
Thermal Properties of Plasticized Cellulose Acetate and Its β-Relaxation Phenomenon



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Sample Preparation
2.3. Characterization and Sample Preparation
2.3.1. Differential Scanning Calorimetry
2.3.2. Dynamic Mechanical Thermal Analysis
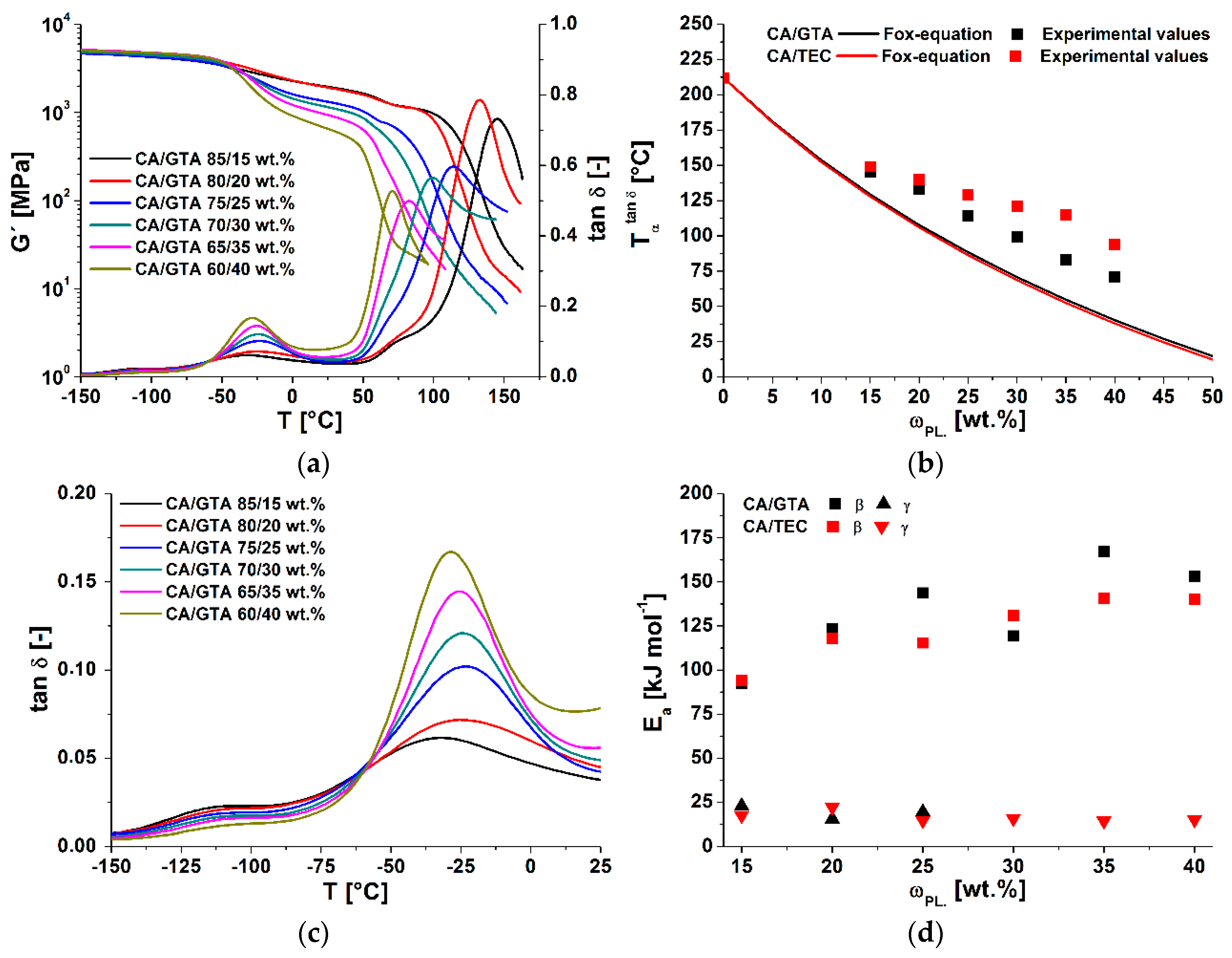
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

References
- Guo, J.-H. Effects of Plasticizers on Water Permeation and Mechanical Properties of Cellulose Acetate: Antiplasticization in Slightly Plasticized Polymer Film. Drug Dev. Ind. Pharm. 1993, 19, 1541–1555. [Google Scholar] [CrossRef]
- Lee, S.-H.; Shiraishi, N. Plasticization of cellulose diacetate by reaction with maleic anhydride, glycerol, and citrate esters during melt processing. J. Appl. Polym. Sci. 2001, 81, 243–250. [Google Scholar] [CrossRef]
- Fridman, O.A.; Sorokina, A.V. Criteria of efficiency of cellulose acetate plasticization. Polym. Sci. Ser. B 2006, 48, 233–236. [Google Scholar] [CrossRef] [Green Version]
- Charvet, A.; Vergelati, C.; Long, D.R. Mechanical and ultimate properties of injection molded cellulose acetate/plasticizer materials. Carbohydr. Polym. 2019, 204, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Bao, C.Y.; Long, D.R.; Vergelati, C. Miscibility and dynamical properties of cellulose acetate/plasticizer systems. Carbohydr. Polym. 2015, 116, 95–102. [Google Scholar] [CrossRef]
- Ghiya, V.P.; Dave, V.; Gross, R.A.; Mccarthy, S.P. Biodegradability of Cellulose Acetate Plasticized with Citrate Esters. J. Macromol. Sci. Part A 1996, 33, 627–638. [Google Scholar] [CrossRef]
- Mohanty, A.K.; Wibowo, A.; Misra, M.; Drzal, L.T. Development of renewable resource-based cellulose acetate bioplastic: Effect of process engineering on the performance of cellulosic plastics. Polym. Eng. Sci. 2003, 43, 1151–1161. [Google Scholar] [CrossRef]
- Wibowo, A.C.; Misra, M.; Park, H.-M.; Drzal, L.T.; Schalek, R.; Mohanty, A.K. Biodegradable nanocomposites from cellulose acetate: Mechanical, morphological, and thermal properties. Compos. Part A Appl. Sci. Manuf. 2006, 37, 1428–1433. [Google Scholar] [CrossRef]
- Kamide, K.; Saito, M. Thermal Analysis of Cellulose Acetate Solids with Total Degrees of Substitution of 0.49, 1.75, 2.46, and 2.92. Polym. J. 1985, 17, 919–928. [Google Scholar] [CrossRef] [Green Version]
