
Anticoagulant Activity of Cellulose Nanocrystals from Isora Plant Fibers Assembled on Cellulose and SiO2 Substrates via a Layer-by-Layer Approach

 ,
, 

 , ,
, , 

Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Cellulose Nanocrystals (CNCs) Preparation
2.3. Inductively Coupled Plasma Mass Spectrometry (ICPMS)
2.4. UV−Vis Absorption Spectroscopy
2.5. Dynamic and Electrophoretic Light Scattering
2.6. Transmission Electron Microscopy (TEM)
2.7. Substrate Cleaning and Cellulose Thin Film Preparation
2.8. Sample Preparation for Multilayer Coatings
2.9. Creation of Multilayers from CNCs and PEI Using QCM-D
2.9.1. Layer-by-Layer (LBL) Adsorption of PEI and CNCs on SiO2 and Cellulose Surface
2.9.2. Anticoagulant Activity
2.9.3. Plasma Adsorption
2.10. Profilometry
2.11. Atomic Force Microscopy (AFM)
3. Results and Discussion
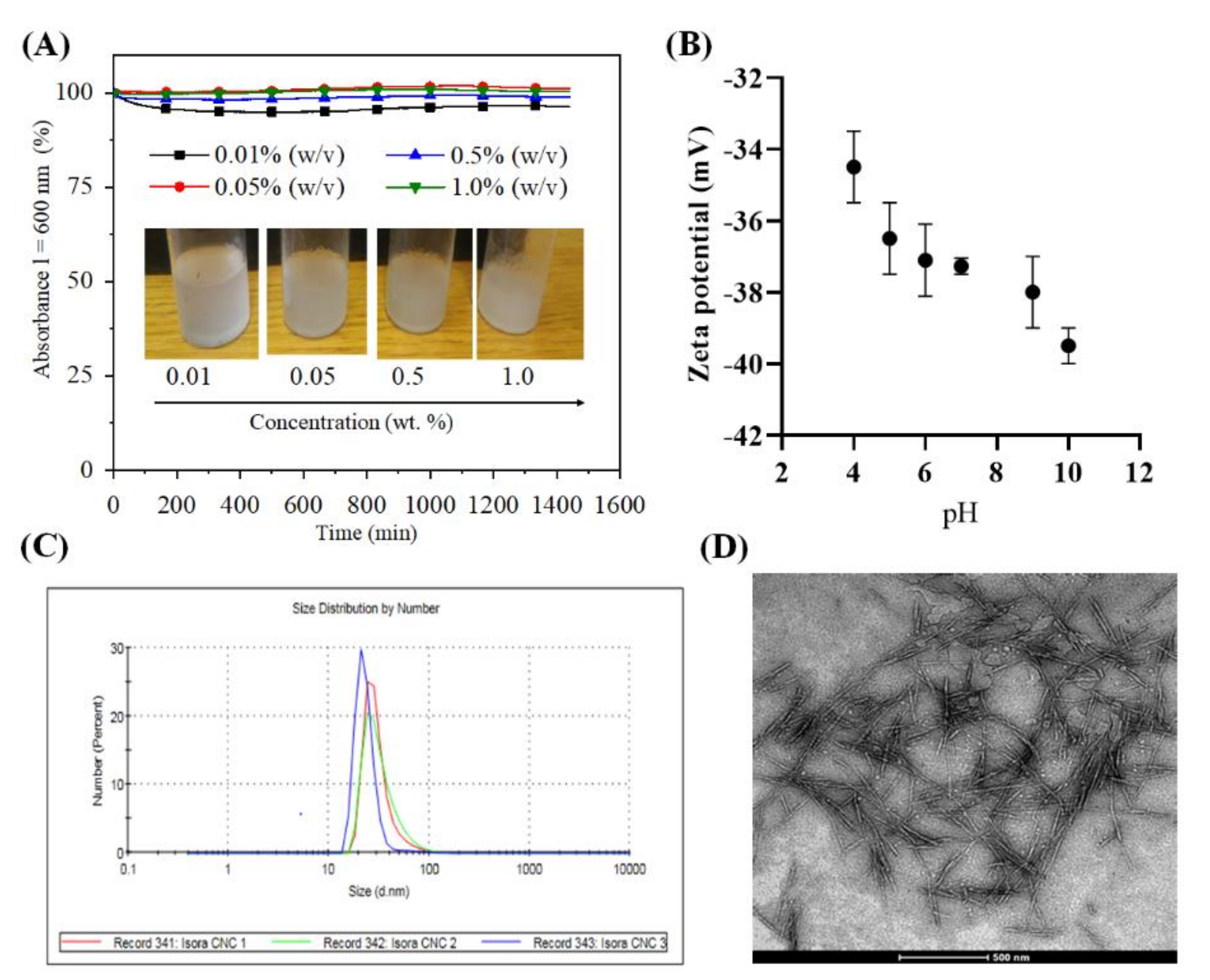
3.1. Stability, Zetapotential and Size of Cellulose Nanocrystals (CNCs)
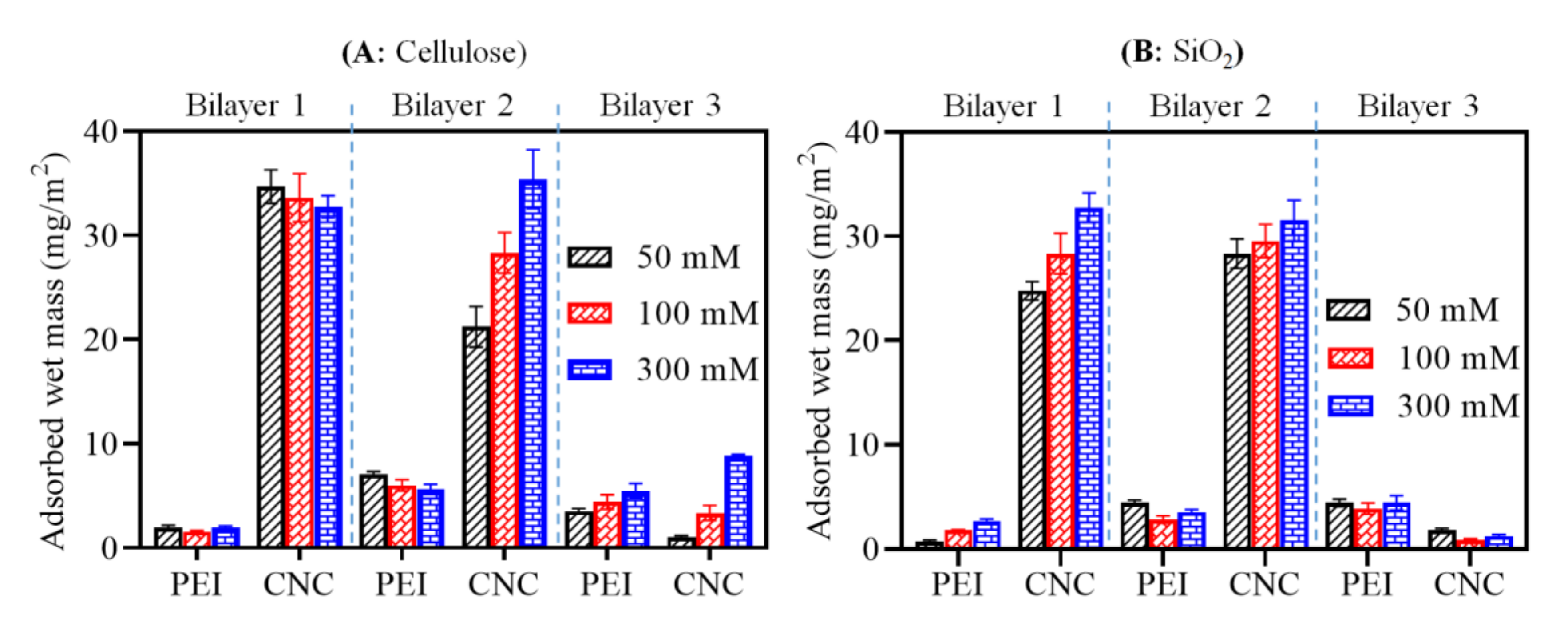
3.2. Multilayer Formation
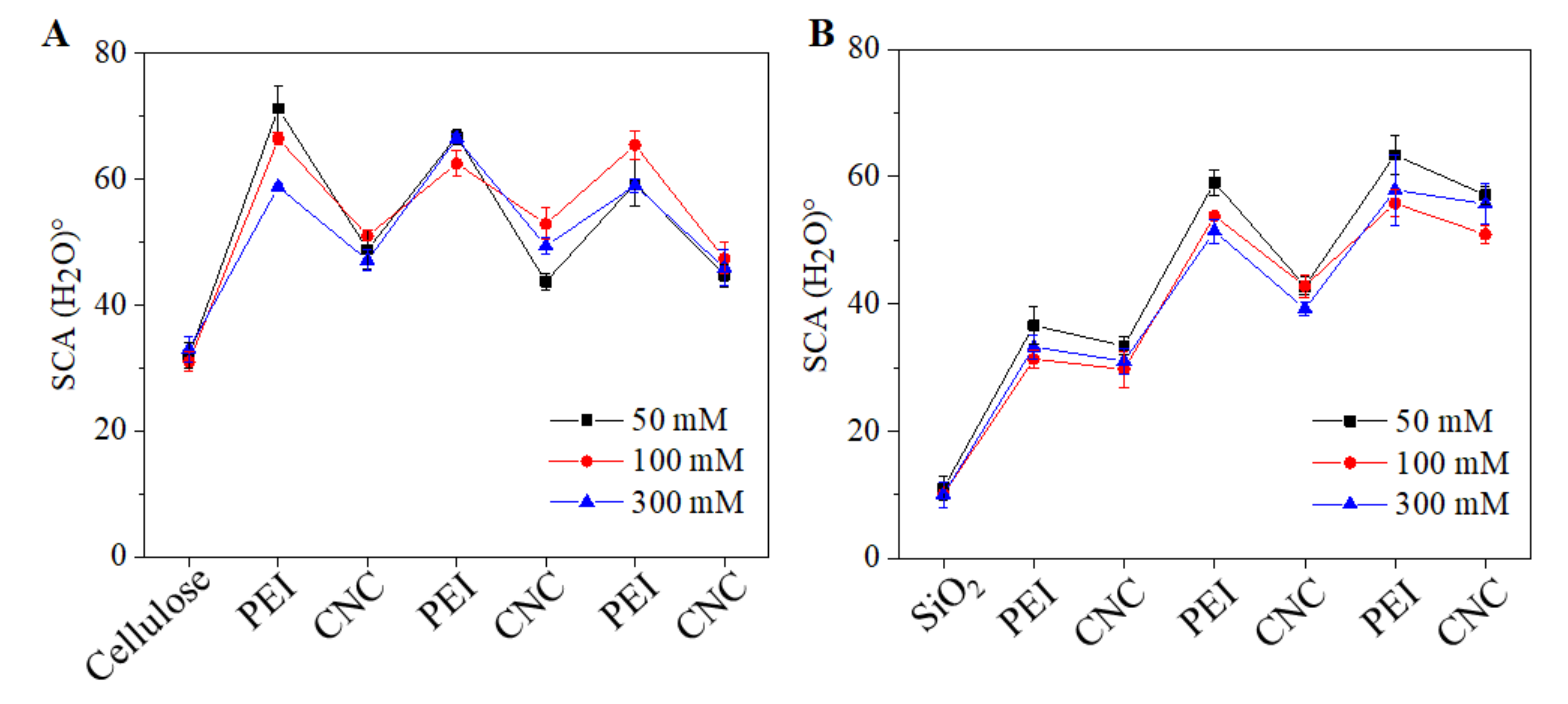
3.3. Surface Morphology, Roughness, Wettability and Layer Thickness
3.4. Anticoagulant Activity and Plasma Adsorption
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, D.; Peng, F.; Liu, X. Protection of magnesium alloys: From physical barrier coating to smart self-healing coating. J. Alloy. Compd. 2021, 853, 157010. [Google Scholar] [CrossRef]
- Hoseinpour, V.; Noori, L.; Mahmoodpour, S.; Shariatinia, Z. A review on surface modification methods of poly(arylsulfone) membranes for biomedical applications. J. Biomater. Sci. Polym. Ed. 2021, 10, 1–61. [Google Scholar] [CrossRef]
- Mohan, T.; Kargl, R.; Tradt, K.E.; Kulterer, M.R.; Braćić, M.; Hribernik, S.; Stana-Kleinschek, K.; Ribitsch, V. Antifouling coating of cellulose acetate thin films with polysaccharide multilayers. Carbohydr. Polym. 2015, 116, 149–158. [Google Scholar] [CrossRef]
- Wågberg, L.; Erlandsson, J. The Use of Layer-by-Layer Self-Assembly and Nanocellulose to Prepare Advanced Functional Materials. Adv. Mater. 2020, 2001474. [Google Scholar] [CrossRef]
