
Influence of the Polymer Microstructure over the Phase Separation of Thermo-Responsive Nanoparticles



Abstract
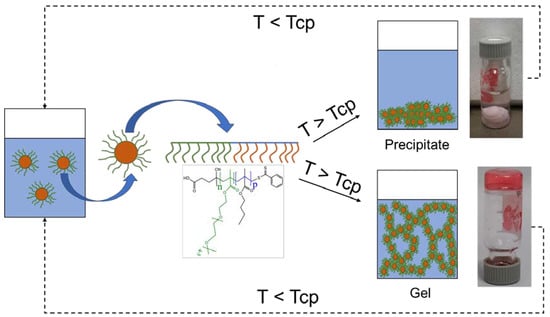
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of the Thermo-Responsive Macro CTAs
2.3. Synthesis of Amphiphilic Block Copolymers Self-Assembled into NPs
2.4. Study of the Thermo-Responsive Properties
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bennet, D.; Kim, S. Polymer Nanoparticles for Smart Drug Delivery. Appl. Nanotechnol. Drug Deliv. 2014. [Google Scholar] [CrossRef] [Green Version]
- Birrenbach, G.; Speiser, P.P. Polymerized Micelles and Their Use as Adjuvants in Immunology. J. Pharm. Sci. 1976, 65, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, J. Nanoparticles and Nanocapsules-New Dosage Forms in the Nanometer Size Range. Pharm. Acta Helv. 1978, 53, 33–39. [Google Scholar] [PubMed]
- Devonport, I.W.; Even, R.C.; Hermes, A.R.; Lorah, D.P.; Tanzer, J.D.; VanDyk, A.K. Aqueous Composition Containing Polymeric Nanoparticles. U.S. Patent No. US7071261B2, 4 July 2006. [Google Scholar]
- Serhan, M.; Sprowls, M.; Jackemeyer, D.; Long, M.; Perez, I.D.; Maret, W.; Tao, N.; Forzani, E. Industrial Applications of Nanoparticles. AIChE Annu. Meet. Conf. Proc. 2019. [Google Scholar] [CrossRef]
- Manfredini, N.; Ilare, J.; Invernizzi, M.; Polvara, E.; Contreras Mejia, D.; Sironi, S.; Moscatelli, D.; Sponchioni, M. Polymer Nanoparticles for the Release of Fragrances: How the Physicochemical Properties Influence the Adsorption on Textile and the Delivery of Limonene. Ind. Eng. Chem. Res. 2020, 59, 12766–12773. [Google Scholar] [CrossRef]
- Patel, A.; Prajapati, P.; Boghra, R. Overview on Application of Nanoparticles in Cosmetics. Asian J. Pharm. Sci. Clin. Res. 2011, 1, 40–55. [Google Scholar]
- Nasir, A.; Kausar, A.; Younus, A. A Review on Preparation, Properties and Applications of Polymeric Nanoparticle-Based Materials. Polym. Plast. Technol. Eng. 2015, 54, 325–341. [Google Scholar] [CrossRef]
- Asua, J.M. Emulsion Polymerization: From Fundamental Mechanisms to Process Developments. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 1025–1041. [Google Scholar] [CrossRef]
- Chern, C.S. Emulsion Polymerization Mechanisms and Kinetics. Prog. Polym. Sci. 2006, 31, 443–486. [Google Scholar] [CrossRef]
- Monteiro, M.J.; Cunningham, M.F. Polymer Nanoparticles via Living Radical Polymerization in Aqueous Dispersions: Design and Applications. Macromolecules 2012, 45, 4939–4957. [Google Scholar] [CrossRef]
- Chem, R. Reaction Chemistry & Engineering Progress in Reactor Engineering of Controlled Radical Polymerization: A Comprehensive Review. React. Chem. Eng. 2016, 1, 23–59. [Google Scholar] [CrossRef]
- Nicolas, J.; Guillaneuf, Y.; Lefay, C.; Bertin, D.; Gigmes, D.; Charleux, B. Nitroxide-Mediated Polymerization. Prog. Polym. Sci. 2013, 38, 63–235. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Atom Transfer Radical Polymerization (ATRP): Current Status and Future Perspectives. Macromolecules 2012, 45, 4015–4039. [Google Scholar] [CrossRef]