- Fox, T.G.; Flory, P.J. Second-Order Transition Temperatures and Related Properties of Polystyrene. I. Influence of Molecular Weight. J. Appl. Phys. 1950, 21, 581–591. [Google Scholar] [CrossRef]
- Kelley, F.N.; Bueche, F. Viscosity and glass temperature relations for polymer-diluent systems. J. Polym. Sci. 1961, 50, 549–556. [Google Scholar] [CrossRef]
- Couchman, P.R.; Karasz, F.E. A Classical Thermodynamic Discussion of the Effect of Composition on Glass-Transition Temperatures. Macromolecules 1978, 11, 117–119. [Google Scholar] [CrossRef]
- Einfeldt, J.; Meißner, D.; Kwasniewski, A. Polymerdynamics of cellulose and other polysaccharides in solid state-secondary dielectric relaxation processes. Prog. Polym. Sci. 2001, 26, 1419–1472. [Google Scholar] [CrossRef]
- Scandola, M.; Ceccorulli, G. Viscoelastic properties of cellulose derivatives: 2. Effect of diethylphthalate on the dynamic mechanical relaxations of cellulose acetate. Polymer 1985, 26, 1958–1962. [Google Scholar] [CrossRef]
- Montès, H.; Mazeau, K.; Cavaillé, J.Y. Secondary Mechanical Relaxations in Amorphous Cellulose. Macromolecules 1997, 30, 6977–6984. [Google Scholar] [CrossRef]
- Saad, G.R.; Furuhata, K.-I. Effect of Substituents on Dielectric β-Relaxation in Cellulose. Polym. Int. 1997, 42, 356–362. [Google Scholar] [CrossRef]
- Zhbankov, R.G. Infrared spectra of polycarbohydrates and their model systems. J. Polym. Sci. C Polym. Symp. 1967, 16, 4629–4643. [Google Scholar] [CrossRef]
- Crofton, D.J.; Pethrick, R.A. Dielectric studies of cellulose and its derivatives: 2. Effects of pressure and temperature on relaxation behaviour. Polymer 1982, 23, 1609–1614. [Google Scholar] [CrossRef]
- Crofton, D.J.; Pethrick, R.A. Dielectric studies of proton migration and relaxation in wet cellulose and its derivatives. Polymer 1981, 22, 1048–1053. [Google Scholar] [CrossRef]
- Montès, H.; Cavaillé, J.Y. Secondary dielectric relaxations in dried amorphous cellulose and dextran. Polymer 1999, 40, 2649–2657. [Google Scholar] [CrossRef]
- Scandola, M.; Ceccorulli, G.; Pizzoli, M. Molecular motions of polysaccharides in the solid state: Dextran, pullulan and amylose. Int. J. Biol. Macromol. 1991, 13, 254–260. [Google Scholar] [CrossRef]
