
Potential of Mean Force between Bare or Grafted Silica/Polystyrene Surfaces from Self-Consistent Field Theory

 , , and
, , and
Abstract
:
1. Introduction
2. Model and Theoretical Formulation
2.1. Model Systems
2.2. Self-Consistent Field Convergence Scheme
2.3. Thermodynamic Description
2.3.1. Systems with Melt: Grand Canonical Ensemble
2.3.2. Systems without Melt: Canonical Ensemble
2.4. Nonbonded Free Energy Density
2.4.1. Square Gradient Theory
2.4.2. Sanchez-Lacombe
2.4.3. Helfand Free Energy Density
2.5. Interactions with the Solid Surfaces
2.5.1. Polymer–Solid Interactions
2.5.2. Solid–Solid Interactions
3. Calculation Details
4. Results
4.1. Single Surfaces
4.1.1. Vacuum/Melt (VM) and Solid/Melt (SM) Interphases
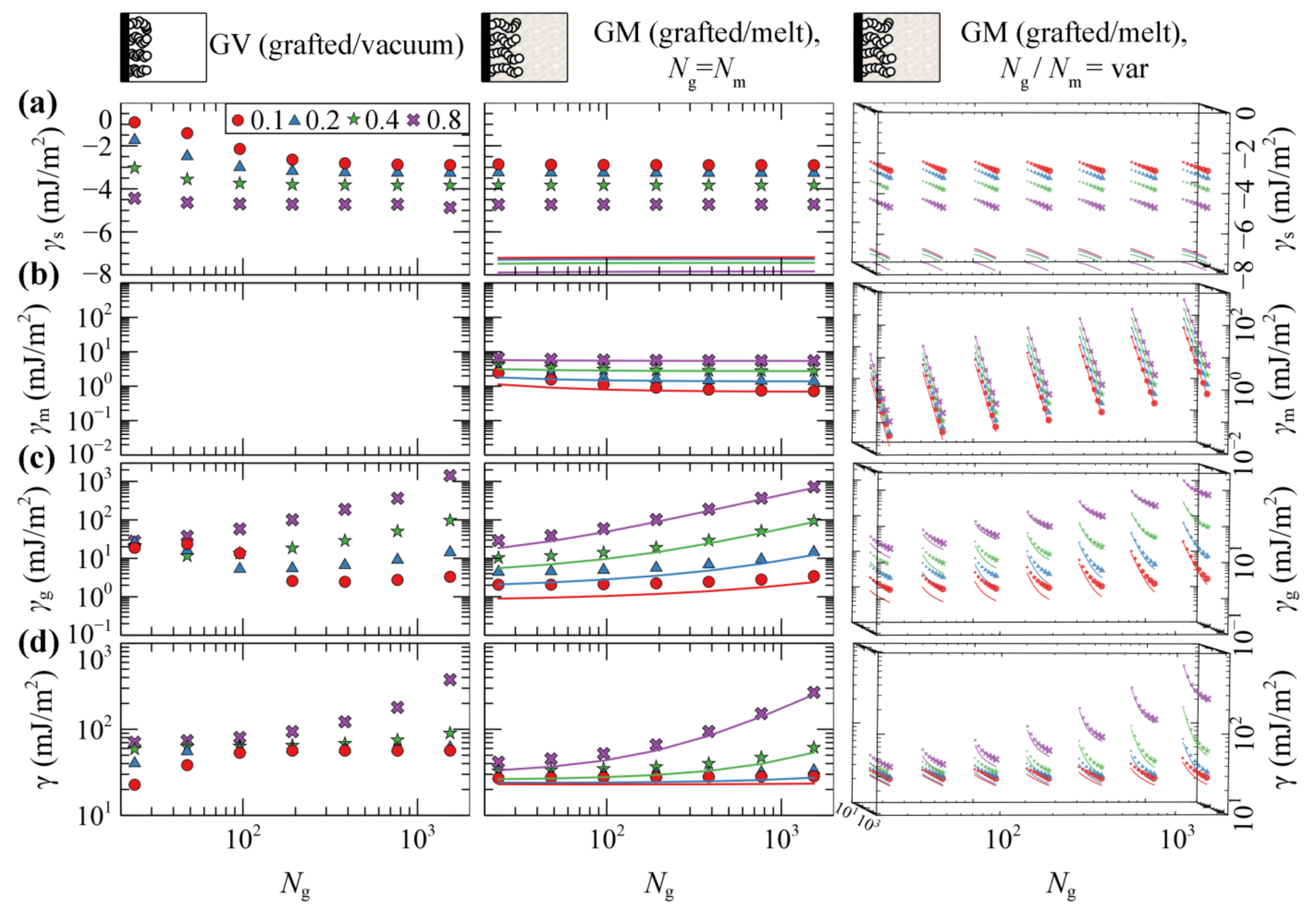
4.1.2. Grafted/Vacuum (GV) and Grafted/Melt (GM) Interphases
- Mushroom regime (low σgNg). The density of the profiles is less than the bulk density and chains assume randomly coiled conformations [63]. Increasing σg and Ng has a minor effect on the thickness of the profiles but rather makes them more pronounced.
- Dense brush regime (high σgNg). The brushes become stretched [46,63] and feature extended regions with bulk density. The thickness of the profiles depends strongly on both σg and Ng, reaching the limiting scaling behavior ~σg1Ng1; i.e., the dimensions of the brushes become proportional to their mass.
- Crowding regime (very high σg). In this regime, the crowding of the chains becomes so intense that the density of the grafted chains surpasses slightly the bulk one, as observed in our previous work [46]. This happens because the entropic penalty due to stretching overcomes the enthalpic penalty due to deviations from the bulk density.
- is purely of enthalpic origin and thus it is a functional of the total density profiles. becomes more attractive with increasing σg, since the profiles become more pronounced in the vicinity of the solid (e.g., see Figure 3). In GV, decreases with increasing Ng, since increasing Ng increases the amount of material near the solid. In GM, is independent of Ng, since the total density profiles are also invariant to Ng, e.g., compare profiles in Figure 2c–f for different chain lengths.
- describes the entropic contribution of the matrix chains. In GM, it decreases precipitously with increasing ratio, since the grafted chains occupy more space in the vicinity of the Interphase leaving the matrix chains with fewer available conformations. Note that, by keeping Ng fixed, γm scales with Nm about as γm ~ Nm−1; see Equation (8). In GV systems, this term is of course zero since there are no matrix chains at all.