- Findenig, G.; Leimgruber, S.; Kargl, R.; Spirk, S.; Stana-Kleinschek, K.; Ribitsch, V. Creating Water Vapor Barrier Coatings from Hydrophilic Components. ACS Appl. Mater. Interfaces 2012, 4, 3199–3206. [Google Scholar] [CrossRef] [PubMed]
- Ai, H.; Jones, S.A.; Lvov, Y.M. Biomedical Applications of Electrostatic Layer-by-Layer Nano-Assembly of Polymers, Enzymes, and Nanoparticles. Cell Biophys. 2003, 39, 23–44. [Google Scholar] [CrossRef]
- Olszewska, A.M.; Kontturi, E.; Laine, J.; Österberg, M. All-cellulose multilayers: Long nanofibrils assembled with short nanocrystals. Cellulose 2013, 20, 1777–1789. [Google Scholar] [CrossRef]
- Bračič, M.; Mohan, T.; Kargl, R.; Grießer, T.; Heinze, T.; Kleinschek, K.S. Protein repellent anti-coagulative mixed-charged cellulose derivative coatings. Carbohydr. Polym. 2021, 254, 117437. [Google Scholar] [CrossRef]
- Delechiave, G.; Naves, A.F.; Kolanthai, E.; Da Silva, R.A.; Vlasman, R.C.; Petri, D.F.; Torresi, R.M.; Catalani, L.H. Tuning protein delivery from different architectures of layer-by-layer assemblies ona polymer films. Mater. Adv. 2020, 1, 2043–2056. [Google Scholar] [CrossRef]
- De Mesquita, J.P.; Patricio, P.S.; Donnici, C.L.; Petri, D.F.S.; De Oliveira, L.C.A.; Pereira, F.V. Hybrid layer-by-layer assembly based on animal and vegetable structural materials: Multilayered films of collagen and cellulose nanowhiskers. Soft Matter 2011, 7, 4405–4413. [Google Scholar] [CrossRef]
- Qi, Z.-D.; Saito, T.; Fan, Y.; Isogai, A. Multifunctional Coating Films by Layer-by-Layer Deposition of Cellulose and Chitin Nanofibrils. Biomacromolecules 2012, 13, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Podsiadlo, P.; Sui, L.; Elkasabi, Y.; Burgardt, P.; Lee, J.; Miryala, A.; Kusumaatmaja, W.; Carman, M.R.; Shtein, M.; Kieffer, J.; et al. Layer-by-Layer Assembled Films of Cellulose Nanowires with Antireflective Properties. Langmuir 2007, 23, 7901–7906. [Google Scholar] [CrossRef]
- Tardy, B.L.; Ago, M.; Guo, J.; Borghei, M.; Kämäräinen, T.; Rojas, O.J. Optical Properties of Self-Assembled Cellulose Nanocrystals Films Suspended at Planar-Symmetrical Interfaces. Small 2017, 13, 1702084. [Google Scholar] [CrossRef]
