
Carbon Capture Utilization for Biopolymer Foam Manufacture: Thermal, Mechanical and Acoustic Performance of PCL/PHBV CO2 Foams



Abstract
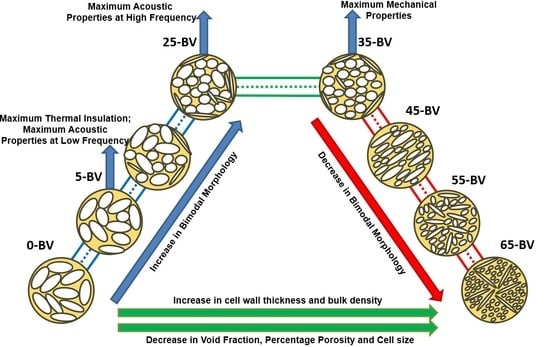
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Materials Preparation
2.3. Differential Scanning Calorimetry
2.4. Foam Density and Porosity Analysis
2.5. Scanning Electron Microscopy (SEM)
2.6. Foam Morphological Analysis
2.7. Acoustic Properties Measurement
2.8. Thermal Conductivity Measurement
2.9. Mechanical Testing (Compression)
3. Results
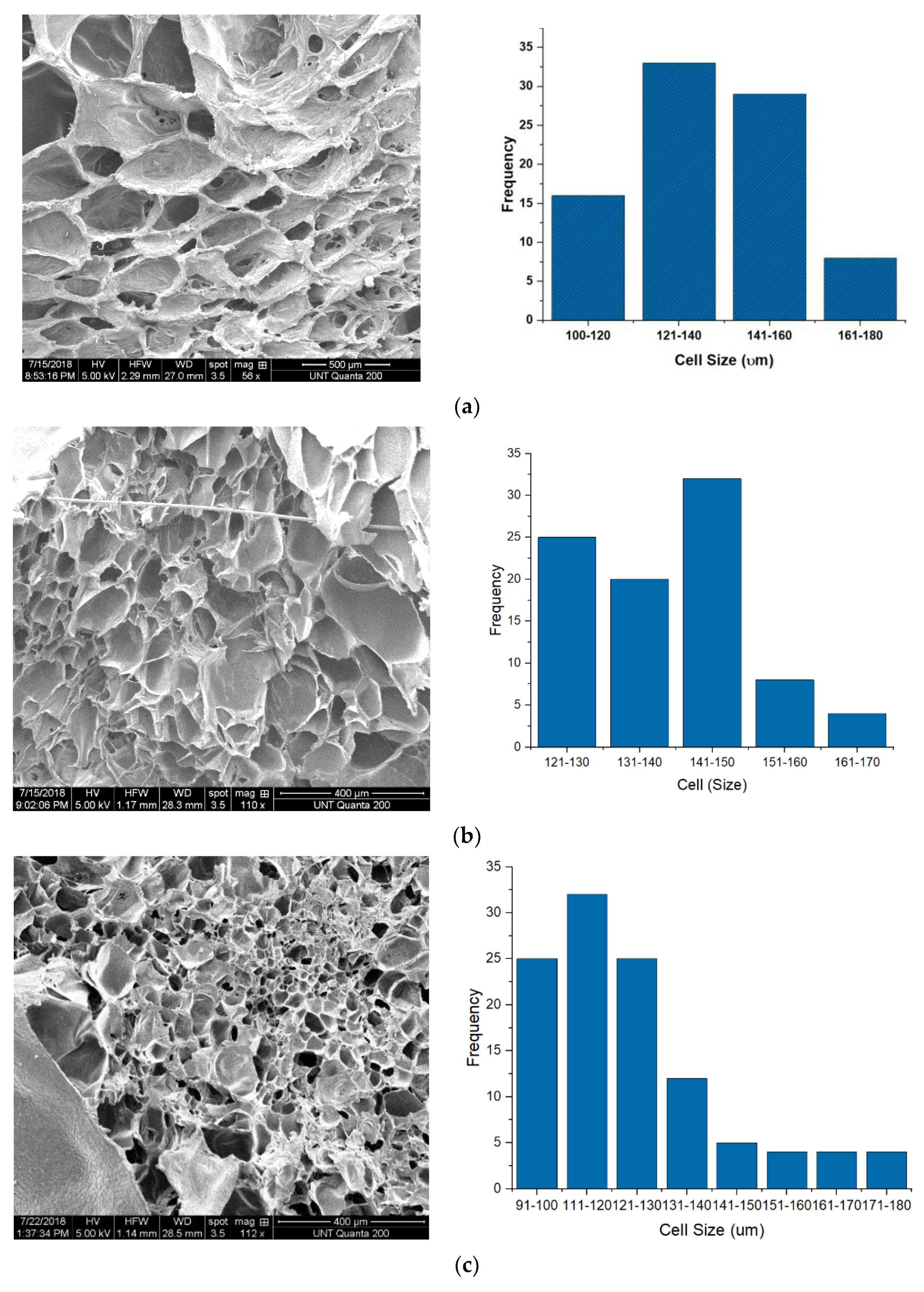
3.1. Foam Morphology
3.2. Differential Scanning Calorimetry (DSC)
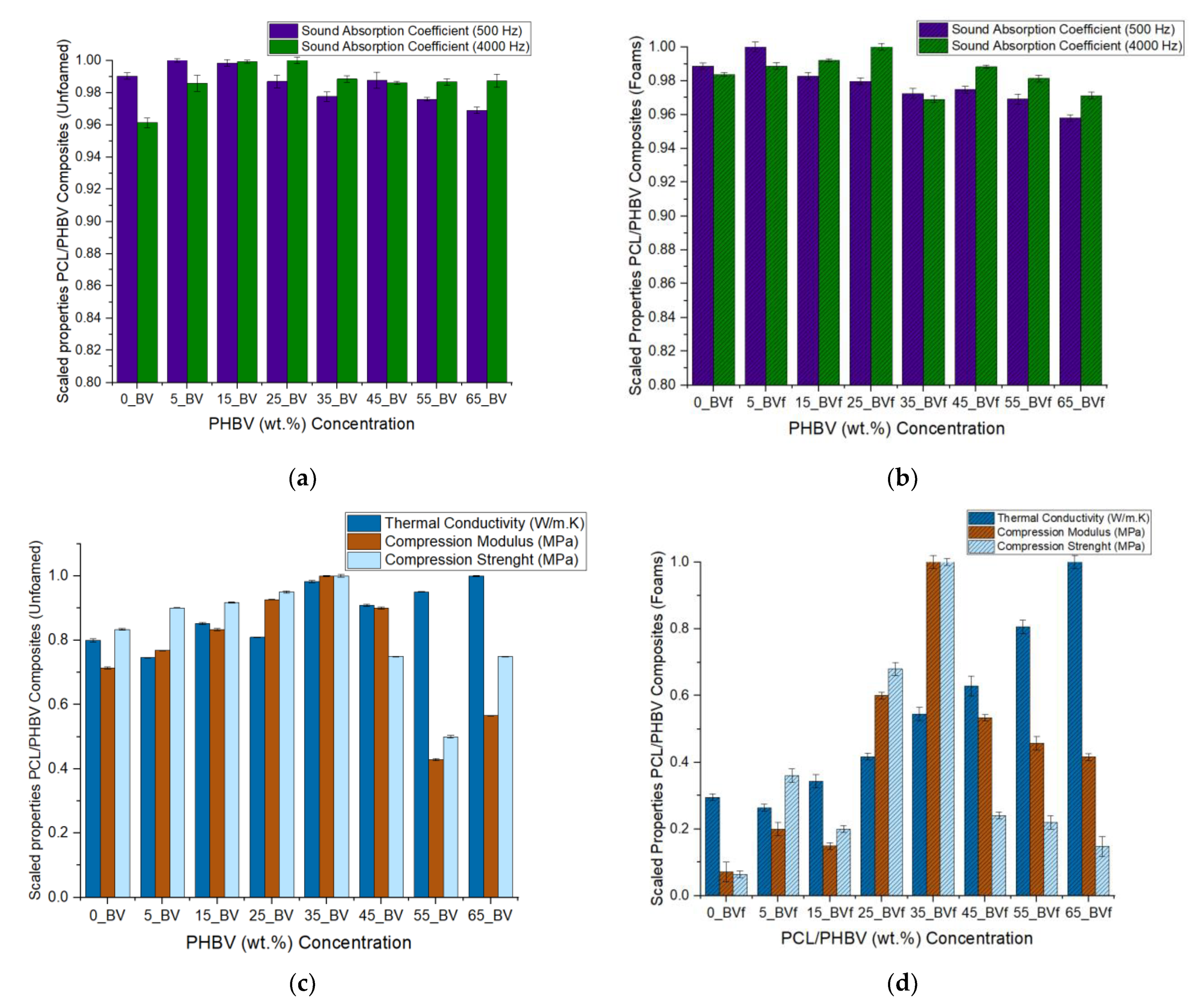
3.3. Acoustic Properties
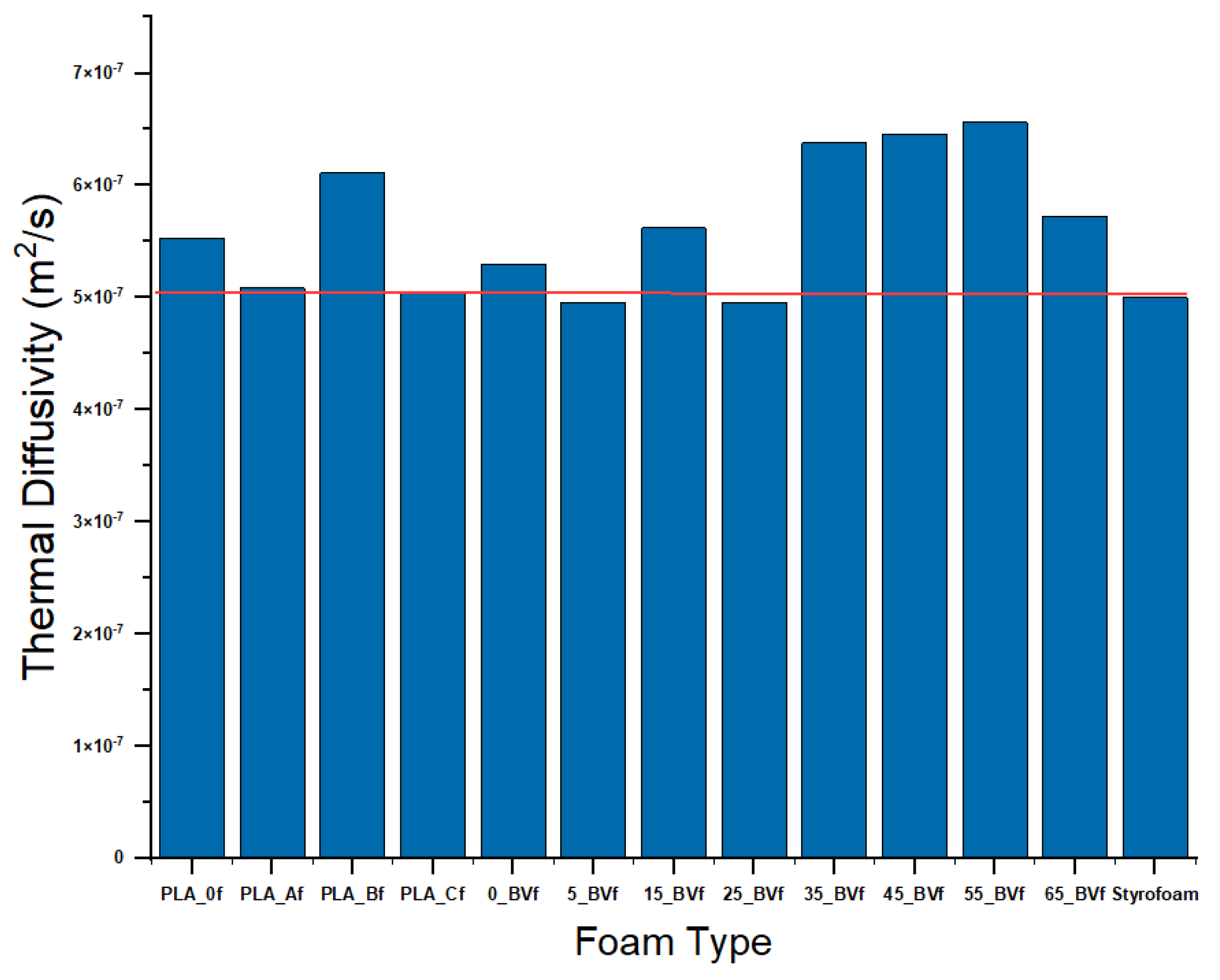
3.4. Thermal Conductivity
3.5. Mechanical Property
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Nomenclature
| Units | ||
| A | Sample area | cm2 |
| c | Particle velocity | m/S |
| Cp | Specific heat capacity at constant pressure | J/Kg·K |
| Cwt | Cell wall thickness | µm |
| Er | Expansion ratio | |
| f | Frequency | Hz |
| F | foam | |
| H°m | Heat of Fusion | J/g |
| Hm | Enthalpy of Melting | J/g |
| M | Magnification | µm |
| N | number of cells | |
| Nf | Cell density | cell/µm3 |
| Ns | Cell size | µm |
| Pfoam | Foaming pressure | MPa |
| Psat | Saturation pressure | MPa |
| R | Thermal resistance value | m·K/W |
| t | sample thickness | m, mm |
| Tc | Crystallization temperature | °C |