- quantifies the entropic contribution of the grafted chains and it is an indicator of the stretching of the brush [46]. It increases monotonically with increasing Ng and σg, since the grafted chains expand and stretch further towards the bulk region. In addition, increases with decreasing Nm, since the grafted chain-melt interactions are enhanced and, as a result, the brushes swell as shown in Figure 4b,c (rightmost column).
- is the total free energy of the interfacial systems per unit area. It increases with increasing molecular weight of grafted chains and appears to be dominated by the conformational entropy term of grafted chains, .
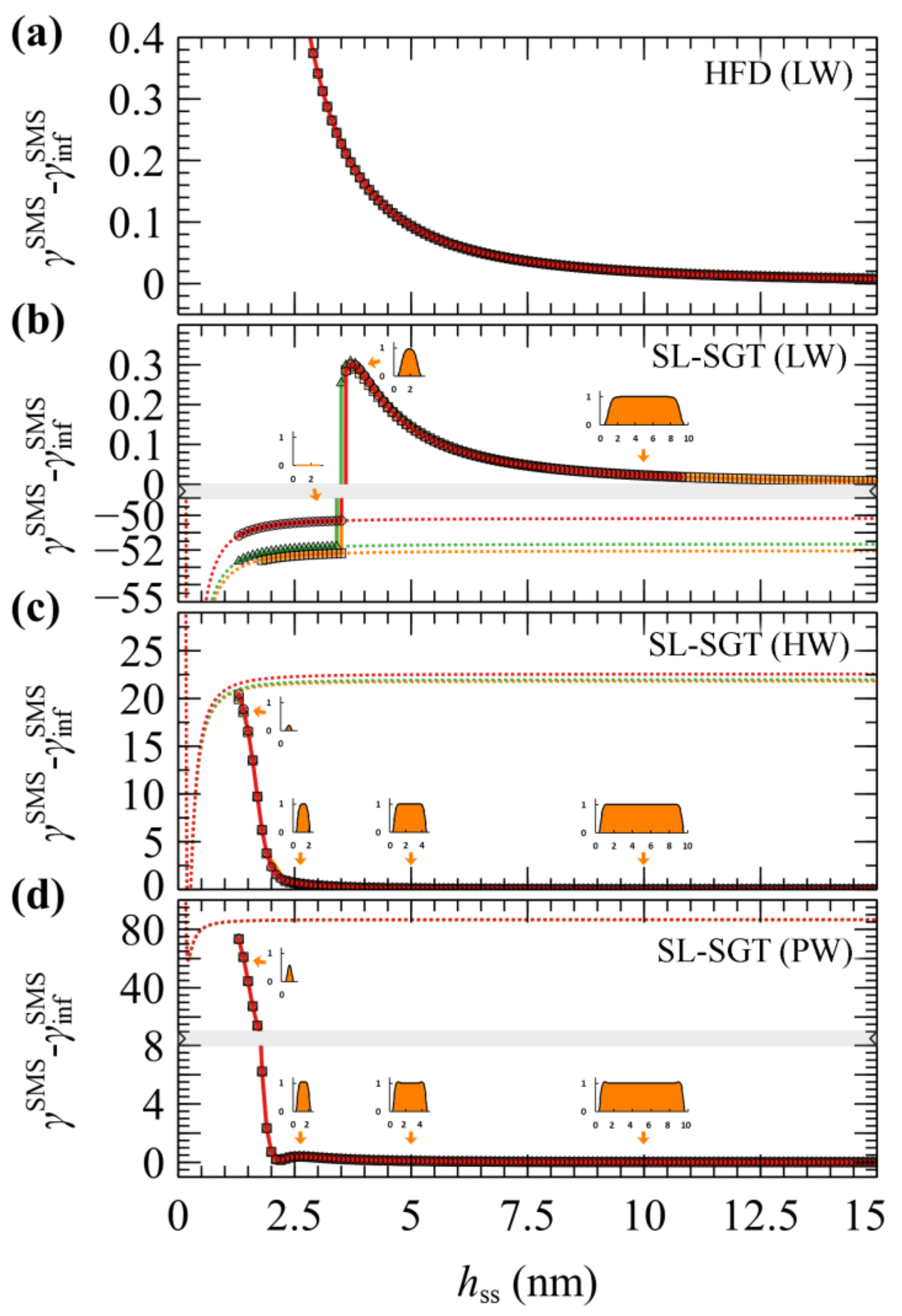
4.2. PS Melt Capped between Two Bare Silica Surfaces (SMS)
- In the first method, the calculations were performed in a decremental fashion, in which the initial configuration of the field was set to the field corresponding to the converged calculation for a slightly larger domain,
- In the calculations corresponding to the second method, the initial configuration of the field was set to zero across the domain, .
