
Anion Exchange Affinity-Based Controllable Surface Imprinting Synthesis of Ultrathin Imprinted Films for Protein Recognition

Abstract
:1. Introduction
2. Experimental
2.1. Materials
2.2. Characterization
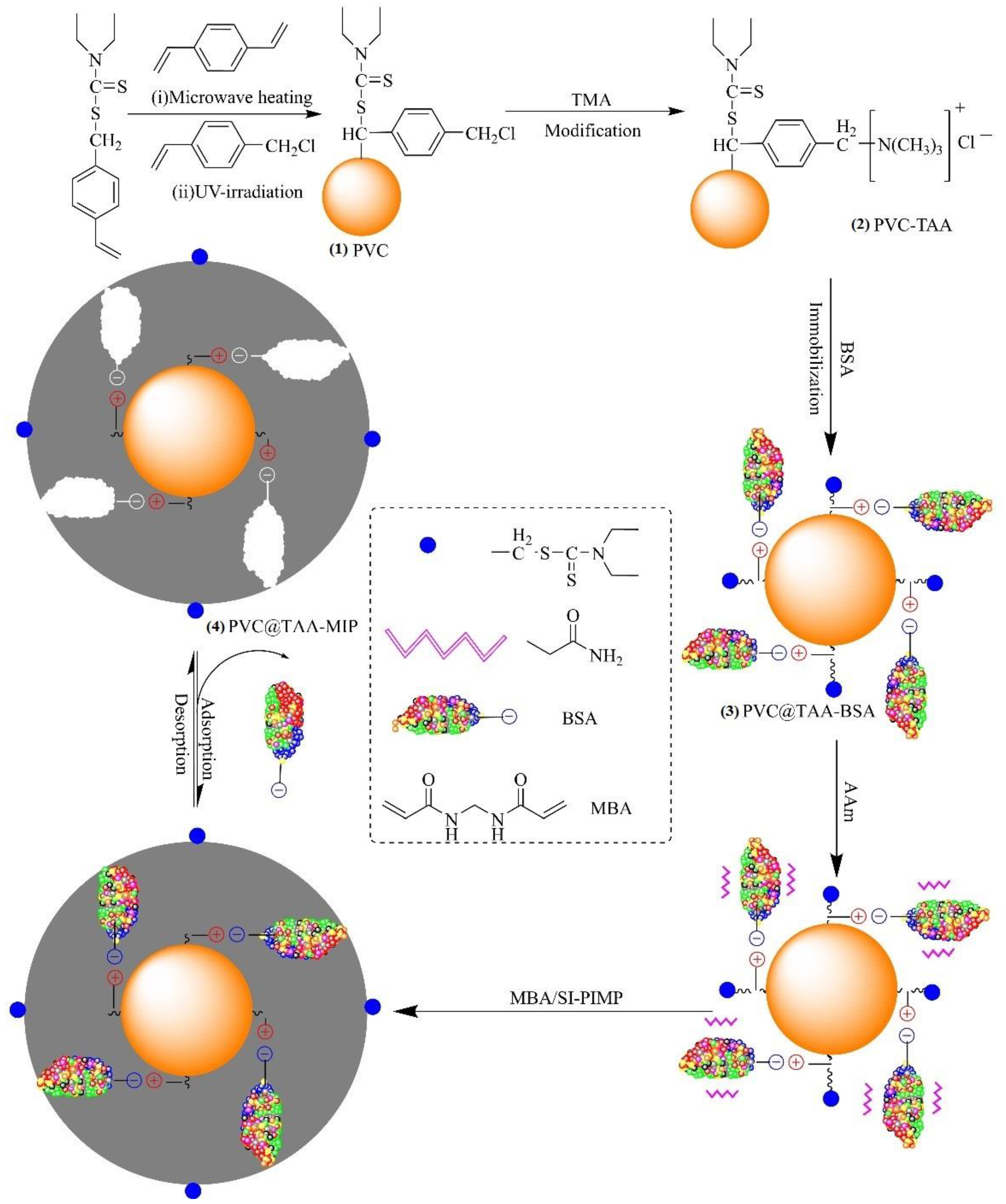
2.3. Preparation of Substrate Nanoparticles
2.4. Immobilization of Template Protein
2.5. Synthesis of BSA-Imprinted Films