- Habibi, Y.; Lucia, L.A.; Rojas, O.J. Cellulose Nanocrystals: Chemistry, Self-Assembly, and Applications. Chem. Rev. 2010, 110, 3479–3500. [Google Scholar] [CrossRef] [PubMed]
- Ehmann, H.M.A.; Mohan, T.; Koshanskaya, M.; Scheicher, S.; Breitwieser, D.; Ribitsch, V.; Stana-Kleinschek, K.; Spirk, S. Design of anticoagulant surfaces based on cellulose nanocrystals. Chem. Commun. 2014, 50, 13070–13072. [Google Scholar] [CrossRef] [PubMed]
- Vanderfleet, O.M.; Cranston, E.D. Production routes to tailor the performance of cellulose nanocrystals. Nat. Rev. Mater. 2021, 6, 124–144. [Google Scholar] [CrossRef]
- Nagarajan, K.J.; Ramanujam, N.R.; Sanjay, M.R.; Siengchin, S.; Rajan, B.S.; Basha, K.S.; Madhu, P.; Raghav, G.R. A comprehensive review on cellulose nanocrystals and cellulose nanofibers: Pretreatment, preparation, and characterization. Polym. Compos. 2021. [Google Scholar] [CrossRef]
- Moon, R.J.; Martini, A.; Nairn, J.; Simonsen, J.; Youngblood, J. Cellulose nanomaterials review: Structure, properties and nanocomposites. Chem. Soc. Rev. 2011, 40, 3941–3994. [Google Scholar] [CrossRef]
- Faßl, H.; Stana, J.; Stropnik, D.; Strnad, S.; Stana-Kleinschek, K.; Ribitsch, V. Improvement of the Hemocompatibility of PET Surfaces Using Different Sulphated Polysaccharides as Coating Materials. Biomacromolecules 2010, 11, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.; Thuy, L.T.; Ki, S.H.; Ko, S.; Kim, S.; Cho, W.K.; Choi, J.S.; Kang, S.M. Multipurpose Antifouling Coating of Solid Surfaces with the Marine-Derived Polymer Fucoidan. Macromol. Biosci. 2018, 18, e1800137. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Bao, L.; Li, X.; Chen, L.; Hong, F.F. Potential of PVA-doped bacterial nano-cellulose tubular composites for artificial blood vessels. J. Mater. Chem. B 2015, 3, 8537–8547. [Google Scholar] [CrossRef]
- Cranston, E.D.; Gray, D.G. Morphological and Optical Characterization of Polyelectrolyte Multilayers Incorporating Nanocrystalline Cellulose. Biomacromolecules 2006, 7, 2522–2530. [Google Scholar] [CrossRef] [PubMed]
