D.; Ejfler, J.; Gulia, N.; John, Ł.; Szafert, S. Designing Ancillary Ligands for Heteroleptic/Homoleptic Zinc Complex Formation: Synthesis, Structures and Application in ROP of Lactides. Dalton Trans. 2015, 44, 13700–13715. [Google Scholar] [CrossRef] [PubMed]
- Ejfler, J.; Szafert, S.; Mierzwicki, K.; Jerzykiewicz, L.B.; Sobota, P. Homo- and heteroleptic zinc aminophenolates as initiators for lactide polymerization. Dalton Trans. 2008, 46, 6556–6562. [Google Scholar] [CrossRef] [PubMed]
- Jędrzkiewicz, D.; Adamus, G.; Kwiecień, M.; John, Ł.; Ejfler, J. Lactide as the Playmaker of the ROP Game: Theoretical and Experimental Investigation of Ring-opening Polymerization of Lactide Initiated by Aminonaphtholate Zinc complexes. Inorg. Chem. 2017, 56, 1349–1365. [Google Scholar] [CrossRef]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef]
- CrysAlisRED Software; Oxford Diffraction: Wrocław, Poland, 1995–2004.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; Mccabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Van De Streek, J.; Wood, P.A. Mercury CSD 2.0-New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Farwell, J.D.; Hitchcock, P.B.; Lappert, M.F.; Luinstra, G.A.; Protchenko, A.V.; Wei, X.-H. Synthesis and structures of some sterically hindered zinc complexes containing 6-membered ZnNCCCN and ZnOCCCN rings. J. Organomet. Chem. 2008, 693, 1861–1869. [Google Scholar] [CrossRef]