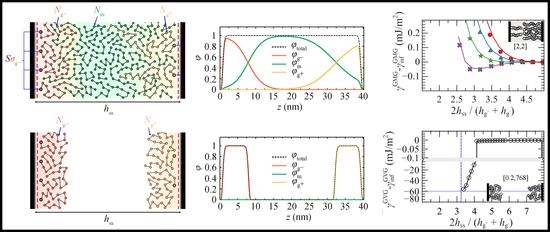
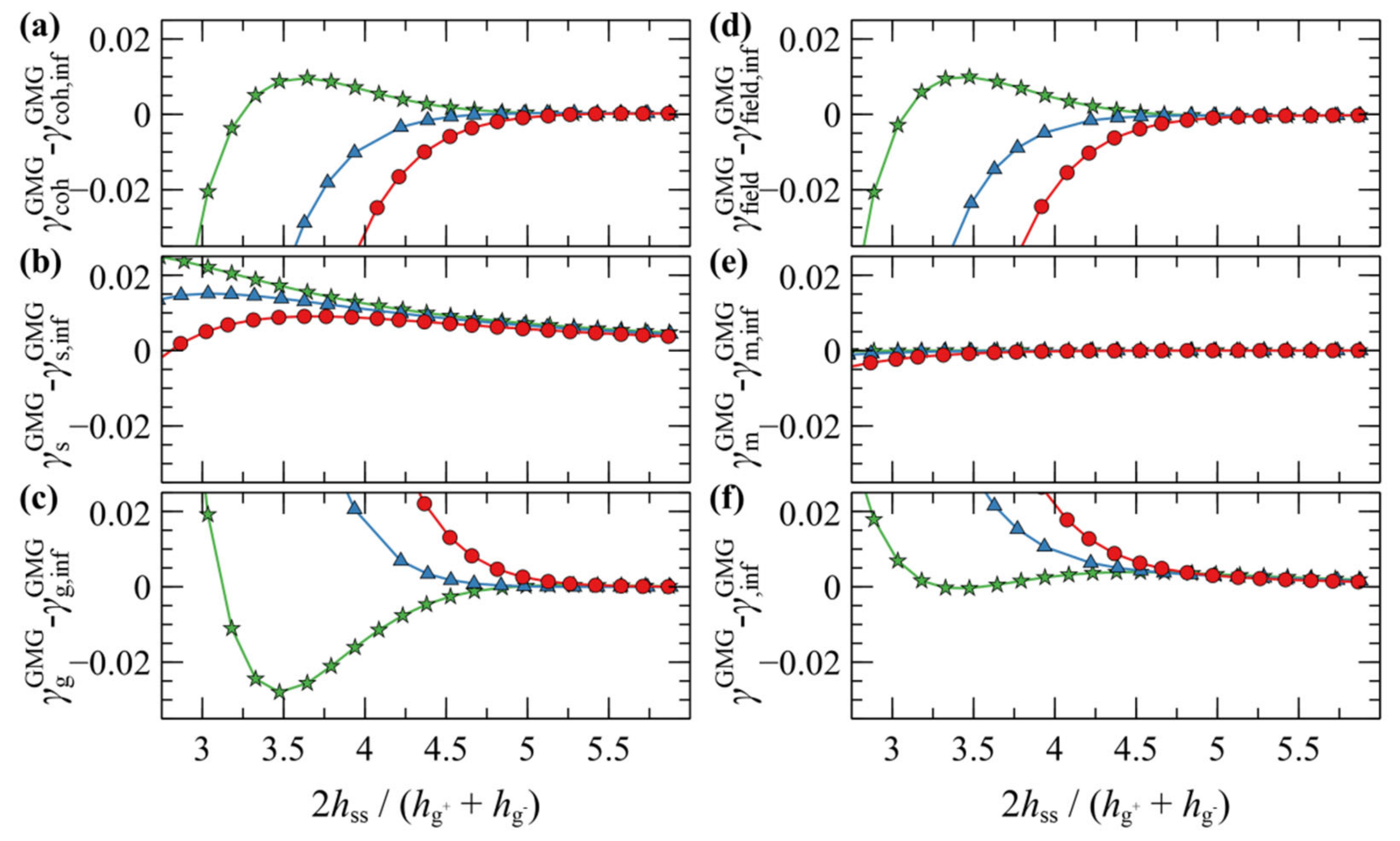
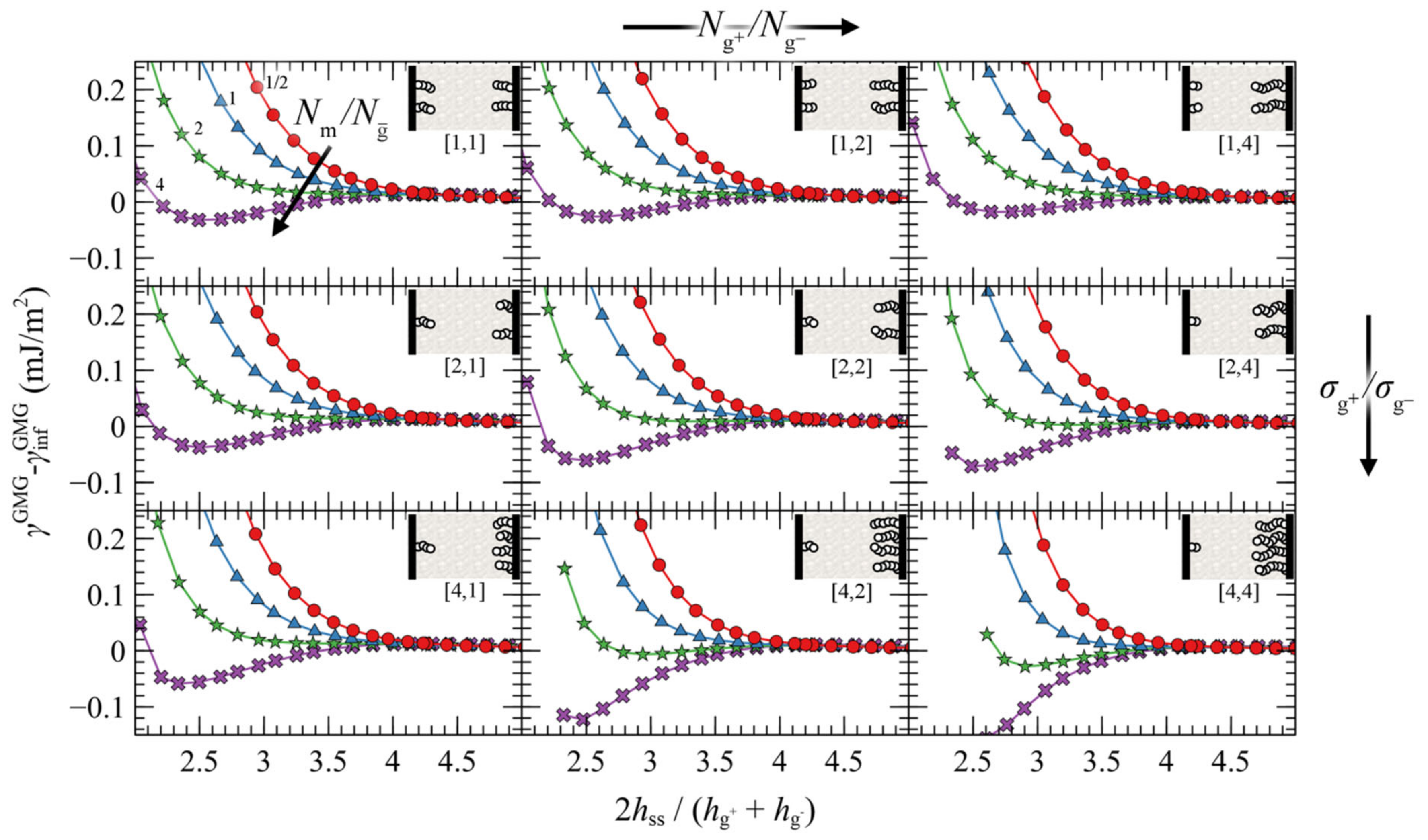
4.3. Interacting Grafted Surfaces in Melt (GMG)
4.3.1. Symmetric Surfaces
4.3.2. Asymmetric Surfaces
4.3.3. Surface Energy
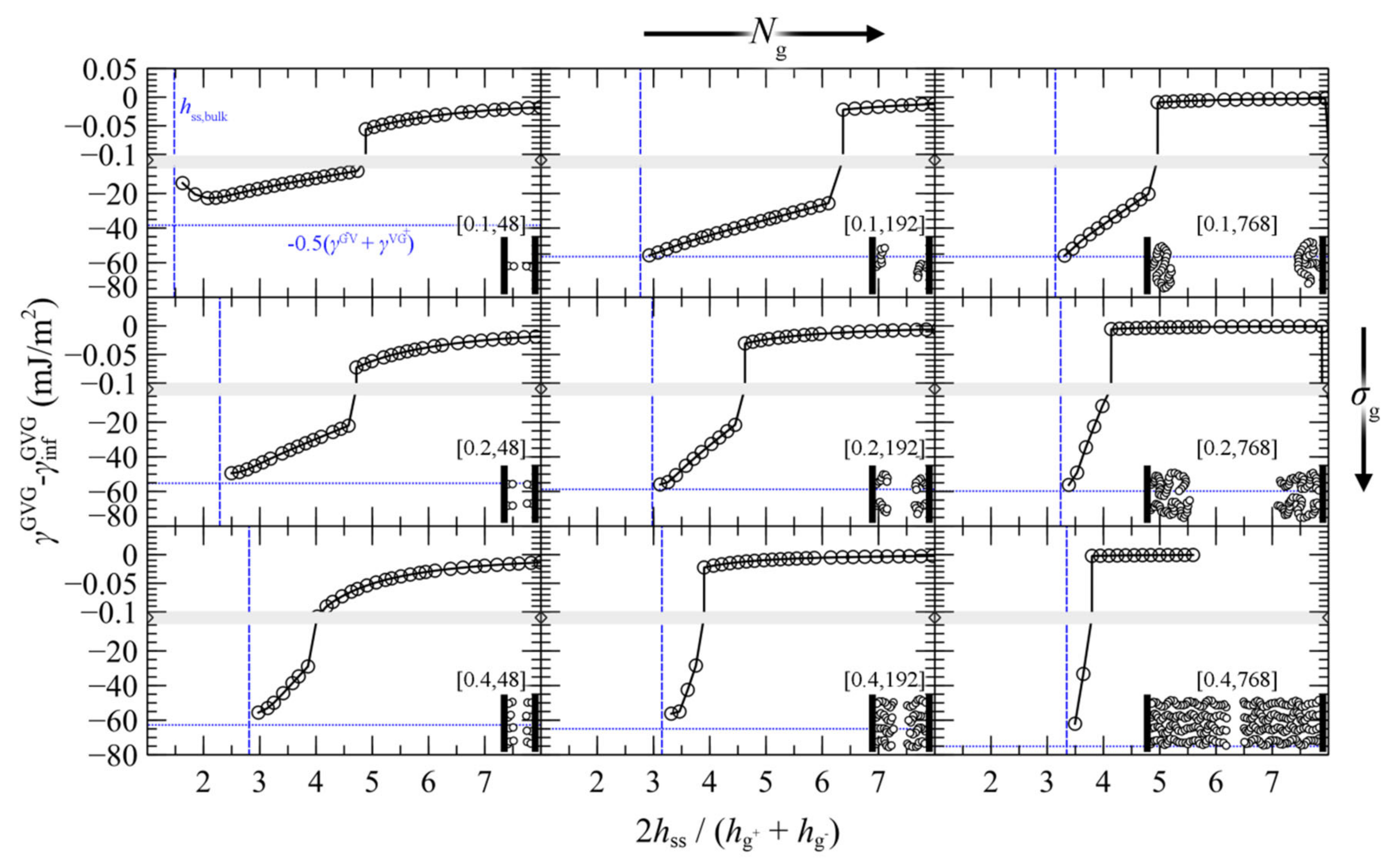
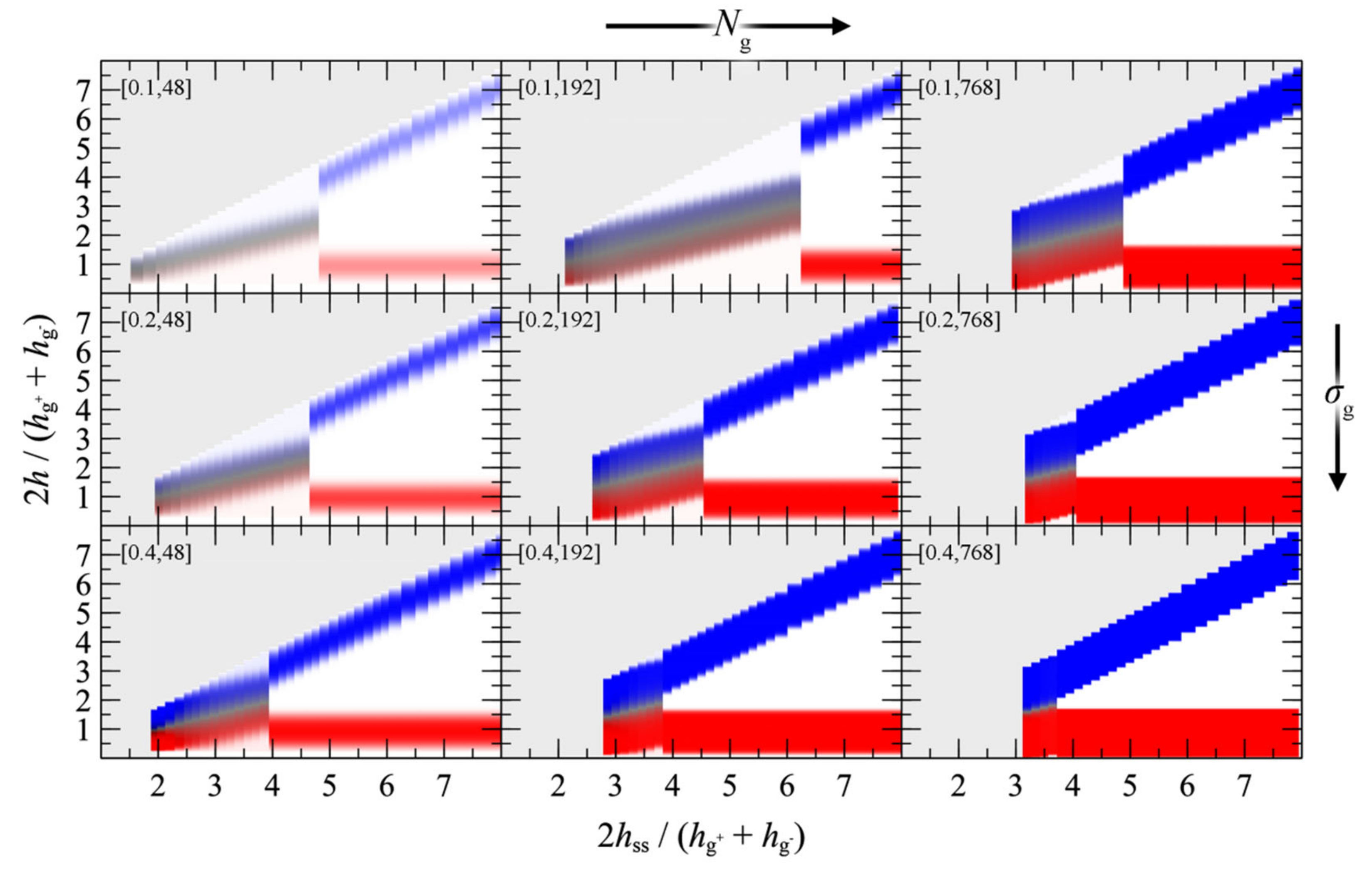
4.4. Interacting Grafted Surfaces in Vacuum (GVG)
- For large separation distances the adjacent polymer brushes interact weakly with each other and the dominant contribution to PMFGVG arises due to polymer/solid and solid/solid Hamaker interactions (e.g., compare with the dotted lines in Figure 6b–d).
- Below a critical plate-plate distance, PMFGVG decreases abruptly, indicating the manifestation of a phase transition where the adjacent brushes interpenetrate each other and form a single film in the central region of the system. In addition, low density regions are formed in the vicinity of the solid surface, indicating that the brushes have been stretched significantly towards the bulk region.
- Decreasing the plate-plate separation further makes the brushes more compact. The low-density regions next to the solid become suppressed, until the free energy becomes commensurate to minus the mean surface free energy of the isolated brushes, . Further squeezing of the brushes leads to increased reduced densities above unity, as indicated by the vertical dashed lines in Figure 13.
- An enthalpic gain due to the lower surface area of the merged brushes that increases with increasing surface tension.
- An enthalpic loss due to the detachment of the grafted film from the surface that depends on the polymer/solid interactions.
- A conformational penalty due to chain stretching.
5. Discussion
6. Conclusions
- Grafting densities; very low (σg = 0, bare surfaces), moderate and high.
- Asymmetry regarding the grafting densities and grafted chain lengths.
- Solvent conditions; good (), theta (), bad ( ), and very bad (vacuum) solvents.
- Wetting degree; low, high and perfectly wetted interfaces.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
References
- Hore, M.J.A. Polymers on nanoparticles: Structure & dynamics. Soft Matter 2019, 15, 1120–1134. [Google Scholar] [CrossRef]
- Kumar, S.K.; Benicewicz, B.C.; Vaia, R.A.; Winey, K.I. 50th Anniversary Perspective: Are Polymer Nanocomposites Practical for Applications? Macromolecules 2017, 50, 714–731. [Google Scholar] [CrossRef]
- Hore, M.J.A.; Korley, L.T.J.; Kumar, S.K. Polymer-Grafted Nanoparticles. J. Appl. Phys. 2020, 128, 030401. [Google Scholar] [CrossRef]
- Liu, P.; Su, Z. Preparation of polystyrene grafted silica nanoparticles by two-steps UV induced reaction. J. Photochem. Photobiol. A Chem. 2004, 167, 237–240. [Google Scholar] [CrossRef]
- Liu, C.H.; Pan, C.Y. Grafting polystyrene onto silica nanoparticles via RAFT polymerization. Polymer 2007, 48, 3679–3685. [Google Scholar] [CrossRef]
- Tumnantong, D.; Rempel, G.L.; Prasassarakich, P. Synthesis of polystyrene-silica nanoparticles via RAFT emulsifier-free emulsion polymerization. Eur. Polym. J. 2016, 80, 145–157. [Google Scholar] [CrossRef]