- Moreau, C.; Beury, N.; Delorme, N.; Cathala, B. Tuning the Architecture of Cellulose Nanocrystal–Poly(allylamine hydrochloride) Multilayered Thin Films: Influence of Dipping Parameters. Langmuir 2012, 28, 10425–10436. [Google Scholar] [CrossRef] [PubMed]
- Chirayil, C.J.; Joy, J.; Mathew, L.; Mozetic, M.; Koetz, J.; Thomas, S. Isolation and characterization of cellulose nanofibrils from Helicteres isora plant. Ind. Crop. Prod. 2014, 59, 27–34. [Google Scholar] [CrossRef]
- Delgado, A.; González-Caballero, F.; Hunter, R.; Koopal, L.; Lyklema, J. Measurement and interpretation of electrokinetic phenomena. J. Colloid Interface Sci. 2007, 309, 194–224. [Google Scholar] [CrossRef]
- Ohshima, H. Electrophoretic mobility of soft particles. Colloids Surfaces A Physicochem. Eng. Asp. 1995, 103, 249–255. [Google Scholar] [CrossRef]
- Kargl, R.; Mohan, T.; Bračič, M.; Kulterer, M.; Doliška, A.; Stana-Kleinschek, K.; Ribitsch, V. Adsorption of Carboxymethyl Cellulose on Polymer Surfaces: Evidence of a Specific Interaction with Cellulose. Langmuir 2012, 28, 11440–11447. [Google Scholar] [CrossRef] [PubMed]
- Mohan, T.; Kargl, R.; Doliska, A.; Ehmann, H.M.; Ribitsch, V.; Stana-Kleinschek, K. Enzymatic digestion of partially and fully regenerated cellulose model films from trimethylsilyl cellulose. Carbohydr. Polym. 2013, 93, 191–198. [Google Scholar] [CrossRef]
- Mohan, T.; Kargl, R.; Doliška, A.; Vesel, A.; Köstler, S.; Ribitsch, V.; Stana-Kleinschek, K. Wettability and surface composition of partly and fully regenerated cellulose thin films from trimethylsilyl cellulose. J. Colloid Interface Sci. 2011, 358, 604–610. [Google Scholar] [CrossRef]
- Ristić, T.; Mohan, T.; Kargl, R.; Hribernik, S.; Doliška, A.; Stana-Kleinschek, K.; Fras, L. A study on the interaction of cationized chitosan with cellulose surfaces. Cellulose 2014, 21, 2315–2325. [Google Scholar] [CrossRef]