- Ikpo, N.; Saunders, L.N.; Walsh, J.L.; Smith, J.M.B.; Dawe, L.N.; Kerton, F.M. Zinc Complexes of Piperazinyl-Derived Aminephenolate Ligands: Synthesis, Characterization and Ring–Opening Polymerization Activity. Eur. J. Inorg. Chem. 2011, 35, 5347–5359. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Zhao, G.; Fablet, R.; Bouyahyi, M.; Thomas, C.M.; Roisnel, T.; Casagrande Jr., O.; Carpentier, J.-F. Zinc and enolato-magnesium complexes based on bi-, tri- and tetradentate aminophenolate ligands. New J. Chem. 2008, 32, 2279–2291. [Google Scholar] [CrossRef]
- Grala, A.; Ejfler, J.; Jerzykiewicz, L.B.; Sobota, P. Chemoselective alcoholysis of lactide mediated by a magnesium catalyst: An efficient route to alkyl lactyllactate. Dalton Trans. 2011, 40, 4042–4044. [Google Scholar] [CrossRef] [PubMed]
- Ejfler, J.; Krauzy-Dziedzic, K.; Szafert, S.; Jerzykiewicz, L.B.; Sobota, P. Synthesis, characterization, and catalytic studies of (aryloxido)magnesium complexes. Eur. J. Inorg. Chem. 2010, 23, 3602–3609. [Google Scholar] [CrossRef]
- Shere, H.; McKeown, P.; Mahon, M.F.; Jones, M.D. Making the cut: Monopyrrolidine-based complexes for the ROP of lactide. Eur. Polym. J. 2019, 114, 319–325. [Google Scholar] [CrossRef]
- Kowalski, A.; Libiszowski, J.; Duda, A.; Penczek, S. Polymerization of L,L-Dilactide Initiated by Tin (II) Butoxide. Macromolecules 2000, 33, 1964–1971. [Google Scholar] [CrossRef]