- Hamaker, H.C. The London—van der Waals attraction between spherical particles. Physica 1937, 4, 1058–1072. [Google Scholar] [CrossRef]
- Abbas, Z.M.; Tawfilas, M.; Khani, M.M.; Golian, K.; Marsh, Z.M.; Jhalaria, M.; Simonutti, R.; Stefik, M.; Kumar, S.K.; Benicewicz, B.C. Reinforcement of polychloroprene by grafted silica nanoparticles. Polymer 2019, 171, 96–105. [Google Scholar] [CrossRef]
- Moll, J.F.; Akcora, P.; Rungta, A.; Gong, S.; Colby, R.H.; Benicewicz, B.C.; Kumar, S.K. Mechanical reinforcement in polymer melts filled with polymer grafted nanoparticles. Macromolecules 2011, 44, 7473–7477. [Google Scholar] [CrossRef]
- Askar, S.; Li, L.; Torkelson, J.M. Polystyrene-Grafted Silica Nanoparticles: Investigating the Molecular Weight Dependence of Glass Transition and Fragility Behavior. Macromolecules 2017, 50, 1589–1598. [Google Scholar] [CrossRef]
- Giovino, M.; Buenning, E.; Jimenez, A.; Kumar, S.K.; Schadler, L. Polymer Grafted Nanoparticle Viscosity Modifiers. Macromol. Chem. Phys. 2019, 220, 1–7. [Google Scholar] [CrossRef]
- Zhao, D.; Ge, S.; Senses, E.; Akcora, P.; Jestin, J.; Kumar, S.K. Role of Filler Shape and Connectivity on the Viscoelastic Behavior in Polymer Nanocomposites. Macromolecules 2015, 48, 5433–5438. [Google Scholar] [CrossRef]
- Harton, S.E.; Kumar, S.K. Mean-Field Theoretical Analysis of Brush-Coated Nanoparticle Dispersion in Polymer Matrices. J. Polym. Sci. 2008, 46, 351–358. [Google Scholar] [CrossRef]
- Pépin, M.P.; Whitmore, M.D. Monte Carlo and numerical self-consistent field study of systems with end-grafted and free polymers in good solvent. J. Chem. Phys. 2001, 114, 8181–8195. [Google Scholar] [CrossRef]
- Vogiatzis, G.G.; Theodorou, D.N. Structure of polymer layers grafted to nanoparticles in silica-polystyrene nanocomposites. Macromolecules 2013, 46, 4670–4683. [Google Scholar] [CrossRef] [Green Version]
- Kalb, J.; Dukes, D.; Kumar, S.K.; Hoy, R.S.; Grest, G.S. End grafted polymer nanoparticles in a polymeric matrix: Effect of coverage and curvature. Soft Matter 2011, 7, 1418–1425. [Google Scholar] [CrossRef] [Green Version]
- Ndoro, T.V.M.; Voyiatzis, E.; Ghanbari, A.; Theodorou, D.N.; Böhm, M.C.; Müller-Plathe, F. Interface of grafted and ungrafted silica nanoparticles with a polystyrene matrix: Atomistic molecular dynamics simulations. Macromolecules 2011, 44, 2316–2327. [Google Scholar] [CrossRef]
- Lo Verso, F.; Egorov, S.A.; Milchev, A.; Binder, K. Spherical polymer brushes under good solvent conditions: Molecular dynamics results compared to density functional theory. J. Chem. Phys. 2010, 133, 184901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munaò, G.; Pizzirusso, A.; Kalogirou, A.; De Nicola, A.; Kawakatsu, T.; Müller-Plathe, F.; Milano, G. Molecular structure and multi-body potential of mean force in silica-polystyrene nanocomposites. Nanoscale 2018, 10, 21656–21670. [Google Scholar] [CrossRef] [Green Version]
- Trombly, D.M.; Ganesan, V. Curvature effects upon interactions of polymer-grafted nanoparticles in chemically identical polymer matrices. J. Chem. Phys. 2010, 133, 154904. [Google Scholar] [CrossRef]