- Andersson, M.; Andersson, J.; Sellborn, A.; Berglin, M.; Nilsson, B.; Elwing, H. Quartz crystal microbalance-with dissipation monitoring (QCM-D) for real time measurements of blood coagulation density and immune complement activation on artificial surfaces. Biosens. Bioelectron. 2005, 21, 79–86. [Google Scholar] [CrossRef]
- Mohan, T.; Nagaraj, C.; Nagy, B.M.; Bračič, M.; Maver, U.; Olschewski, A.; Kleinschek, K.S.; Kargl, R.J. Nano- and Micropatterned Polycaprolactone Cellulose Composite Surfaces with Tunable Protein Adsorption, Fibrin Clot Formation, and Endothelial Cellular Response. Biomacromolecules 2019, 20, 2327–2337. [Google Scholar] [CrossRef]
- Nečas, D.; Klapetek, P. Gwyddion: An open-source software for SPM data analysis. Open Phys. 2012, 10, 181–188. [Google Scholar] [CrossRef]
- Marchessault, R.; Morehead, F.; Koch, M. Some hydrodynamic properties of neutral suspensions of cellulose crystallites as related to size and shape. J. Colloid Sci. 1961, 16, 327–344. [Google Scholar] [CrossRef]
- Ehmann, H.M.A.; Spirk, S.; Doliška, A.; Mohan, T.; Gössler, W.; Ribitsch, V.; Sfiligoj-Smole, M.; Stana-Kleinschek, K. Generalized Indirect Fourier Transformation as a Valuable Tool for the Structural Characterization of Aqueous Nanocrystalline Cellulose Suspensions by Small Angle X-ray Scattering. Langmuir 2013, 29, 3740–3748. [Google Scholar] [CrossRef]
- Beck, S.; Bouchard, J.; Berry, R. Dispersibility in Water of Dried Nanocrystalline Cellulose. Biomacromolecules 2012, 13, 1486–1494. [Google Scholar] [CrossRef]
- Trilokesh, C.; Uppuluri, K.B. Isolation and characterization of cellulose nanocrystals from jackfruit peel. Sci. Rep. 2019, 9, 16709. [Google Scholar] [CrossRef] [PubMed]
- Boluk, Y.; Danumah, C. Analysis of cellulose nanocrystal rod lengths by dynamic light scattering and electron microscopy. J. Nanoparticle Res. 2014, 16, 1–7. [Google Scholar] [CrossRef]
- Moud, A.A.; Arjmand, M.; Yan, N.; Nezhad, A.S.; Hejazi, S.H. Colloidal Behavior of Cellulose Nanocrystals in Presence of Sodium Chloride. ChemistrySelect 2018, 3, 4969–4978. [Google Scholar] [CrossRef]