| Tc2 | Second cycle Tc (unfoamed polymer) | °C |
| Tcf1 | First cycle Tc (foam) | °C |
| Tfoam | Foaming temperature | °C |
| Tm | Melting temperature | °C |
| Tm° | Equilibrium Melting Point | °C |
| Tm1 | First melting temperature (unfoamed polymer) | °C |
| Tm2 | Second melting temperature (unfoamed polymer) | °C |
| Tm3 | Third melting temperature (unfoamed polymer) | °C |
| Tmf1 | First melting temperature (foams) | °C |
| Tmf2 | Second melting temperature (foams) | °C |
| Tmf3 | Third melting temperature (foams) | °C |
| Tsat | Saturation temperature | °C |
| Vf | Void fraction | % |
| X_BV | Percentage weight concentration of PHBV (unfoamed) | % |
| X_BVf | Percentage weight concentration of PHBV (foam) | % |
| Greek Symbols | ||
| Δ(.) | Increment of a quantity | |
| ϴ | Volume fraction | % |
| λ | Individual acoustic wavelength | m |
| ƛ | Thermal Conductivity | W/m·K |
| ρ | Density | g/cm3 |
| ϕ | Weight percentage of dispersed phase | % |
| ꭓ | Percentage crystallinity | % |
| Subscript | ||
| cv | convection | |
| f | foam | |
| g | gas | |
| p | polymer | |
| r | radiation | |
| s | size | |
| sat | saturation | |
| sol | solid phase | |
| 1, 2 | indices for cycles |
References
- Folger, P. Carbon Capture and Sequestration (CCS) in the United States. Congr. Res. Serv. 2018, 3–8. Available online: http://large.stanford.edu/courses/2020/ph240/villanueva1/docs/R44902.pdf (accessed on 20 July 2021).