- Chiefari, J.; Chong, Y.K.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T.P.T.; Mayadunne, R.T.A.; Meijs, G.F.; Moad, C.L.; Moad, G.; et al. Living Free-Radical Polymerization by Reversible Addition—Fragmentation Chain Transfer: The RAFT Process. Macromolecules 1998, 31, 5559–5562. [Google Scholar] [CrossRef]
- Capasso Palmiero, U.; Maraldi, M.; Manfredini, N.; Moscatelli, D. Zwitterionic Polyester-Based Nanoparticles with Tunable Size, Polymer Molecular Weight, and Degradation Time. Biomacromolecules 2018, 19, 1314–1323. [Google Scholar] [CrossRef] [PubMed]
- Sponchioni, M.; Ferrari, R.; Morosi, L.; Moscatelli, D. Influence of the Polymer Structure over Self-Assembly and Thermo-Responsive Properties: The Case of PEG-b-PCL Grafted Copolymers via a Combination of RAFT and ROP. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 2919–2931. [Google Scholar] [CrossRef]
- Sponchioni, M.; Palmiero, U.C.; Moscatelli, D. HPMA-PEG Surfmers and Their Use in Stabilizing Fully Biodegradable Polymer Nanoparticles. Macromol. Chem. Phys. 2017, 218, 1–12. [Google Scholar] [CrossRef]
- Sponchioni, M.; Morosi, L.; Lupi, M.; Capasso Palmiero, U. Poly(HPMA)-Based Copolymers with Biodegradable Side Chains Able to Self Assemble into Nanoparticles. RSC Adv. 2017, 7, 50981–50992. [Google Scholar] [CrossRef] [Green Version]
- Zanoni, A.; Gardoni, G.; Sponchioni, M.; Moscatelli, D. Valorisation of Glycerol and CO2 to Produce Biodegradable Polymer Nanoparticles with a High Percentage of Bio-Based Components. J. CO2 Util. 2020, 40, 101192. [Google Scholar] [CrossRef]
- Zanetti, M.; Carniel, T.K.; Dalcanton, F.; dos Anjos, R.S.; Gracher Riella, H.; de Araújo, P.H.H.; de Oliveira, D.; Antônio Fiori, M. Use of Encapsulated Natural Compounds as Antimicrobial Additives in Food Packaging: A Brief Review. Trends Food Sci. Technol. 2018, 81, 51–60. [Google Scholar] [CrossRef]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable Polymeric Nanoparticles Based Drug Delivery Systems. Colloids Surf. B Biointerfaces 2010, 75, 1–18. [Google Scholar] [CrossRef]
- Wang, F.; Luo, Y.; Li, B.G.; Zhu, S. Synthesis and Redispersibility of Poly(Styrene- Block—N -Butyl Acrylate) Core-Shell Latexes by Emulsion Polymerization with Raft Agent-Surfactant Design. Macromolecules 2015, 48, 1313–1319. [Google Scholar] [CrossRef]
- Sponchioni, M.; Capasso Palmiero, U.; Moscatelli, D. Thermo-Responsive Polymers: Applications of Smart Materials in Drug Delivery and Tissue Engineering. Mater. Sci. Eng. C 2019, 102, 589–605. [Google Scholar] [CrossRef] [PubMed]
- Sponchioni, M.; Rodrigues Bassam, P.; Moscatelli, D.; Arosio, P.; Capasso Palmiero, U. Biodegradable Zwitterionic Nanoparticles with Tunable UCST-Type Phase Separation under Physiological Conditions. Nanoscale 2019, 11, 16582–16591. [Google Scholar] [CrossRef]
- Sponchioni, M.; O’Brien, C.T.; Borchers, C.; Wang, E.; Rivolta, M.N.; Penfold, N.J.W.; Canton, I.; Armes, S.P. Probing the Mechanism for Hydrogel-Based Stasis Induction in Human Pluripotent Stem Cells: Is the Chemical Functionality of the Hydrogel Important? Chem. Sci. 2020, 11, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Sponchioni, M.; Manfredini, N.; Zanoni, A.; Scibona, E.; Morbidelli, M.; Moscatelli, D. Readily Adsorbable Thermoresponsive Polymers for the Preparation of Smart Cell-Culturing Surfaces on Site. ACS Biomater. Sci. Eng. 2020, 6, 5337–5345. [Google Scholar] [CrossRef] [PubMed]