- Mohan, T.; Čas, A.; Bračič, M.; Plohl, O.; Vesel, A.; Rupnik, M.; Zemljič, L.F.; Rebol, J. Highly Protein Repellent and Antiadhesive Polysaccharide Biomaterial Coating for Urinary Catheter Applications. ACS Biomater. Sci. Eng. 2018, 5, 5825–5832. [Google Scholar] [CrossRef] [Green Version]
- Barick, P.; Saha, B.P.; Mitra, R.; Joshi, S.V. Effect of concentration and molecular weight of polyethylenimine on zeta potential, isoelectric point of nanocrystalline silicon carbide in aqueous and ethanol medium. Ceram. Int. 2015, 41, 4289–4293. [Google Scholar] [CrossRef]
- Bračič, M.; Mohan, T.; Kargl, R.; Griesser, T.; Hribernik, S.; Köstler, S.; Stana-Kleinschek, K.; Fras-Zemljič, L. Preparation of PDMS ultrathin films and patterned surface modification with cellulose. RSC Adv. 2014, 4, 11955–11961. [Google Scholar] [CrossRef]
- Mivehi, L.; Bordes, R.; Holmberg, K. Adsorption of Cationic Gemini Surfactants at Solid Surfaces Studied by QCM-D and SPR: Effect of the Rigidity of the Spacer. Langmuir 2011, 27, 7549–7557. [Google Scholar] [CrossRef] [PubMed]
- Nagalakshmaiah, M.; El Kissi, N.; Dufresne, A. Ionic Compatibilization of Cellulose Nanocrystals with Quaternary Ammonium Salt and Their Melt Extrusion with Polypropylene. ACS Appl. Mater. Interfaces 2016, 8, 8755–8764. [Google Scholar] [CrossRef]
- Bruel, C.; Queffeulou, S.; Carreau, P.J.; Tavares, J.R.; Heuzey, M.-C. Orienting Cellulose Nanocrystal Functionalities Tunes the Wettability of Water-Cast Films. Langmuir 2020, 36, 12179–12189. [Google Scholar] [CrossRef]
- Doliska, A.; Strnad, S.; Stana, J.; Martinelli, E.; Ribitsch, V.; Stana-Kleinschek, K. In Vitro Haemocompatibility Evaluation of PET Surfaces Using the Quartz Crystal Microbalance Technique. J. Biomater. Sci. Polym. Ed. 2012, 23, 697–714. [Google Scholar] [CrossRef]
- Müller, L.; Sinn, S.; Drechsel, H.; Ziegler, C.; Wendel, H.-P.; Northoff, H.; Gehring, F.K. Investigation of Prothrombin Time in Human Whole-Blood Samples with a Quartz Crystal Biosensor. Anal. Chem. 2010, 82, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Okutucu, B.; Dınçer, A.; Habib, Ö.; Zıhnıoglu, F. Comparison of five methods for determination of total plasma protein concentration. J. Biochem. Biophys. Methods 2007, 70, 709–711. [Google Scholar] [CrossRef] [PubMed]