- Li, A.; Mi, Z.; Coffman, D.M.; Wei, Y.-M. Assessing the policy impacts on non-ferrous metals industry’s CO2 reduction: Evidence from China. J. Clean. Prod. 2018, 192, 252–261. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Wang, Y.; Shi, L.; Klemeš, J.J. Uncovering energy use, carbon emissions and environmental burdens of pulp and paper industry: A systematic review and meta-analysis. Renew Sustain. Energy Rev. 2018, 92, 823–833. [Google Scholar] [CrossRef]
- Ketzer, M.; Iglesias, R.; Einloft, S. Reducing Greenhouse Gas Emissions with CO2 Capture and Geological Storage. In Handbook of Climate Change Mitigation; Springer: New York, NY, USA, 2012. [Google Scholar] [CrossRef]
- Romasheva, N.V.; Kruk, M.N.; Cherepovitsyn, A.E. Propagation perspectives of CO2 sequestration in the world. Int. J. Mech. Eng. Technol. 2018, 9, 1877–1885. [Google Scholar]
- Jiang, K.; Ashworth, P.; Zhang, S.; Liang, X.; Sun, Y.; Angus, D. China’s carbon capture, utilization and storage (CCUS) policy: A critical review. Renew Sustain. Energy Rev. 2019, 119, 10960. [Google Scholar] [CrossRef]
- Robert, F. A tertiary injectant refers to the use of CO2 for EOR or Enhanced Natural Gas Recovery. www.crs.gov. R44902; pp. 5–7. Available online: https://www.energy.gov/fe/science-innovation/office-clean-coal-and-carbon-management (accessed on 20 July 2021).
- DOE’s Office of Fossil Energy Refers to Its CCS Program Activities as the Carbon Capture, Utilization, and Storage. Available online: https://www.energy.gov/fe/science-innovation/office-clean-coal-and-carbon-management/carbon-capture-utilization-and-storage (accessed on 30 March 2021).
- U.S. DOE, National Energy Technology Laboratory, CO2 Utilization Focus Area. Available online: https://www.netl.doe.gov/research/coal/carbon-storage/research-and-development/CO2-utilization (accessed on 30 June 2021).
- Liu, P.S.; Chen, G.F. Application of Polymer Foams Chapter Eight—Applications of Polymer Foams. In Porous Materials. Processing and Applications; 2014; pp. 383–410. [Google Scholar] [CrossRef]
- Austin, A.; Hicks, D.A. A review of the global PU industry 2016 and outlook for 2017. PU Mag. 2017, 14, 1. [Google Scholar]
- Forest, P.; Chaumonta, P.; Cassagnau, P.; Swobodab, B.; Sonntag, P. Polymer nano-foams for insulating applications prepared from CO2 foaming. Prog. Polym. Sci. 2015, 41, 122–145. [Google Scholar] [CrossRef]
- Jost, V.; Kopitzky, R. Blending of Polyhydroxybutyrate-co-valerate with Polylactic Acid for Packaging Applications—Reflections on Miscibility and Effects on the Mechanical and Barrier Properties. Chem. Biochem. Eng. Q. 2015, 29, 221–246. [Google Scholar] [CrossRef]
- Auras, R.; Harte, B.; Selke, S. An overview of polylactides as packaging materials. Macromol. Biosci. 2004, 4, 835–864. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, M.; Hirami, M. Structural effects on biodegradation of aliphatic polyesters. Polym. Adv. Technol. 1997, 8, 203. [Google Scholar] [CrossRef]
- Tu, C.; Cai, Q.; Yang, J.; Wan, Y.; Bei, J.; Wang, S. The fabrication and characterization of poly (lactic acid) scaffolds for tissue engineering by improved solid–liquid phase separation. Polym. Adv. Technol. 2003, 14, 565–573. [Google Scholar] [CrossRef]
- Lee, J.H.; Park, T.G.; Park, H.S.; Lee, D.S.; Lee, Y.K.; Yoon, S.C.; Nam, J.D. Thermal and mechanical characteristics of poly (l-lactic acid) nanocomposite scaffold. Biomaterials 2005, 24, 2773–2778. [Google Scholar] [CrossRef]
- Karageorgiou, V.; Kaplan, D. Porosity of 3D biomaterial scaffolds and osteogenesis. Biomaterials 2005, 26, 5474–5491. [Google Scholar] [CrossRef]
- Jung, Y.; Kim, S.S.; Kim, Y.H.; Kim, S.H.; Kim, B.S.; Kim, S.; Choi, C.Y.; Kim, S.H. A poly (lactic acid)/calcium metaphosphate composite for bone tissue engineering. Biomaterials 2005, 26, 6314–6322. [Google Scholar] [CrossRef]
- Yildirim, N. Performance comparison of bio-based thermal insulation foam boars with petroleum-based foam boards on the market. Bioresources Resour. 2018, 13, 3395–3403. [Google Scholar]
- Mosanenzadeh, S.G.; Naguib, H.E.; Park, C.B.; Atalla, N. Development of Polylactide Open-Cell Foams with Bimodal Structure for High-Acoustic Absorption. J. Appl. Polym. Sci. 2013, 131. [Google Scholar] [CrossRef]
- Di Maio, E.; Kiran, E. Foaming of polymers with supercritical fluids and perspectives on the current knowledge and gaps. J. Supercrit. Fluids 2018, 134, 157–166. [Google Scholar] [CrossRef]
- Liu, W.J.; Yang, H.L.; Wang, Z.; Dong, L.S.; Liu, J.J. Effect of nucleating agents on the crystallization of poly(3-hydroxybutyrate-co-3-hydroxyvalerate). J. Appl. Polym. Sci. 2002, 86, 2145. [Google Scholar] [CrossRef]
- Gama, N.V.; Soares, B.; Freire, C.S.R.; Silva, R.; Neto, C.P.; Timmons, A.B.-; Ferreira, A. Bio-based polyurethane foams toward applications beyond thermal Insulation. Mater. Des. 2015, 76, 77–85. [Google Scholar] [CrossRef]