- Veeravazhuthi, V.; Narayandass, S.K.; Mangalaraj, D. Dielectric behaviour of pure and nickel-doped cellulose acetate films. Polym. Int. 1998, 45, 383–388. [Google Scholar] [CrossRef]
- Butler, M.F.; Cameron, R.E. A study of the molecular relaxations in solid starch using dielectric spectroscopy. Polymer 2000, 41, 2249–2263. [Google Scholar] [CrossRef]
- McBrierty, V.J.; Keely, C.M.; Coyle, F.M.; Xu, H.; Vij, J.K. Hydration and plasticization effects in cellulose acetate: Molecular motion and relaxation. Faraday Disc. 1996, 103, 255. [Google Scholar] [CrossRef]
- Bizot, H.; Le Bail, P.; Leroux, B.; Davy, J.; Roger, P.; Buleon, A. Calorimetric evaluation of the glass transition in hydrated, linear and branched polyanhydroglucose compounds. Carbohydr. Polym. 1997, 32, 33–50. [Google Scholar] [CrossRef]
- Gordon, M.; Taylor, J.S. Ideal copolymers and the second-order transitions of synthetic rubbers. i. non-crystalline copolymers. J. Appl. Chem. 1952, 2, 493–500. [Google Scholar] [CrossRef]
- Simha, R.; Boyer, R.F. On a General Relation Involving the Glass Temperature and Coefficients of Expansion of Polymers. J. Chem. Phys. 1962, 37, 1003–1007. [Google Scholar] [CrossRef]
- Kalichevsky, M.T.; Jaroszkiewicz, E.M.; Blanshard, J.M.V. A study of the glass transition of amylopectin—Sugar mixtures. Polymer 1993, 34, 346–358. [Google Scholar] [CrossRef]
- Lourdin, D.; Ring, S.G.; Colonna, P. Study of plasticizer–oligomer and plasticizer–polymer interactions by dielectric analysis: Maltose–glycerol and amylose–glycerol–water systems. Carbohydr. Res. 1998, 306, 551–558. [Google Scholar] [CrossRef]
- Bottenbruch, L.; Binsack, R.; Becker, G.W.; Braun, D. (Eds.) Kunststoff-Handbuch; 1. Aufl.; Hanser: München, Germany, 1993; ISBN 3446163689. [Google Scholar]
- Zugenmaier, P. 4. Characteristics of cellulose acetates—4.1 Characterization and physical properties of cellulose acetates. Macromol. Symp. 2004, 208, 81–166. [Google Scholar] [CrossRef]
- Mandelkern, L.; Flory, P.J. Melting and Glassy State Transitions in Cellulose Esters and their Mixtures with Diluents 1,2. J. Am. Chem. Soc. 1951, 73, 3206–3212. [Google Scholar] [CrossRef]
- Stefan, Z. Untersuchungen zur Weichmachung und Verschäumung von Thermoplastischen Celluloseestern. Ph.D. Thesis, Martin-Luther-Universität Halle-Wittenberg, Halle, Germany, 2015. [Google Scholar]