- Petibois, C.; Cazorla, G.; Cassaigne, A.; Déléris, G. Plasma Protein Contents Determined by Fourier-Transform Infrared Spec-trometry. Clin. Chem. 2001, 47, 730. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cellulose | SiO2 | |||||||
|---|---|---|---|---|---|---|---|---|
| 0 mM | 50 mM | 100 mM | 300 mM | 0 mM | 50 mM | 100 mM | 300 mM | |
| PEI | 0.9 | 6.5 | 6.1 | 6.7 | 0.4 | 1 | 1.1 | 1.1 |
| CNC3 | - | 9.2 | 7.2 | 6.8 | - | 7.1 | 5.6 | 5.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohan, T.; Chirayil, C.J.; Nagaraj, C.; Bračič, M.; Steindorfer, T.A.; Krupa, I.; Maadeed, M.A.A.A.; Kargl, R.; Thomas, S.; Stana Kleinschek, K. Anticoagulant Activity of Cellulose Nanocrystals from Isora Plant Fibers Assembled on Cellulose and SiO2 Substrates via a Layer-by-Layer Approach. Polymers 2021, 13, 939. https://doi.org/10.3390/polym13060939
Mohan T, Chirayil CJ, Nagaraj C, Bračič M, Steindorfer TA, Krupa I, Maadeed MAAA, Kargl R, Thomas S, Stana Kleinschek K. Anticoagulant Activity of Cellulose Nanocrystals from Isora Plant Fibers Assembled on Cellulose and SiO2 Substrates via a Layer-by-Layer Approach. Polymers. 2021; 13(6):939. https://doi.org/10.3390/polym13060939
Chicago/Turabian StyleMohan, Tamilselvan, Cintil Jose Chirayil, Chandran Nagaraj, Matej Bračič, Tobias Alexander Steindorfer, Igor Krupa, Mariam Al Ali Al Maadeed, Rupert Kargl, Sabu Thomas, and Karin Stana Kleinschek. 2021. "Anticoagulant Activity of Cellulose Nanocrystals from Isora Plant Fibers Assembled on Cellulose and SiO2 Substrates via a Layer-by-Layer Approach" Polymers 13, no. 6: 939. https://doi.org/10.3390/polym13060939
APA StyleMohan, T., Chirayil, C. J., Nagaraj, C., Bračič, M., Steindorfer, T. A., Krupa, I., Maadeed, M. A. A. A., Kargl, R., Thomas, S., & Stana Kleinschek, K. (2021). Anticoagulant Activity of Cellulose Nanocrystals from Isora Plant Fibers Assembled on Cellulose and SiO2 Substrates via a Layer-by-Layer Approach. Polymers, 13(6), 939. https://doi.org/10.3390/polym13060939