- Jenkins, M.J.; Cao, Y.; Howell, L.; Leeke, G.A. Miscibility in blends of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) and poly(3-caprolactone) induced by melt blending in the presence of supercritical CO2. Polymer 2007, 48, e6304–e6310. [Google Scholar] [CrossRef]
- Numata, K.; Yamashita, K.; Fujita, M.; Tsuge, T.; Kasuya, K.; Iwata, T. Adsorption and hydrolysis reactions of poly (hydroxybutyric acid) depolymerases secreted from ralstonia picketti T1 and penicillium funculosum onto poly[(R)-3-hydroxybutyric acid]. Biomacromolecules 2007, 8, 2276. [Google Scholar] [CrossRef] [PubMed]
- Favaro, L.; Basaglia, M.; Casella, S. Improving polyhydroxyalkanoate production from inexpensive carbon sources by genetic approaches; a review. Biofuels Bioprod. Bioref. 2018, 13, 208–227. [Google Scholar] [CrossRef] [Green Version]
- Mozumder, M.S.I.; Garcia-Gonzalez, L.; De Wever, H.; Volcke, E.I.P. Poly(3-hydroxybutyrate) (PHB) production from CO2: Model development and process optimization. Biochem. Eng. J. 2015, 98, 107–116. [Google Scholar] [CrossRef]
- Garcia-Gonzalez, L.; Mozumder, M.S.I.; Dubreuil, M.; Volcke, E.I.P.; De Wever, H. Sustainable autotrophic production of polyhydroxybutyrate (PHB) from CO2 using a two-stage cultivation system. Catal. Today 2015, 257, 237–245. [Google Scholar] [CrossRef]
- Ishizaki, A.; Tanaka, K. Production of poly-β-hydroxybutyric acid from carbon dioxide by Alcaligenes eutrophus ATCC 17697T. J. Ferment. Bioeng. 1991, 71, 254–257. [Google Scholar] [CrossRef]
- Pan, P.; Inoue, Y. Polymorphism and isomorphism in biodegradable polyesters. Prog. Polym. Sci. 2009, 34, 605–640. [Google Scholar] [CrossRef]
- Kachrimanidou, V.; Kopsahelis, N.; Papanikolaou, S.; Kookos, I.K.; Bruyn, M.D.; Clark, J.H.; Koutinas, A.A. Sunflower-based biorefinery: Poly(3-hydroxybutyrate) and poly(3-hydroxybutyrate-co-3-hydroxyvalerate) production from crude glycerol, sunflower meal and levulinic acid. Bioresour. Technol. 2014, 172, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Li, K.; Cui, Z.; Turng, L.S.; Peng, X.-F. Comparisons of Microcellular PHBV/PBAT Parts Injection Molded with Supercritical Nitrogen and Expandable Thermoplastic Microspheres: Surface Roughness, Tensile Properties, and Morphology. Cell. Polym. 2010, 29, 327–342. [Google Scholar] [CrossRef]
- Moigne, N.L.; Sauceau, M.; Benyakhlef, M.; Jemai, R.; Benezet, J.C.; Rodier, E.; Lopez-Cuesta, J.M.; Fages, J. Foaming of poly(3-hydroxybutyrate-co-3-hydroxyvalerate)/ organo-clays nano-biocomposites by a continuous supercritical CO2 assisted extrusion process. Eur. Polym. J. 2014, 61, 157–171. [Google Scholar] [CrossRef] [Green Version]
- Richard, E.; Rizvi, R.; Chow, A. Biodegradable Composite Foams of PLA and PHBV using Subcritical CO2. J. Polym. Environ. 2008, 16, 258–266. [Google Scholar] [CrossRef]
- Szegda, D.; Duangphet, S.; Song, S.; Tarverdi, K. Extrusion foaming of PHBV. J. Cell. Plast. 2014, 50, 145–162. [Google Scholar] [CrossRef]
- Wright, Z.C.; Frank, C.W. Increasing cell homogeneity of semi crystalline, biodegradable polymer foams with a narrow processing window via rapid quenching. Polym. Eng. Sci. 2014, 54, 2877–2886. [Google Scholar] [CrossRef]
- Xu, J.K.; Li, D.; Bao, J.; Wang, Z. Foaming of poly(3-hydroxybutyrate-co-3-hydroxyvalerate) with supercritical carbon dioxide: Foaming performance and crystallization behavior. ACS Omega 2020, 5, 9839–9845. [Google Scholar] [CrossRef] [Green Version]
- Evlashin, S.; Dyakonov, P.; Tarkhov, M.; Dagaesyan, S.; Rodionov, S.; Shpichka, A.; Kostenko, M.; Konev, S.; Sergeichev, I.; Timashev, I.; et al. Flexible polycaprolactone and polycaprolactone/graphene scaffolds for tissue engineering. Materials 2019, 12, 2991. [Google Scholar] [CrossRef] [Green Version]
- Nair, L.S.; Laurencin, C.T. Biodegradable polymers as biomaterials. Progr. Polym. Sci. 2007, 32, 762–798. [Google Scholar] [CrossRef]
- Woodruff, M.A.; Hutmacher, D.W. The return of a forgotten polymer polycaprolactone in the 21st century. Prog. Polym. Sci. 2010, 35, 1217–1256. [Google Scholar] [CrossRef] [Green Version]
- Marrazzo, C.; Di Maio, E.; Iannace, S. Conventional and nanometric nucleating agents in poly(epsilon-caprolactone) foaming: Crystals vs. bubbles nucleation. Polym. Eng. Sci. 2008, 48, 336–344. [Google Scholar] [CrossRef]
- Huang, H.; Oizumi, S.; Kojima, N.; Niino, T.; Sakai, Y. Avidin–biotin binding-based cell seeding and perfusion culture of liver-derived cells in a porous scaffold with a three-dimensional interconnected flow-channel network. Biomaterials 2007, 28, 3815–3823. [Google Scholar] [CrossRef]
- Zein, I.; Hutmacher, D.W.; Tan, K.C.; Teoh, S.H. Fused deposition modeling of novel scaffold architectures for tissue engineering applications. Biomaterials 2002, 23, 1169–1185. [Google Scholar] [CrossRef]