- Roan, J.R.; Kawakatsu, T. Self-consistent-field theory for interacting polymeric assemblies. II. Steric stabilization of colloidal particles. J. Chem. Phys. 2002, 116, 7295–7310. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, H.; Yang, Y. Morphology and Interactions of Polymer Brush-Coated Spheres. J. Polym. Sci. 2006, 44, 2811–2820. [Google Scholar] [CrossRef]
- Martin, T.B.; Dodd, P.M.; Jayaraman, A. Polydispersity for tuning the potential of mean force between polymer grafted nanoparticles in a polymer matrix. Phys. Rev. Lett. 2013, 110, 018301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, D.; Kumar, S.K.; Lane, J.M.D.; Grest, G.S. Effective interactions between grafted nanoparticles in a polymer matrix. Soft Matter 2012, 8, 5002–5010. [Google Scholar] [CrossRef]
- Hasegawa, R.; Aoki, Y.; Doi, M. Optimum graft density for dispersing particles in polymer melts. Macromolecules 1996, 29, 6656–6662. [Google Scholar] [CrossRef]
- Fredrickson, G.H. The Equilibrium Theory of Inhomogeneous Polymers; International Series of Monographs on Physics; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Schmid, F. Self-consistent-field theories for complex fluids. J. Phys. Condens. Matter 1998, 10, 8105–8138. [Google Scholar] [CrossRef]
- Muller, M.; Schmid, F. Incorporating fluctuations and dynamics in self-consistent field theories for polymer blends. Adv. Polym. Sci. 2005, 185, 1–58. [Google Scholar]
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: New York, NY, USA, 1953; ISBN 0801401348. [Google Scholar]
- Schneck, E.; Sedlmeier, F.; Netz, R.R. Hydration repulsion between biomembranes results from an interplay of dehydration and depolarization. Proc. Natl. Acad. Sci. USA 2012, 109, 14405–14409. [Google Scholar] [CrossRef] [Green Version]
- Schneck, E.; Netz, R.R. From simple surface models to lipid membranes: Universal aspects of the hydration interaction from solvent-explicit simulations. Curr. Opin. Colloid Interface Sci. 2011, 16, 607–611. [Google Scholar] [CrossRef]
- Jhalaria, M.; Buenning, E.; Huang, Y.; Tyagi, M.; Zorn, R.; Zamponi, M.; García-Sakai, V.; Jestin, J.; Benicewicz, B.C.; Kumar, S.K. Accelerated Local Dynamics in Matrix-Free Polymer Grafted Nanoparticles. Phys. Rev. Lett. 2019, 123, 158003. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Dong, H.; Matyjaszewski, K.; Bockstaller, M.R. Flexible particle array structures by controlling polymer graft architecture. J. Am. Chem. Soc. 2010, 132, 12537–12539. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, C.A.; Koerner, H.; Meth, J.S.; Dang, A.; Hui, C.M.; Matyjaszewski, K.; Bockstaller, M.R.; Durstock, M.F.; Vaia, R.A. Performance of dielectric nanocomposites: Matrix-free, hairy nanoparticle assemblies and amorphous polymer-nanoparticle blends. ACS Appl. Mater. Interfaces 2014, 6, 21500–21509. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Hsiao, M.-S.; Vaia, R.A.; Tibbits, A.; Drummy, L.F.; Gillman, A.; Buskohl, P. Deformation Behavior of Polystyrene-Grafted Nanoparticle Assemblies with Low Grafting Density. Macromolecules 2018, 51, 7257–7265. [Google Scholar] [CrossRef]