2.6. Removal of Template Protein
2.7. Binding Experiments
3. Results and Discussion
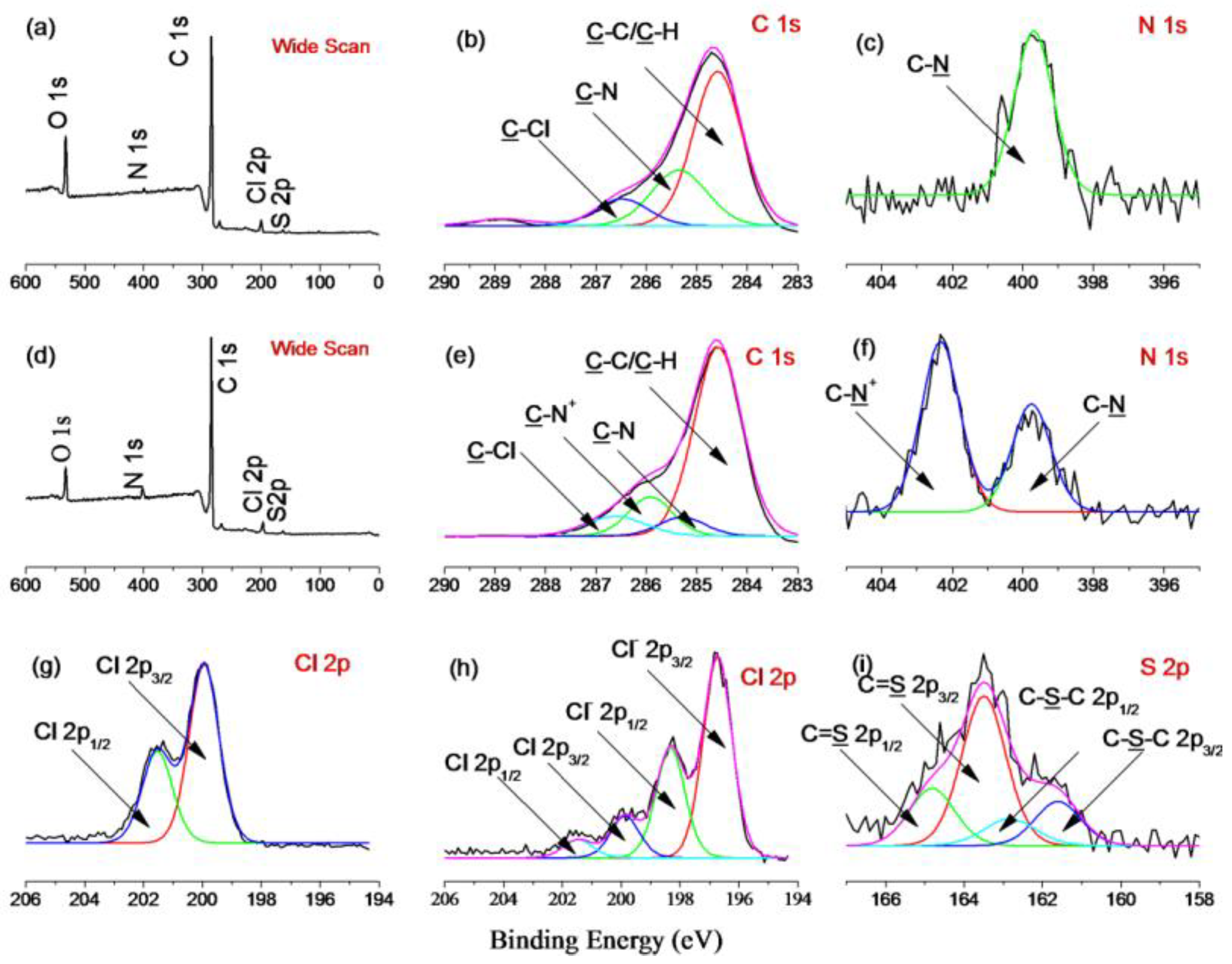
3.1. Surface Modification of Substrate Nanoparticles