- Tay, B.Y.; Zhang, S.X.; Myint, M.H.; Ng, F. Processing of polycaprolactone porous structure for scaffold development. J. Mater. Process. Technol. 2007, 182, 117–121. [Google Scholar] [CrossRef]
- Rezgui, F.; Swistek, M.; Hiver, J.M. Deformation and damage upon stretching of degradable polymers (PLA and PCL). Polymer 2005, 46, 7370–7385. [Google Scholar] [CrossRef]
- Lezcano, E.; Coll, C.S.; Prolongo, M. Melting behavior and miscibility of poly (e-caprolactone) and poly (4-hydroxystyrene) blends. Polymer 1996, 37, 3603–3609. [Google Scholar] [CrossRef]
- Aroguz, A.Z.; Baysal, B.M.; Engin, H.H. The assessment of miscibility and morphology of poly (e-caprolactone) and poly (para-chlorostyrene) blends. Eur. Polym. J. 2006, 43, 403–409. [Google Scholar] [CrossRef]
- Wang, J.; Cheung, M.K.; Mi, Y. Miscibility and morphology in crystalline/amorphous blends of poly(caprolactone)/poly(4-vinylphenol) as studied by DSC, FTIR and 13C solid state NMR. Polymer 2002, 43, 1357. [Google Scholar] [CrossRef]
- De Kesel, C.; Lefèvre, C.; Nagy, J.B.; David, C. Blends of polycaprolactone with polyvinyl alcohol: A DSC, optical microscopy and solid-state NMR study. Polymer 1999, 40, 1969. [Google Scholar] [CrossRef]
- Bisso, G.; Casarino, P.; Pedemonte, E. Poly(e-caprolactone)/poly (vinyl methyl ether) blends: Miscibility by calorimetric measurements. Thermochim. Acta 1998, 321, 81. [Google Scholar] [CrossRef]
- Rosa, D.S.; Guedes, C.G.F.; Pedroso, A.G. Gelatinized and nongelatinized corn starch/poly(є-caprolactone) blends: Characterization by rheological, mechanical and morphological properties. Polimeros Cienciae Tecnologia 2004, 14, 181–186. [Google Scholar] [CrossRef]
- Verma, N.; Zafar, S.; Talha, M. Influence of nano-hydroxyapatite on mechanical behavior of microwave processed polycaprolactone composite foams. Mater. Res. Express 2019, 6, 181–186. [Google Scholar] [CrossRef]
- Huang, J.M.; Yang, S.J. Studying the miscibility and thermal behavior of polybenzoxazine/ poly(“-caprolactone) blends using DSC, DMA, and solid state 13C NMR spectroscopy. Polymer 2005, 46, 8068–8078. [Google Scholar] [CrossRef]
- Botlhoko, O.J.; Ramontja, J.; Ray, S.S. A new insight into morphological, thermal, and mechanical properties of melt-processed polylactide/poly(ε-caprolactone) blends. Polym. Degrad. Stab. 2018, 154, e84–e95. [Google Scholar] [CrossRef]
- Salerno, A.; Maio, E.D.; Iannace, S.; Netti, P.A. Solid-state supercritical CO2 foaming of PCL and PCL-HA nano-composite: Effect of composition, thermal history and foaming process on foam pore structure. J. Supercrit. Fluids 2011, 28, 158–167. [Google Scholar] [CrossRef]
- Shieh, Y.; Yang, H. Morphological changes of polycaprolactone with high-pressure CO2 treatment. J. Supercrit. Fluids 2005, 33, 183–192. [Google Scholar] [CrossRef]
- Lian, Z.; Epstein, S.A.; Blenk, C.W.; Shine, A.D. Carbon dioxide-induced melting point depression of biodegradable semicrystalline polymers. J. Supercrit. Fluids 2006, 39, 107–117. [Google Scholar] [CrossRef]
- Kiran, E.; Liu, K.; Ramsdell, K. Morphological changes in poly(3-caprolactone) in dense carbon dioxide. Polymer 2008, 49, 1853–1859. [Google Scholar] [CrossRef]
- Ogunsona, E.; D’Souza, N. Characterization and mechanical properties of foamed poly(“-caprolactone) and Mater-Bi blends using CO2 as blowing agent. J. Cell Plast 2015, 51, 245–268. [Google Scholar] [CrossRef]
- Reignier, J.; Gendron, R.; Champagne, M.F. Extrusion foaming of poly (lactic acid) blown with CO2: Toward 100% green material. Cell. Polym. 2007, 26, 83–115. [Google Scholar] [CrossRef] [Green Version]
- Reignier, J.; Gendron, R.; Champagne, M.F. Autoclave Foaming of Poly(e-Caprolactone) Using Carbon Dioxide: Impact of Crystallization on Cell Structure. J. Cell. Plast. 2007, 43, 459–489. [Google Scholar] [CrossRef] [Green Version]
- Qui, Z.; Yang, W.; Ikehara, T.; Nishi, T. Miscibility and crystallization behavior of biodegradable blends of two aliphatic polyesters; Poly(3-hydroxybutyrate-co-valerate) and poly(ε-caprolactone). Polymer 2005, 46, 11814–11819. [Google Scholar]
- Oliver, S.; Fleming, K.L.; Chan, A.; Kazarian, S.G. Applications of ATR-FTIR spectroscopic imaging to biomedical samples. Biochim. Biophys. Acta Biomembr. 2006, 1758, 858–867. [Google Scholar]
- Sharples, A. Introduction to polymer crystallization. EJ Arnold 1966, 23, 872. [Google Scholar]
- Dagnon, K.L.; Robinson, C.; Chen, H.H.; Garrett, D.C.; Innocentini-Mei, L.H.; D’Souza, N.A. Layer Double Hydroxides for Enhanced Poly(3-hydroxybutyrate-co-3-hydroxyvalarate) crystallization. J. Appl. Polym. Sci. 2013, 127, 3395–3406. [Google Scholar] [CrossRef]
- ASTM Standard. Standard Test Method for apparent density of rigid cellular plastics; ASTM Int: West Conshohocken, PA, USA, 2013; pp. 20–23. [Google Scholar]
- ASTM D1505-18. Standard Test Method for Density of Plastics by the Density-Gradient Technique; ASTM International: West Conshohocken, PA, USA, 2018; pp. 1–7. [Google Scholar]