- Wei, Y.; Xu, Y.; Faraone, A.; Hore, M.J.A. Local Structure and Relaxation Dynamics in the Brush of Polymer-Grafted Silica Nanoparticles. ACS Macro Lett. 2018, 7, 699–704. [Google Scholar] [CrossRef]
- Barnett, J.W.; Kumar, S.K. Modeling gas transport in polymer-grafted nanoparticle membranes. Soft Matter 2019, 15, 424–432. [Google Scholar] [CrossRef]
- Bilchak, C.R.; Buenning, E.; Asai, M.; Zhang, K.; Durning, C.J.; Kumar, S.K.; Huang, Y.; Benicewicz, B.C.; Gidley, D.W.; Cheng, S.; et al. Polymer-Grafted Nanoparticle Membranes with Controllable Free Volume. Macromolecules 2017, 50, 7111–7120. [Google Scholar] [CrossRef]
- Bilchak, C.R.; Jhalaria, M.; Huang, Y.; Abbas, Z.; Midya, J.; Benedetti, F.M.; Parisi, D.; Egger, W.; Dickmann, M.; Minelli, M.; et al. Tuning Selectivities in Gas Separation Membranes Based on Polymer-Grafted Nanoparticles. ACS Nano 2020, 14, 17174–17183. [Google Scholar] [CrossRef]
- Midya, J.; Rubinstein, M.; Kumar, S.K.; Nikoubashman, A. Structure of polymer-grafted nanoparticle melts. ACS Nano 2020, 14, 15505–15516. [Google Scholar] [CrossRef]
- Matsen, M.W.; Gardiner, J.M. Autophobic dewetting of homopolymer on a brush and entropic attraction between opposing brushes in a homopolymer matrix. J. Chem. Phys. 2001, 115, 2794–2804. [Google Scholar] [CrossRef]
- Deng, S.; Zhang, L.; Zhou, X.; Fan, C.; Lin, Q.; Lin, J. Exploring Microstructures and Interphase Properties of Surface- Grafted Diblock Copolymers in a Homopolymer Melt by Self-Consistent Field Theory Simulations. J. Macromol. Sci. Part B Phys. 2015, 54, 348–364. [Google Scholar] [CrossRef]
- Kim, J.U.; Matsen, M.W. Finite-stretching corrections to the Milner-Witten-Cates theory for polymer brushes. Eur. Phys. J. E 2007, 23, 135–144. [Google Scholar] [CrossRef]
- Lakkas, A.T.; Sgouros, A.P.; Theodorou, D.N. Self-Consistent Field Theory Coupled with Square Gradient Theory of Free Surfaces of Molten Polymers and Compared to Atomistic Simulations and Experiment. Macromolecules 2019, 52, 5337–5356. [Google Scholar] [CrossRef]
- Helfand, E.; Tagami, Y. Theory of the interface between immiscible polymers. II. J. Chem. Phys. 1972, 56, 3592–3601. [Google Scholar] [CrossRef]
- Lakkas, A.T.; Sgouros, A.P.; Revelas, C.J.; Theodorou, D.N. Structure and Thermodynamics of Grafted Silica/Polystyrene Nanocomposites Investigated Through Self-Consistent Field Theory. Soft Matter 2021. [Google Scholar] [CrossRef] [PubMed]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Clarendon Press: Oxford, UK, 1986. [Google Scholar]
- Lin, H.; Duan, Y.Y.; Min, Q. Gradient theory modeling of surface tension for pure fluids and binary mixtures. Fluid Phase Equilib. 2007, 254, 75–90. [Google Scholar] [CrossRef]
- Sgouros, A.P.; Lakkas, A.T.; Megariotis, G.; Theodorou, D.N. Mesoscopic Simulations of Free Surfaces of Molten Polyethylene: Brownian Dynamics/kinetic Monte Carlo Coupled with Square Gradient Theory and Compared to Atomistic Calculations and Experiment. Macromolecules 2018, 51, 9798–9815. [Google Scholar] [CrossRef]
- Sanchez, I.C.; Lacombe, R.H. An elementary molecular theory of classical fluids. Pure fluids. J. Phys. Chem. 1976, 80, 2352–2362. [Google Scholar] [CrossRef]