{kind=link}
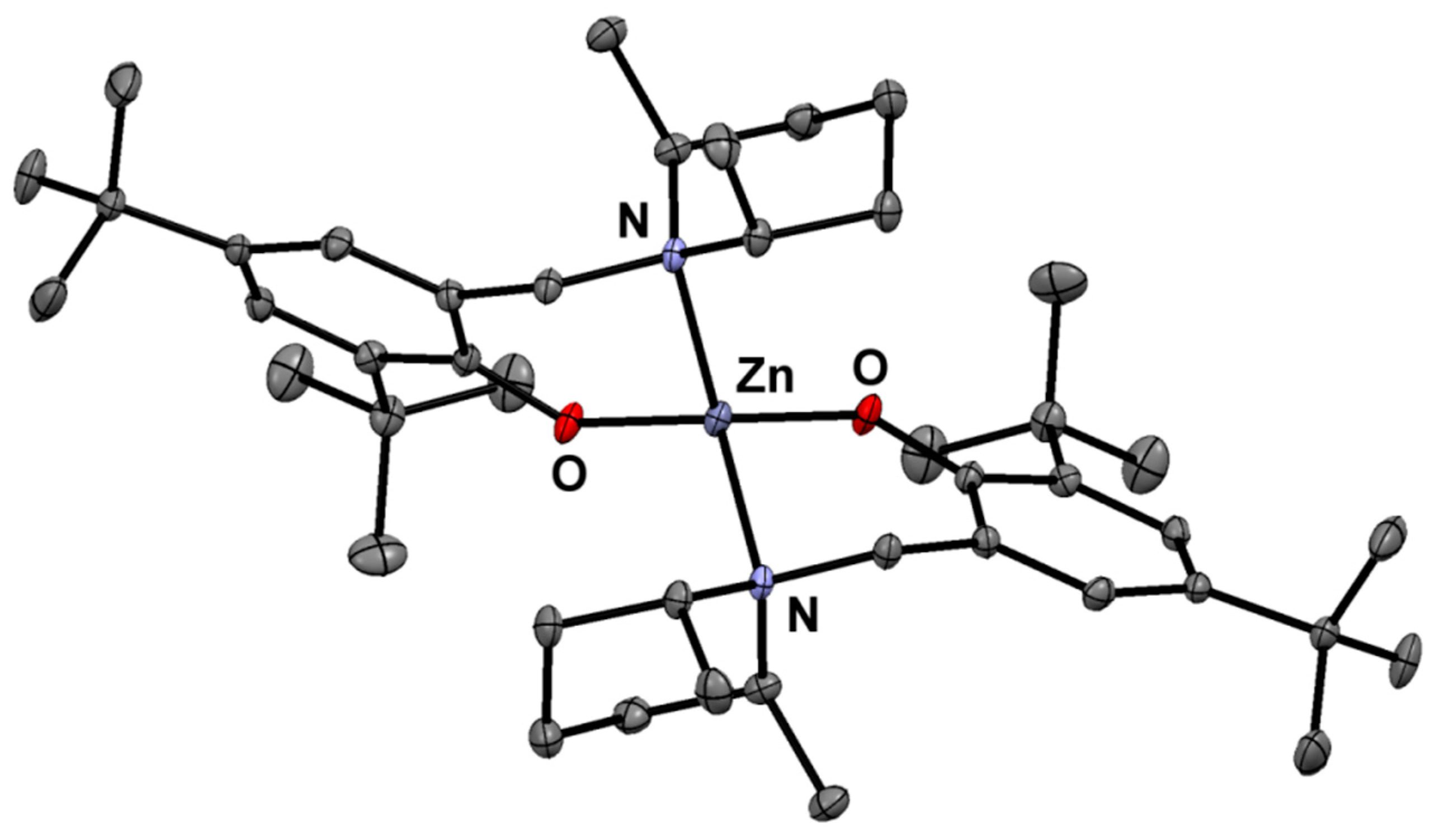{kind=link}
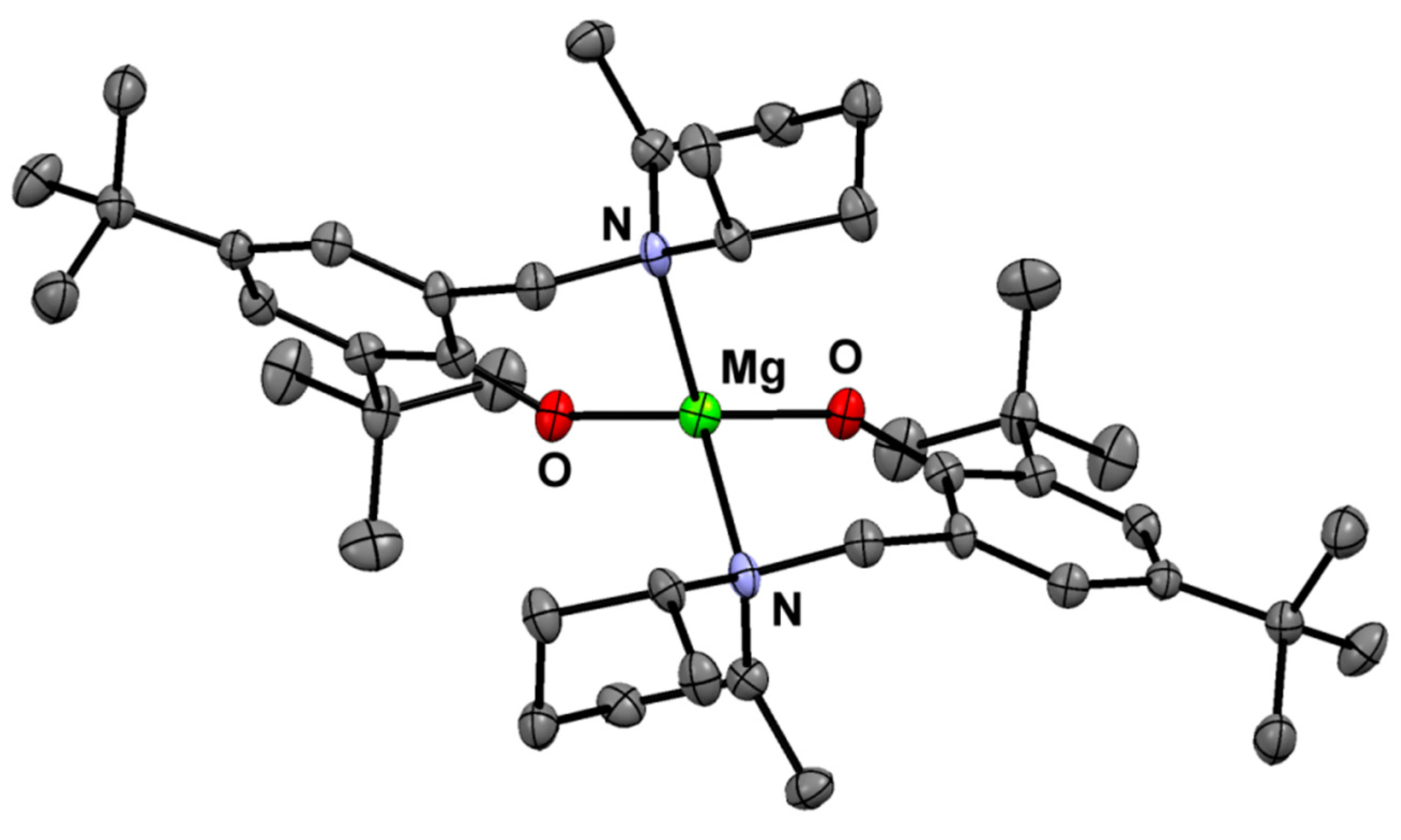{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Initiator [I] | ROH | Molar Ratios a | Time (min.) | pL-LA (%) b | DPn c | Mn,cal d | CC-RI-GPCe | TDA-GPC f | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mn,PS | ƉM,PS | Mn,TDA | ƉM,TDA | ||||||||
| 1 | (Ldmp)2Zn | DR13 | 1/10/1 | 5 | 100 | 10 | 1.79 | 1.78 | 1.90 | 6.87 | 1.10 |
| 2 | (Ldmp)2Zn | DR13 | 1/40/1 | 5 | 100 | 40 | 6.11 | 6.79 | 1.81 | 12.9 | 1.04 |
| 3 | (Ldmp)2Zn | DR13 | 1/100/1 | 5 | 99.1 | 102 | 14.6 | 19.8 | 1.52 | 14.6 | 1.28 |
| 4 | (Ldmp)2Zn | DR13 | 1/200/1 | 10 | 97.3 | 192 | 28.4 | 34.8 | 1.55 | 31.6 | 1.10 |
| 5 | (Ldmp)2Mg | DR13 | 1/250/1 | 15 | 99.1 | 248 | 36.1 | 43.7 | 1.51 | 29.8 | 1.31 |
| 6 | (Ldmp)2Zn | DR1 | 1/10/1 | 5 | 100 | 10 | 1,76 | 1.39 | 2.00 | 3.77 | 1.05 |
| 7 | (Ldmp)2Zn | DR1 | 1/30/1 | 5 | 100 | 30 | 4.64 | 4.81 | 2.29 | 9.29 | 1.02 |
| 8 | (Ldmp)2Zn | DR1 | 1/100/1 | 5 | 99.8 | 100 | 14.7 | 10.4 | 3.12 | 15.5 | 1.25 |
| 9 | (Ldmp)2Mg | DR1 | 1/100/1 | 5 | 97.8 | 94 | 14.4 | 10.3 | 3.74 | 52.5 | 1.50 |
| 10 | (Ldmp)2Mg | DR1 | 1/200/1 | 15 | 99.6 | 190 | 29.0 | 38.9 | 1.97 | 21.6 | 1.63 |
| 11 | (Ldmp)2Mg | DR1 | 1/300/1 | 30 | 99.9 | 289 | 43.5 | 55.8 | 2.06 | 36.2 | 1.72 |
| No. (Corresp. to Table 1) | Fraction of Number of Molecules in Population (%) a | |||||||