- Sponchioni, M.; Capasso Palmiero, U.; Manfredini, N.; Moscatelli, D. RAFT Copolymerization of Oppositely Charged Monomers and Its Use to Tailor the Composition of Nonfouling Polyampholytes with an UCST Behaviour. React. Chem. Eng. 2019, 4, 436–446. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, G.; Wang, W.J.; Yuan, H.; Li, B.G.; Zhu, S. Switchable Block Copolymer Surfactants for Preparation of Reversibly Coagulatable and Redispersible Poly(Methyl Methacrylate) Latexes. Macromolecules 2013, 46, 1261–1267. [Google Scholar] [CrossRef]
- Jessop, P.G.; Mercer, S.M.; Heldebrant, D.J. CO2-Triggered Switchable Solvents, Surfactants, and Other Materials. Energy Environ. Sci. 2012, 5, 7240–7253. [Google Scholar] [CrossRef]
- Li, Y.; Liu, L.; Liu, X.; Chen, S.; Fang, Y. Reversibly Responsive Microemulsion Triggered by Redox Reactions. J. Colloid Interface Sci. 2019, 540, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Halperin, A.; Kröger, M.; Winnik, F.M. Poly(N-Isopropylacrylamide) Phase Diagrams: Fifty Years of Research. Angew. Chem. Int. Ed. 2015, 54, 15342–15367. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Weber, C.; Schubert, U.S.; Hoogenboom, R. Thermoresponsive Polymers with Lower Critical Solution Temperature: From Fundamental Aspects and Measuring Techniques to Recommended Turbidimetry Conditions. Mater. Horiz. 2017, 4, 109–116. [Google Scholar] [CrossRef]
- Ward, M.A.; Georgiou, T.K. Thermoresponsive Polymers for Biomedical Applications. Polymers 2011, 3, 1215–1242. [Google Scholar] [CrossRef] [Green Version]
- Sanoj Rejinold, N.; Muthunarayanan, M.; Divyarani, V.V.; Sreerekha, P.R.; Chennazhi, K.P.; Nair, S.V.; Tamura, H.; Jayakumar, R. Curcumin-Loaded Biocompatible Thermoresponsive Polymeric Nanoparticles for Cancer Drug Delivery. J. Colloid Interface Sci. 2011, 360, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Sponchioni, M. Polymeric Nanoparticles for Controlled Drug Delivery. In Nanomaterials for Theranostics and Tissue Engineering; Rossi, F., Rainer, A.B.T.-N., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1–28. [Google Scholar] [CrossRef]
- Cao, P.-F.; Mangadlao, J.D.; Advincula, R.C. Stimuli-Responsive Polymers and Their Potential Applications in Oil-Gas Industry. Polym. Rev. 2015, 55, 706–733. [Google Scholar] [CrossRef]
- Winninger, J.; Iurea, D.M.; Atanase, L.I.; Salhi, S.; Delaite, C.; Riess, G. Micellization of Novel Biocompatible Thermo-Sensitive Graft Copolymers Based on Poly(ε-Caprolactone), Poly(N-Vinylcaprolactam) and Poly(N-Vinylpyrrolidone). Eur. Polym. J. 2019, 119, 74–82. [Google Scholar] [CrossRef]
- Palmiero, U.C.; Agostini, A.; Gatti, S.; Sponchioni, M.; Valenti, V.; Brunel, L.; Moscatelli, D. RAFT Macro-Surfmers and Their Use in the Ab Initio RAFT Emulsion Polymerization To Decouple Nanoparticle Size and Polymer Molecular Weight. Macromolecules 2016, 49, 8387–8396. [Google Scholar] [CrossRef]