- Israelachvili, J.N. Intermolecular and Surface Forces, 3rd ed.; Elsevier: Berkeley, CA, USA, 2011; ISBN 9780123919274. [Google Scholar]
- Revelas, C.J.; Sgouros, A.P.; Lakkas, A.T.; Theodorou, D.N. A Three-Dimensional Finite Element Methodology for Addressing Heterogeneous Polymer Systems with Simulations Based on Self-Consistent Field Theory. AIP Conf. Proc. 2021, 2343, 130002. [Google Scholar] [CrossRef]
- Theodorou, D.N. Polymers at Surfaces and Interfaces. Comput. Simul. Surfaces Interfaces 2003, 329–419. [Google Scholar] [CrossRef]
- Mortezaei, M.; Famili, M.H.N.; Kokabi, M. The role of interfacial interactions on the glass-transition and viscoelastic properties of silica/polystyrene nanocomposite. Compos. Sci. Technol. 2011, 71, 1039–1045. [Google Scholar] [CrossRef]
- Hong, K.M.; Noolandi, J. Conformational Entropy Effects in a Compressible Lattice Fluid Theory of Polymers. Macromolecules 1981, 14, 1229–1234. [Google Scholar] [CrossRef]
- Gaines, G.L. Surface and interfacial tension of polymer liquids—A review. Polym. Eng. Sci. 1972, 12, 1–11. [Google Scholar] [CrossRef]
- Sanchez, I.C.; Lacombe, R.H. Statistical Thermodynamics of Polymer Solutions. Macromolecules 1978, 11, 1145–1156. [Google Scholar] [CrossRef] [Green Version]
- Müller, M. Phase diagram of a mixed polymer brush. Phys. Rev. E Stat. 2002, 65, 030802. [Google Scholar] [CrossRef]
- Poser, C.I.; Sanchez, I.C. Surface tension theory of pure liquids and polymer melts. J. Colloid Interface Sci. 1979, 69, 539–548. [Google Scholar] [CrossRef]
- Rolo, L.I.; Caço, A.I.; Queimada, A.J.; Marrucho, I.M.; Coutinho, J.A.P. Surface tension of heptane, decane, hexadecane, eicosane, and some of their binary mixtures. J. Chem. Eng. Data 2002, 47, 1442–1445. [Google Scholar] [CrossRef]
- Sgouros, A.P.; Vogiatzis, G.G.; Kritikos, G.; Boziki, A.; Nikolakopoulou, A.; Liveris, D.; Theodorou, D.N. Molecular Simulations of Free and Graphite Capped Polyethylene Films: Estimation of the Interfacial Free Energies. Macromolecules 2017, 50, 8827–8844. [Google Scholar] [CrossRef]
- Mansfield, K.F.; Theodorou, D.N. Atomistic Simulation of a Glassy Polymer/Graphite Interface. Macromolecules 1991, 24, 4295–4309. [Google Scholar] [CrossRef]
- Aubouy, M.; Fredrickson, G.H.; Pincus, P.; Raphael, E. End-Tethered Chains. Macromolecules 1995, 28, 2979–2981. [Google Scholar] [CrossRef]
- Alexander, S. Adsorption of Chain Molecules with a Polar Head—A Scaling Description. LE J. Phys. 1977, 38, 983–987. [Google Scholar] [CrossRef]
- De Gennes, P.G. Conformations of Polymers Attached to an Interface. Macromolecules 1980, 13, 1069–1075. [Google Scholar] [CrossRef]
- Theodorou, D.N. Variable-Density Model of Polymer Melt/Solid Interfaces: Structure, Adhesion Tension, and Surface Forces. Macromolecules 1989, 22, 4589–4597. [Google Scholar] [CrossRef]
- Ferreira, P.G.; Ajdari, A.; Leibler, L. Scaling law for entropic effects at interfaces between grafted layers and polymer melts. Macromolecules 1998, 31, 3994–4003. [Google Scholar] [CrossRef]
- Green, D.L.; Mewis, J. Connecting the wetting and rheological behaviors of poly(dimethylsiloxane)-grafted silica spheres in poly(dimethylsiloxane) melts. Langmuir 2006, 22, 9546–9553. [Google Scholar] [CrossRef] [PubMed]