- Stickney, P.B.; Cheyney, L.E. Plasticizers for rubbers and resins. J. Polym. Sci. 1948, 3, 231–245. [Google Scholar] [CrossRef]
- Bao, C. Cellulose Acetate/Plasticizers: Structure, Morphology and Dynamics. Ph.D. Thesis, Universite Claude Bernard Lyon, Lyon, France, 2015. [Google Scholar]
- Scandola, M.; Ceccorulli, G. Viscoelastic properties of cellulose derivatives: 1. Cellulose acetate. Polymer 1985, 26, 1953–1957. [Google Scholar] [CrossRef]
- Guo, J.-H. An Investigation Into the Formation of Plasticizer Channels in Plasticized Polymer Films. Drug Dev. Ind. Pharm. 1994, 20, 1883–1893. [Google Scholar] [CrossRef]
- Ferry, J.D. Viscoelastic Properties of Polymers, 3rd ed.; John Wiley & Sons: New York, NY, USA; Chichester, UK; Brisbane, Australia; Toronto, ON, Canada; Singapore, 1980; ISBN 978-0-471-04894-7. [Google Scholar]
- Seymour, R.W.; Weinhold, S.; Haynes, S.K. Mechanical and dielectric relaxation in cellulose esters. J. Macromol. Sci. Part B 1979, 16, 337–353. [Google Scholar] [CrossRef]
- Gloor, W.E.; Gilbert, C.B. Plasticizers for Cellulose Organic Ester Plastics. Ind. Eng. Chem. 1941, 33, 597–601. [Google Scholar] [CrossRef]
- Sousa, M.; Brás, A.R.; Veiga, H.I.M.; Ferreira, F.C.; de Pinho, M.N.; Correia, N.T.; Dionísio, M. Dynamical characterization of a cellulose acetate polysaccharide. J. Phys. Chem. B 2010, 114, 10939–10953. [Google Scholar] [CrossRef]
- Montes, H.; Cavaillé, J.Y.; Mazeau, K. Secondary relaxations in amorphous cellulose. J. Non-Cryst. Solids 1994, 172–174, 990–995. [Google Scholar] [CrossRef]
- Jafarpour, G.; Dantras, E.; Boudet, A.; Lacabanne, C. Study of dielectric relaxations in cellulose by combined DDS and TSC. J. Non-Cryst. Solids 2007, 353, 4108–4115. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasticizer | Acidity | APHA | NTU | n | ||||
|---|---|---|---|---|---|---|---|---|
| [g cm−3] | [%] | [-] | [-] | [-] | [°C] | [°C] | ||
| GTA | 1.157–1.159 1 | 0.045 | 21–24 1 | 15 | - | 1.4307–1.4319 1 | −78 | 258–259 |
| TEC | 1.135–1.139 2 | 0.02 | 35.2 2 | 30 | 2 | 1.439–1.441 2 | −45.5 | 127 3 |
| Compound | CA/PL.-Ratio | |||||||
|---|---|---|---|---|---|---|---|---|
| [wt%] | [°C] | [°C] | [°C] | [J g−1 K−1] | [°C] | [°C] | [°C] | |
| CA | 100 | 191.4 | 196.6 | 201.6 | 0.233 | - | - | - |
| CA/GTA | 85/15 | 109.6 | 122.8 | 137.1 | 0.143 | 120.2 | 112.0 | 98.4 |