|---|---|---|---|---|---|---|---|---|
| Macrocycles | H–(LA)n–OH | DR1–(LA)n–OH | DR1–(LA)n–OH | |||||
| Even | Odd | Even | Odd | Even | Odd | Even | Odd | |
| 1 | 1.3 | 62.1 | 36.6 | |||||
| 2 | 1.9 | 2.1 | 1.1 | 0.9 | 52.3 | 41.7 | ||
| 3 | 11.4 | 11.7 | 39.1 | 37.8 | ||||
| 4 | 12.8 | 13.5 | 38.0 | 35.7 | ||||
| 6 | 60.4 | 39.6 | ||||||
| 7 | 1.2 | 1.2 | 48.7 | 48.9 | ||||
| 8 | 5.6 | 5.3 | 44.4 | 44.7 | ||||
| 9 | 9.1 | 9.1 | 41.9 | 39.9 | ||||
| 10 | 6.9 | 7.1 | 43.0 | 43.0 | ||||
| 11 | 20.2 | 21.2 | 29.6 | 29.0 | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jędrzkiewicz, D.; Kowalczyk, S.; Plichta, A.; Ejfler, J. Lastingly Colored Polylactide Synthesized by Dye-Initiated Polymerization. Polymers 2020, 12, 1980. https://doi.org/10.3390/polym12091980
Jędrzkiewicz D, Kowalczyk S, Plichta A, Ejfler J. Lastingly Colored Polylactide Synthesized by Dye-Initiated Polymerization. Polymers. 2020; 12(9):1980. https://doi.org/10.3390/polym12091980
Chicago/Turabian StyleJędrzkiewicz, Dawid, Sebastian Kowalczyk, Andrzej Plichta, and Jolanta Ejfler. 2020. "Lastingly Colored Polylactide Synthesized by Dye-Initiated Polymerization" Polymers 12, no. 9: 1980. https://doi.org/10.3390/polym12091980
APA StyleJędrzkiewicz, D., Kowalczyk, S., Plichta, A., & Ejfler, J. (2020). Lastingly Colored Polylactide Synthesized by Dye-Initiated Polymerization. Polymers, 12(9), 1980. https://doi.org/10.3390/polym12091980