- Sunday, D.; Ilavsky, J.; Green, D.L. A phase diagram for polymer-grafted nanoparticles in homopolymer matrices. Macromolecules 2012, 45, 4007–4011. [Google Scholar] [CrossRef]
- Kumar, S.K.; Jouault, N.; Benicewicz, B.; Neely, T. Nanocomposites with polymer grafted nanoparticles. Macromolecules 2013, 46, 3199–3214. [Google Scholar] [CrossRef]
- Sunday, D.F.; Green, D.L. Thermal and Rheological Behavior of Polymer Grafted Nanoparticles. Macromolecules 2015, 48, 8651–8659. [Google Scholar] [CrossRef]
- Green, P.F. The structure of chain end-grafted nanoparticle/homopolymer nanocomposites. Soft Matter 2011, 7, 7914–7926. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value | Reference | |
|---|---|---|---|
| System | T | 500 K | |
| 0.05 nm | - | ||
| 0.05 nm | - | ||
| Chain stiffness | bk | 1.83 nm | [15] |
| lC-C | 0.154 nm | - | |
| 0.829 | [53] | ||
| mmonomer | 52.08 g/mol | - | |
| Hamaker | hHS | ~0.4 nm | - |
| 0.37 nm | [15] | ||
| 0.30 nm | [15] | ||
| 5.84 × 10−20 J | [15] | ||
| 6.43 × 10−20 J | [15] | ||
| Ramp potential | vramp (LW) | 0.0 | |
| vramp (HW) | –2.481 × 10−20 J | Fitted to WA = 38.8 mJ/m2 from [54] | |
| vramp (PW) | –3.975 × 10−20 J | Fitted to WA = 71.1 mJ/m2 from [54] | |
| σramp | 1.28 nm | ||
| Helfand | 3.97 (GPa)−1 | [55,56] | |
| 953 kg/m3 | [15] | ||
| Sanchez Lacombe | ρ* | 1105 kg/m3 | [57] |
| P* | 357 MPa | [57] | |
| T* | 735 K | [57] | |
| Square Gradient | κ | 0.2233·10–66 J m5 0.55 | [44] |
| Edwards Diffusion | 0.05 nm | [46] | |
| 0.25 | [46] | ||
| 10−6 | - |
| EoS | Wetting | γVM | γSM | σSV–σSM | WA | WS | WI | WC | θ (°) |
|---|---|---|---|---|---|---|---|---|---|
| HFD | low (LW) | 28.85 | 21.97 | –21.97 | 6.88 | –50.81 | –21.97 | 57.70 | 139.6 |
| SL-SGT | low (LW) | 27.89 | 26.02 | –26.02 | 1.86 | –53.91 | –26.02 | 55.77 | 158.9 |
| SL-SGT | high (HW) | 27.89 | –10.91 | 10.91 | 38.8 [54] | –16.97 | 10.91 | 55.77 | 67.0 |
| SL-SGT | Full (FW) | 27.89 | –43.21 | 43.21 | 71.1 [54] | 15.33 | 43.21 | 55.77 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sgouros, A.P.; Revelas, C.J.; Lakkas, A.T.; Theodorou, D.N. Potential of Mean Force between Bare or Grafted Silica/Polystyrene Surfaces from Self-Consistent Field Theory. Polymers 2021, 13, 1197. https://doi.org/10.3390/polym13081197
Sgouros AP, Revelas CJ, Lakkas AT, Theodorou DN. Potential of Mean Force between Bare or Grafted Silica/Polystyrene Surfaces from Self-Consistent Field Theory. Polymers. 2021; 13(8):1197. https://doi.org/10.3390/polym13081197
Chicago/Turabian StyleSgouros, Aristotelis P., Constantinos J. Revelas, Apostolos T. Lakkas, and Doros N. Theodorou. 2021. "Potential of Mean Force between Bare or Grafted Silica/Polystyrene Surfaces from Self-Consistent Field Theory" Polymers 13, no. 8: 1197. https://doi.org/10.3390/polym13081197
APA StyleSgouros, A. P., Revelas, C. J., Lakkas, A. T., & Theodorou, D. N. (2021). Potential of Mean Force between Bare or Grafted Silica/Polystyrene Surfaces from Self-Consistent Field Theory. Polymers, 13(8), 1197. https://doi.org/10.3390/polym13081197