| 80/20 | 108.1 | 130.7 | 140.3 | 0.072 | 100.0 | 90.9 | 76.1 | |
| 75/25 | 86.3 | 104.1 | 122.6 | 0.150 | 81.8 | 72.2 | 57.1 | |
| 70/30 | 87.4 | 102.4 | 115.8 | 0.107 | 65.2 | 55.5 | 40.7 | |
| 65/35 | 63.4 | 85.7 | 92.6 | 0.063 | 50.1 | 40.7 | 26.4 | |
| 60/40 | 73.7 | 84.7 | 93.4 | 0.114 | 36.4 | 27.3 | 13.8 | |
| GTA 1 | 100 | −70.2 | −68.4 | −66.3 | 0.777 | - | - | - |
| CA/TEC | 85/15 | 129.5 | 142.4 | 153.4 | 0.157 | 118.8 | 109.5 | 88.9 |
| 80/20 | 111.2 | 124.0 | 135.1 | 0.141 | 98.3 | 87.9 | 65.9 | |
| 75/25 | 96.5 | 115.6 | 128.7 | 0.121 | 79.9 | 69.0 | 46.8 | |
| 70/30 | 91.4 | 108.3 | 121.1 | 0.193 | 63.2 | 52.2 | 30.5 | |
| 65/35 | 90.1 | 100.0 | 113.2 | 0.071 | 48.0 | 37.2 | 16.6 | |
| 60/40 | 69.7 | 75.8 | 111.1 | 0.151 | 34.1 | 23.8 | 4.5 | |
| TEC 1 | 100 | −74.1 | −70.9 | −67.8 | 0.890 | - | - | - |
| Compound | CA/PL.-Ratio | |||||
|---|---|---|---|---|---|---|
| [wt%] | [°C] | [°C] | [°C] | [kJ mol−1] | [kJ mol−1] | |
| CA/GTA | 85/15 | 145 | −32 | −104 | 92 | 23 |
| 80/20 | 133 | −25 | −102 | 123 | 15 | |
| 75/25 | 114 | −23 | −101 | 144 | 19 | |
| 70/30 | 99 | −24 | −101 | 119 | - | |
| 65/35 | 83 | −26 | −102 | 167 | - | |
| 60/40 | 71 | −29 | - | 153 | - | |
| CA/TEC | 85/15 | 149 | −30 | −110 | 94 | 17 |
| 80/20 | 140 | −24 | −104 | 118 | 22 | |
| 75/25 | 129 | −21 | −112 | 115 | 15 | |
| 70/30 | 121 | −20 | −109 | 131 | 16 | |
| 65/35 | 115 | −20 | −110 | 141 | 15 | |
| 60/40 | 94 | −20 | −106 | 140 | 15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erdmann, R.; Kabasci, S.; Heim, H.-P. Thermal Properties of Plasticized Cellulose Acetate and Its β-Relaxation Phenomenon. Polymers 2021, 13, 1356. https://doi.org/10.3390/polym13091356
Erdmann R, Kabasci S, Heim H-P. Thermal Properties of Plasticized Cellulose Acetate and Its β-Relaxation Phenomenon. Polymers. 2021; 13(9):1356. https://doi.org/10.3390/polym13091356
Chicago/Turabian StyleErdmann, Rafael, Stephan Kabasci, and Hans-Peter Heim. 2021. "Thermal Properties of Plasticized Cellulose Acetate and Its β-Relaxation Phenomenon" Polymers 13, no. 9: 1356. https://doi.org/10.3390/polym13091356
APA StyleErdmann, R., Kabasci, S., & Heim, H. -P. (2021). Thermal Properties of Plasticized Cellulose Acetate and Its β-Relaxation Phenomenon. Polymers, 13(9), 1356. https://doi.org/10.3390/polym13091356