- Pan, J.; Jackson, P. Review of Test Methods for Material Properties of Elastic Porous Materials. SAE Int. J. Mater. Manuf. 2009, 2, 570–579. [Google Scholar] [CrossRef]
- Kumar, K.; Weller, J. Production of microcellular polycarbonate using carbon dioxide for bubble nucleation. ASME J. Eng. Ind 1994, 116, 413–420. [Google Scholar] [CrossRef]
- Guan, Q.; Naguib, E.H. Fabrication and Characterization of PLA/PHBV-Chitin Nanocomposites and Their Foams. J. Polym Environ. 2014, 22, 119–130. [Google Scholar] [CrossRef]
- Buahom, P.; Areerat, S. The estimation of cell density in isotropic microcellular polymeric foams using the critical bubble lattice. J. Cell. Plast. 2011, 47, 133–152. [Google Scholar] [CrossRef]
- Buahom, P.; Ameli, A.; Park, C.B.; Sain, M.; Areerat, S. Multi-scale void fraction analysis for cell density characterization and nuclei density prediction of anisotropic polymeric foams. In Proceedings of the 28th International Conference of The Polymer Processing Society (PPS-28), Pattaya, Thailand, 11–15 December 2012. [Google Scholar]
- Ji, G.; Wang, J.; Zhai, W.; Lin, D.; Zheng, W. Tensile properties of microcellular poly (lactic acid) foams blown by compressed CO2. J. Cell. Plast. 2013, 49, 101–117. [Google Scholar] [CrossRef]
- Standard A. Standard Test Method for Impedance and Absorption of Acoustical Materials Using a Tube, Two Microphones, and a Digital Frequency Analysis System; ASTM Standard E; American Society for Testing and Materials: West Conshohocken, PA, USA, 2019; pp. 1050–1098. [Google Scholar]
- Abbad, A.; Jaboviste, K.; Ouisse, M.; Dauchez, N. Acoustic performances of silicone foams for sound absorption. J. Cell. Plast. 2018, 54, 651–670. [Google Scholar] [CrossRef]
- Allard, J.F.; Atalla, N. Propagation of Sound in Porous Media: Modelling Sound Absorbing Materials, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2009; p. 358. [Google Scholar]
- Alvarez-Lainez, M.; Rodriguez-Perez, M.A.; De Saja, J. Thermal conductivity of open-cell polyolefin foams. J. Polym. Sci. Part B Polym. Phys. 2007, 46, 212–221. [Google Scholar] [CrossRef]
- Ruiz, J.A.; Arroyo, C.S.; Dumon, M.; Rodríguez-Pérez, M.A.; González, G. ‘Production, cellular structure and thermal conductivity of microcellular (methyl methacrylate)–(butyl acrylate)–(methyl methacrylate) triblock copolymers. Polym. Int. Sci. 2010, 60, 1. [Google Scholar]
- ISO. ASTM Standard Test D3574-C and Factory Regulations. Compressive Properties /Compression Stress Value/ Compression Stress-Strain Characteristics. Available online: https://www.zwickroell.com/industries/plastics/polymer-foam/compression-stress-value-iso-3386/ (accessed on 8 June 2021).
- Wypych, G. Handbook of Foaming and Blowing Agents; Chemtec Publishing: Toronto, ON, Canada, 2017. [Google Scholar]
- Jenkins, M.J.; Harrison, K.L.; Silva, M.M.C.G.; Whitaker, M.J.; Shakesheff, K.M.; Howdle, S.M. Characterization of microcellular foams produced from semi-crystalline PCL using supercritical carbon dioxide. Eur. Polym. J. 2006, 42, 3145–3151. [Google Scholar] [CrossRef]
- Yamashita, T.; Suzuki, K.; Nishino, S.; Tomota, Y. Relationship between sound absorption and microscopic structure determined by x-ray computed tomography in urethane foam used as sound absorption materials for automobiles. Mater. Trans. 2008, 49, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Soto, P.F.; Herraez, M.; Gonzalez, A.; de Saja, J.A. Acoustic impedance and absorption coefficient measurement of porous materials used in the automotive industry. Polym. Test. 1994, 13, 77–88. [Google Scholar] [CrossRef]
- Zhang, C.; Junqing, L.; Zhen, H.; Fenglei, Z.; Yudong, H. Correlation between the acoustic and porous cell morphology of polyurethane foam: Effect of interconnected porosity. Mater. Des. 2012, 41, 319–325. [Google Scholar] [CrossRef]
- Huang, C.; Qian, X.; Yang, R. Thermal conductivity of polymers and polymer nanocomposites. Mater. Sci. Eng. R Rep. 2018, 132, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Oluwabunmi, K.; D’Souza, N.A.; Zhao, W.; Choi, T.Y.; Theyson, T. Compostable, fully biobased foams using PLA and micro cellulose for zero energy buildings. Sci. Rep. 2020, 10, 1771. [Google Scholar] [CrossRef] [PubMed]
- Riviere, L.; Causse, N.; Lonjon, A.; Dantras, E.; Lacabanne, C. Specific heat capacity of PEEK/Ag nanoparticles composites determined by modulated—temperature differential scanning calorimetry. Polym. Degrad. Stab. 2015, 127, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Yao, C.; Shao, Y.; Yang, J. Numerical investigation on the influence of areal flow on EGS thermal exploitation based on the 3-D T-H single fracture model. Energies 2018, 11, 3026. [Google Scholar] [CrossRef] [Green Version]
- Heat Transfer Module User’s Guide. COMSOL. Available online: www.comsol.com/patents (accessed on 28 October 2019).