- Maddinelli, G.; Bartosek, M.; Carminati, S.; Moghadasi, L.; Mandredini, N.; Moscatelli, D. Design of Thermoresponsive Polymers to Selective Permeability Reduction in Porous Media. In Proceedings of the Abu Dhabi International Petroleum Exhibition and Conference, Abu Dhabi, United Arab Emirates, 9–12 November 2020. [Google Scholar]
- Boulif, N.; Sebakhy, K.O.; Joosten, H.; Raffa, P. Design and Synthesis of Novel Di- and Triblock Amphiphilic Polyelectrolytes: Improving Salt-Induced Viscosity Reduction of Water Solutions for Potential Application in Enhanced Oil Recovery. J. Appl. Polym. Sci. 2021, 138, 50366. [Google Scholar] [CrossRef]
- Parker, B.R.; Derry, M.J.; Ning, Y.; Armes, S.P. Exploring the Upper Size Limit for Sterically Stabilized Diblock Copolymer Nanoparticles Prepared by Polymerization-Induced Self- Assembly in Non-Polar Media. Langmuir 2020, 36, 3730–3736. [Google Scholar] [CrossRef]
- Wu, H.; Xie, J.; Morbidelli, M. Soft Matter Kinetics of Colloidal Gelation and Scaling of the Gelation point. Soft Matter 2013, 9, 4437–4443. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Dn [nm] | PDI [-] | Tcp [°C] | HLB [-] |
|---|---|---|---|---|
| 40EG4-100BMA | 58.9 ± 0.3 | 0.15 ± 0.01 | 64 | 9.1 |
| 40EG4-200BMA | 72.6 ± 0.7 | 0.11 ± 0.01 | 65 | 5.9 |
| 40EG4-300BMA | 135.9 ± 0.7 | 0.12 ± 0.01 | 63 | 4.5 |
| 40EG4-500BMA | 194.9 ± 3.1 | 0.08 ± 0.01 | 63 | 2.8 |
| 40EG4-1000BMA | 412.5 ± 3.5 | 0.09 ± 0.01 | 64 | 1.6 |
| 40EG4-3000BMA | 1251.8 ± 94.1 | 0.07 ± 0.01 | 62 | 0.6 |
| 60EG4-150BMA | 48.6 ± 0.2 | 0.15 ± 0.03 | 65 | 9.3 |
| 60EG4-275BMA | 73.3 ± 0.5 | 0.05 ± 0.02 | 65 | 6.2 |
| 60EG4-400BMA | 125.2 ± 0.8 | 0.03 ± 0.02 | 64 | 4.8 |
| 60EG4-725BMA | 192.7 ± 0.1 | 0.06 ± 0.02 | 64 | 2.9 |
| 80EG4-450BMA | 110.3 ± 2.4 | 0.22 ± 0.01 | 64 | 5.9 |
| 80EG4-650BMA | 160.6 ± 2.1 | 0.05 ± 0.01 | 63 | 4.2 |
| 80EG4-850BMA | 177.3 ± 1.8 | 0.04 ± 0.08 | 63 | 3.5 |
| 100EG4-275BMA | 57.7 ± 2.1 | 0.05 ± 0.10 | 66 | 9.2 |
| 100EG4-600BMA | 102.1± 0.9 | 0.03 ± 0.01 | 64 | 5.9 |
| 100EG4-850BMA | 126.6 ± 2.5 | 0.03 ± 0.03 | 65 | 4.5 |
| 100EG4-1200BMA | 168.9 ± 4.1 | 0.03 ± 0.04 | 64 | 3.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manfredini, N.; Tomasoni, M.; Sponchioni, M.; Moscatelli, D. Influence of the Polymer Microstructure over the Phase Separation of Thermo-Responsive Nanoparticles. Polymers 2021, 13, 1032. https://doi.org/10.3390/polym13071032
Manfredini N, Tomasoni M, Sponchioni M, Moscatelli D. Influence of the Polymer Microstructure over the Phase Separation of Thermo-Responsive Nanoparticles. Polymers. 2021; 13(7):1032. https://doi.org/10.3390/polym13071032
Chicago/Turabian StyleManfredini, Nicolò, Marco Tomasoni, Mattia Sponchioni, and Davide Moscatelli. 2021. "Influence of the Polymer Microstructure over the Phase Separation of Thermo-Responsive Nanoparticles" Polymers 13, no. 7: 1032. https://doi.org/10.3390/polym13071032
APA StyleManfredini, N., Tomasoni, M., Sponchioni, M., & Moscatelli, D. (2021). Influence of the Polymer Microstructure over the Phase Separation of Thermo-Responsive Nanoparticles. Polymers, 13(7), 1032. https://doi.org/10.3390/polym13071032