- Shieh, Y.-T.; Su, J.-H.; Manivannan, G.; Lee, P.H.C.; Sawan, S.P.; Spall, W.D. Interaction of supercritical carbon dioxide with polymers. II. Amorphous polymers. J. Appl. Polym. Sci. 1996, 59, 707–717. [Google Scholar] [CrossRef]
- Chen, T.; Cai, T.; Jin, C.Q.; Ji, J. Design and fabrication of functional polycaprolactone. e-Polymers 2015, 205, 13. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Zhang, G.; Yang, Q.; Shi, X.; Liu, Y. Mechanical and dielectric properties of microcellular polycarbonate foams with unimodal or bimodal cell-size distributions. J. Cell. Plast. 2015, 51, 307–327. [Google Scholar] [CrossRef]
- Park, J.E.; Todo, M.J. Compressive mechanical properties and deformation behavior of porous polymer blends of poly(ε-caprolactone) and poly(l-lactic acid). Mater. Sci. 2011, 46, 7850. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample (% PHBV) | 0 | 5 | 15 | 25 | 35 | 45 | 55 | 65 |
|---|---|---|---|---|---|---|---|---|
| Density unfoamed (g/cm3) | 1.099 ± 0.006 | 1.106 ± 0.003 | 1.108 ± 0.051 | 1.157 ± 0.057 | 1.16 ± 0.049 | 1.175 ± 0.034 | 1.188 ± 0.019 | 1.19 ± 0.009 |
| Density foam (g/cm3) | 0.33 ± 0.002 | 0.3 ± 0.001 | 0.364 ± 0.003 | 0.512 ± 0.003 | 0.585 ± 0.001 | 0.687 ± 0.001 | 0.88 ± 0.002 | 1.055 ± 0.004 |
| Expansion ratio | 3.4 | 3.65 | 3.26 | 2.25 | 2.17 | 1.74 | 1.31 | 1.127 |
| Percentage porosity (%) (Pycnometer) | 69.06 ± 0.125 | 70.15 ± 0.079 | 65 ± 0.097 | 57 ± 0.313 | 50.25 ± 0.204 | 38.79 ± 0.0405 | 19.81 ± 0.137 | 10.51 ± 0.002 |
| Cell Density (cell/µm3) | 8.59 × 107 | 7.75 × 107 | 1.21 × 106 | 2.73 × 106 | 1.95 × 106 | 3.40 × 107 | 5.66 × 107 | 4.63 × 107 |
| Cell Size (µm) | 134.07 ± 17.07 | 143.26 ± 16.40 | 117.23 ± 30.09 | 81.54 ± 24.59 | 79.97 ± 20.74 | 123.48 ± 12.96 | 78.3 ± 21.05 | 61.65 ± 12.17 |
| Void fraction (%) | 71.5 | 72.8 | 68.7 | 60 | 53.9 | 42.5 | 25.9 | 13 |
| Cell wall thickness (µm) | 26.21 ± 0.017 | 24.55 ± 0.008 | 25.83 ± 0.045 | 27.66 ± 0.037 | 33.6 ± 0.010 | 68.15 ± 0.0064 | 75.5 ± 0.021 | 121.69 ± 0.024 |
| A.T. Conductivity (W/m·K) (unfoamed) | 0.2407 ± 0.034 | 0.2244 ± 0.017 | 0.2561 ± 0.021 | 0.2435 ± 0.007 | 0.2953 ± 0.005 | 0.2731 ± 0.002 | 0.2856 ± 0.0081 | 0.3007 ± 0.0008 |
| Cp (J/Kg·K) (Unfoamed) | 485.2 | 510.67 | 481.08 | 472.11 | 420.47 | 404.6 | 399.46 | 473 |
| Thickness (m) (Unfoamed) | 0.02 ± 0.001 | 0.02 ± 0.003 | 0.02 ± 0.001 | 0.02 ± 0.003 | 0.02 ± 0.002 | 0.02 ± 0.001 | 0.02 ± 0.128 | 0.02 ± 0.424 |
| A.T. Conductivity (W/m·K) (Foams) | 0.085 ± 0.0034 | 0.076 ± 0.0017 | 0.099 ± 0.0021 | 0.120 ± 0.0077 | 0.157 ± 0.0052 | 0.181 ± 0.0029 | 0.232 ± 0.0082 | 0.288 ± 0.0008 |
| Cp (J/Kg·K) (Foams) | 486.33 | 511.06 | 483.8 | 472.76 | 420.59 | 407.81 | 401.74 | 476.75 |
| Thickness (Foams) | 0.02 ± 0.007 | 0.02 ± 0.003 | 0.02 ± 0.005 | 0.02 ± 0.003 | 0.02 ± 0.002 | 0.02 ± 0.001 | 0.02 ± 0.001 | 0.02 ± 0.004 |
| Thermal Dffusivity (m2/s) (Foams) | 5.29 × 107 ± 0.003 | 4.96 × 107 ± 0.002 | 5.62 × 107 ± 0.002 | 4.96 × 107 ± 0.007 | 6.38 × 107 ± 0.005 | 6.46 × 107 ± 0.003 | 6.56 × 107 ± 0.005 | 5.73 × 107 ± 0.001 |
| Compression Modulus (MPa) Unfoamed | 7.14 ± 0.425 | 7.69 ± 0.144 | 8.33 ± 0.342 | 9.26 ± 0.244 | 10 ± 0.420 | 9 ± 1.160 | 4.29 ± 0.470 | 5.56 ± 3.033 |
| Compression Strength (MPa) Unfoamed | 0.5 ± 0.431 | 0.54 ± 1.613 | 0.55 ± 0.13 | 0.57 ± 0.66 | 0.6 ± 0.08 | 0.45 ± 0.03 | 0.3 ± 0.28 | 0.5 ± 0.025 |
| Compression Modulus (MPa) Foams | 0.89 ± 0.008 | 2.5 ± 0.006 | 1.86 ± 0.004 | 7.5 ± 0.005 | 12.5 ± 0.003 | 6.67 ± 0.002 | 5.71 ± 0.005 | 5.2 ± 0.003 |
| Compression Strength (MPa) Foams | 0.16 ± 0.047 | 0.9 ± 0.042 | 0.5 ± 0.022 | 1.7 ± 0.012 | 2.5 ± 0.005 | 0.6 ± 0.004 | 0.55 ± 0.05 | 0.37 ± 0.007 |
| Sound Absorption (500 Hz) Unfoamed | 0.9046 ± 0.056 | 0.9135 ± 0.061 | 0.9120 ± 0.035 | 0.9015 ± 0.035 | 0.8930 ± 0.031 | 0.9022 ± 0.06 | 0.8915 ± 0.034 | 0.8852 ± 0.049 |
| Sound Absorption (4000 Hz) Unfoamed | 0.8740 ± 0.012 | 0.8962 ± 0.005 | 0.9085 ± 0.019 | 0.9092 ± 0.018 | 0.8987 ± 0.027 | 0.8965 ± 0.015 | 0.8971 ± 0.014 | 0.8977 ± 0.004 |
| Sound Absorption (500 Hz) Foam Core | 0.9204 ± 0.016 | 0.9310 ± 0.024 | 0.9149 ± 0.01 | 0.9120 ± 0.06 | 0.9053 ± 0.025 | 0.9075 ± 0.02 | 0.9023 ± 0.013 | 0.8918 ± 0.02 |
| Sound Absorption (4000 Hz) Foam Core | 0.9113 ± 0.015 | 0.9159 ± 0.013 | 0.9191 ± 0.08 | 0.9264 ± 0.015 | 0.8977 ± 0.024 | 0.9155 ± 0.081 | 0.9019 ± 0.05 | 0.8997 ± 0.044 |
| Second DSC Cycle | Melting Temp. (°C) | (J/g) | (J/g) | (J/g) | First DSC Cycle | Melting Temp. (°C) | (J/g) | (J/g) | (J/g) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Unfoamed PCL/PHBV | Tm1 | Tm2 | Tm3 | ΔHm1 | ΔHm2 | ΔHm3 | PCL/PHBV Foams | Tmf1 | Tmf2 | Tmf3 | ΔHm1 | ΔHm2 | ΔHm3 |
| 0_BV | 58.35 | N/A | N/A | 46.90 | N/A | N/A | 0_BV | 59.6 | NA | NA | 10.64 | NA | NA |
| 5_BV | 58.44 | N/A | 164.2 | 71.70 | N/A | 12.69 | 5_BV | 60.39 | NA | 167.82 | 9.25 | NA | 18.67 |
| 15_BV | 58.91 | 158.71 | 168.2 | 51.20 | 15.97 | 15.97 | 15_BV | 61.76 | 159.06 | 168.17 | 17.96 | 23.32 | 26.80 |
| 25_BV | 58.32 | 160.64 | 169.7 | 58.60 | 16.91 | 20.74 | 25_BV | 60.26 | 161.14 | 170.65 | 10.19 | 25.63 | 22.52 |
| 35_BV | 58.78 | 164.09 | 171.28 | 54.83 | 27.90 | 12.26 | 35_BV | 61.94 | 163.45 | 170.53 | 6.17 | 26.45 | 9.41 |
| 45_BV | 58.52 | N/A | 168.1 | 36.34 | N/A | 31.64 | 45_BV | 60.84 | 168.28 | 173.16 | 12.75 | 43.28 | 10.01 |
| 55_BV | 59.47 | N/A | 167.55 | 54.82 | N/A | 68.54 | 55_BV | 60.14 | 167.65 | 172.52 | 4.95 | 31.28 | 5.34 |
| 65_BV | 58.78 | N/A | 168.83 | 49.67 | N/A | 60.35 | 65_BV | 61.48 | 167.99 | 172.86 | 10.31 | 41.43 | 9.77 |
| 75_BV | 57.93 | 166.8 | 171.38 | 34.15 | 28.81 | 16.56 | 75_BV | NA | NA | NA | NA | NA | NA |
| 85_BV | 57.1 | 165.46 | 171.17 | 31.78 | 36.25 | 16.40 | 85_BV | NA | NA | NA | NA | NA | NA |
| 100_BV | N/A | N/A | 174.41 | N/A | N/A | 82.95 | 100_BV | NA | NA | NA | NA | NA | NA |
| First DSC Cycle | Tc1 | Tc2 | Hc1 | Hc2 | First DSC | Tcf1 | Tcf2 | Hcf1 | Hcf2 |
|---|---|---|---|---|---|---|---|---|---|
| Cycle | |||||||||
| Unf. PCL/PHBV | (°C) | (°C) | (J/g) | (J/g) | PCL/PHBV Foams | (°C) | (°C) | (J/g) | (J/g) |
| 0_BV | 33.01 | N/A | −2.00 | N/A | 0_BV | 33.3 | N/A | −2.87 | N/A |
| 5_BV | 33.09 | 73.2 | −2.41 | −2.61 | 5_BV | 34.77 | 74.5 | −2.10 | −2.17 |
| 15_BV | 32.7 | 95.59 | −1.78 | −15.75 | 15_BVf | 32.01 | 101.18 | −4.78 | −36.51 |
| 25_BV | 32.26 | 101.27 | −2.26 | −18.72 | 25_BVf | 32.29 | 101.22 | −3.40 | −22.37 |
| 35_BV | 32.57 | 111.58 | −1.76 | −14.53 | 35_BVf | 31.9 | 112.98 | −1.48 | −14.04 |
| 45_BV | 36.17 | 113.75 | −1.61 | −10.75 | 45_BVf | 35.3 | 112.92 | −5.48 | −34.39 |
| 55_BV | 36.21 | 113.29 | −3.57 | −24.91 | 55_BVf | 34 | 114.04 | −2.73 | −16.08 |
| 65_BV | 34.67 | 113.77 | −2.18 | −14.81 | 65_BVf | 34.03 | 113.1 | −5.09 | −34.75 |
| 75_BV | 36.33 | 114.52 | −3.61 | −21.12 | 75_BVf | N/A | N/A | N/A | N/A |
| 85_BV | 31.9 | 115.12 | −2.29 | −18.31 | 85_BVf | N/A | N/A | N/A | N/A |
| 100_BV | N/A | 121.43 | N/A | −26.02 | 100_BVf | N/A | N/A | N/A | N/A |
| Sample Type | Theoretical Values (W/m.K) | Experimental Values (W/m.K) | Difference (%) |
|---|---|---|---|
| 0_BV | 0.079 | 0.0853 | 7.3 |
| 5_Bv | 0.0691 | 0.0762 | 9.3 |
| 15_BV | 0.0885 | 0.0988 | 10.4 |
| 25_BV | 0.1081 | 0.1201 | 10 |
| 35_BV | 0.1484 | 0.1571 | 5.5 |
| 45_BV | 0.1724 | 0.1806 | 4.5 |
| 55_BV | 0.2314 | 0.232 | 0.3 |
| 65_BV | 0.2848 | 0.2875 | 0.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oluwabunmi, K.E.; Zhao, W.; D’Souza, N.A. Carbon Capture Utilization for Biopolymer Foam Manufacture: Thermal, Mechanical and Acoustic Performance of PCL/PHBV CO2 Foams. Polymers 2021, 13, 2559. https://doi.org/10.3390/polym13152559
Oluwabunmi KE, Zhao W, D’Souza NA. Carbon Capture Utilization for Biopolymer Foam Manufacture: Thermal, Mechanical and Acoustic Performance of PCL/PHBV CO2 Foams. Polymers. 2021; 13(15):2559. https://doi.org/10.3390/polym13152559
Chicago/Turabian StyleOluwabunmi, Kayode E., Weihuan Zhao, and Nandika Anne D’Souza. 2021. "Carbon Capture Utilization for Biopolymer Foam Manufacture: Thermal, Mechanical and Acoustic Performance of PCL/PHBV CO2 Foams" Polymers 13, no. 15: 2559. https://doi.org/10.3390/polym13152559
APA StyleOluwabunmi, K. E., Zhao, W., & D’Souza, N. A. (2021). Carbon Capture Utilization for Biopolymer Foam Manufacture: Thermal, Mechanical and Acoustic Performance of PCL/PHBV CO2 Foams. Polymers, 13(15), 2559. https://doi.org/10.3390/polym13152559