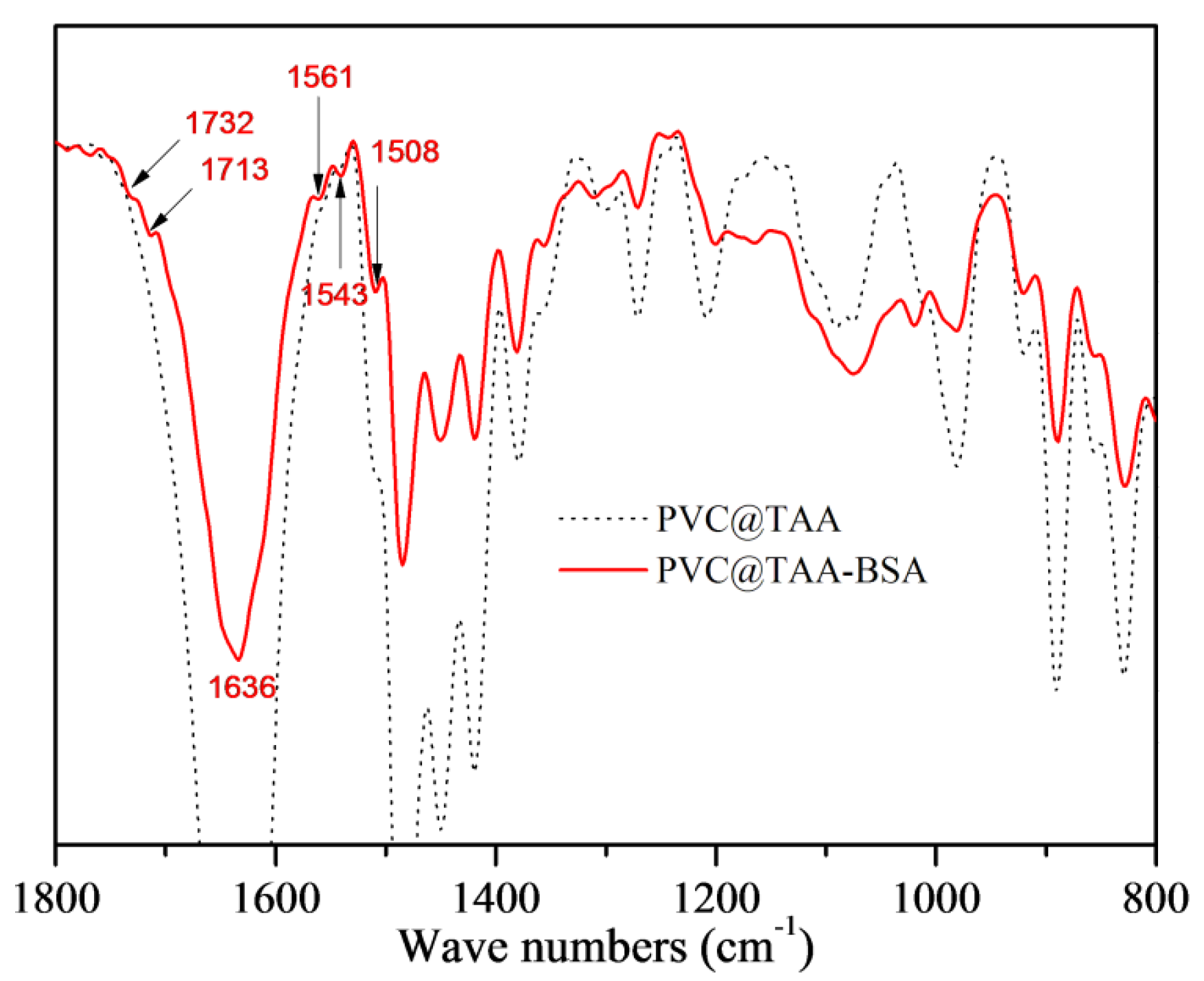
3.2. Surface Immobilization of Template Proteins
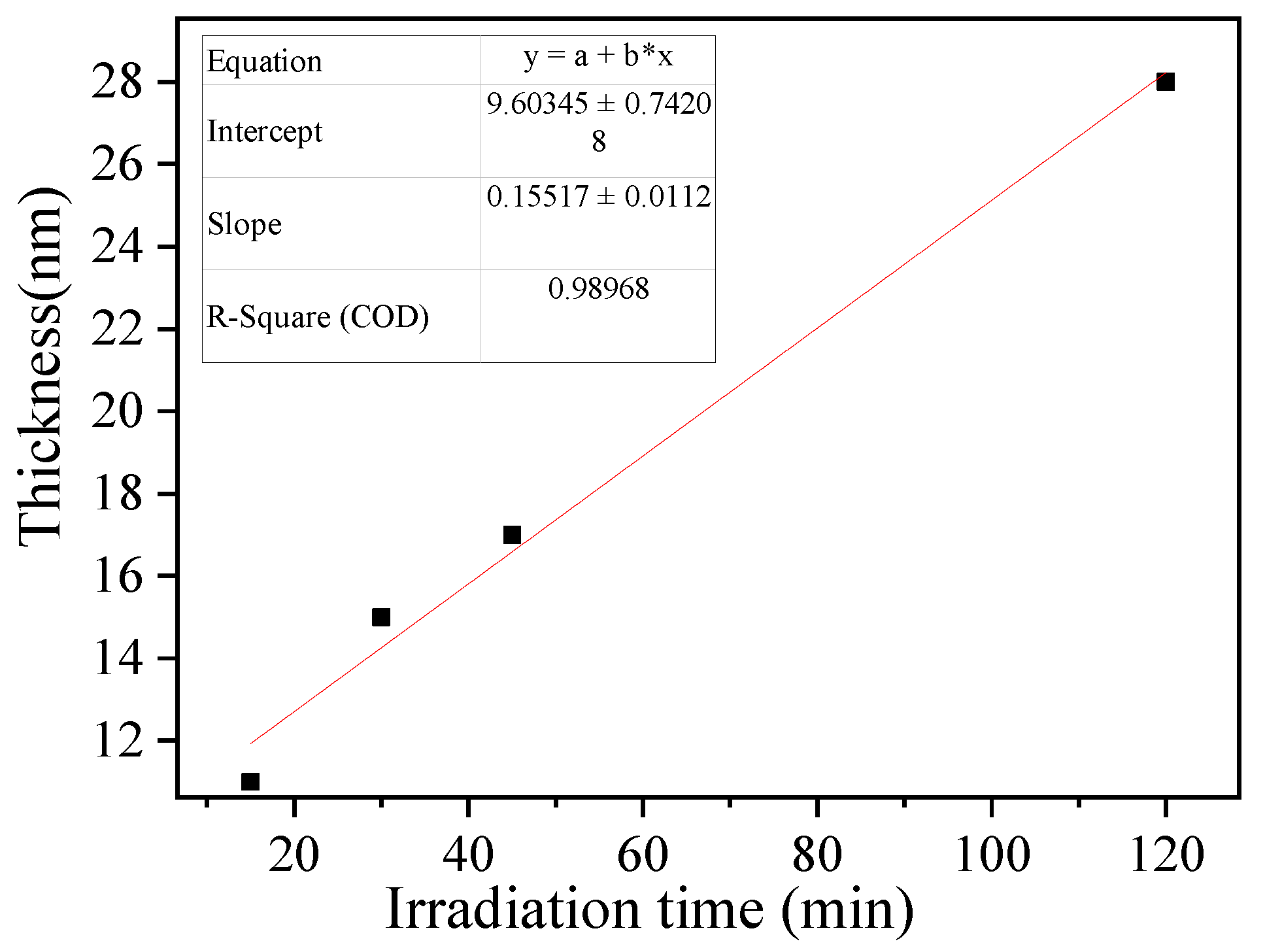
3.3. Synthesis of BSA-Imprinted Thin Films
3.4. Elution of Template Proteins
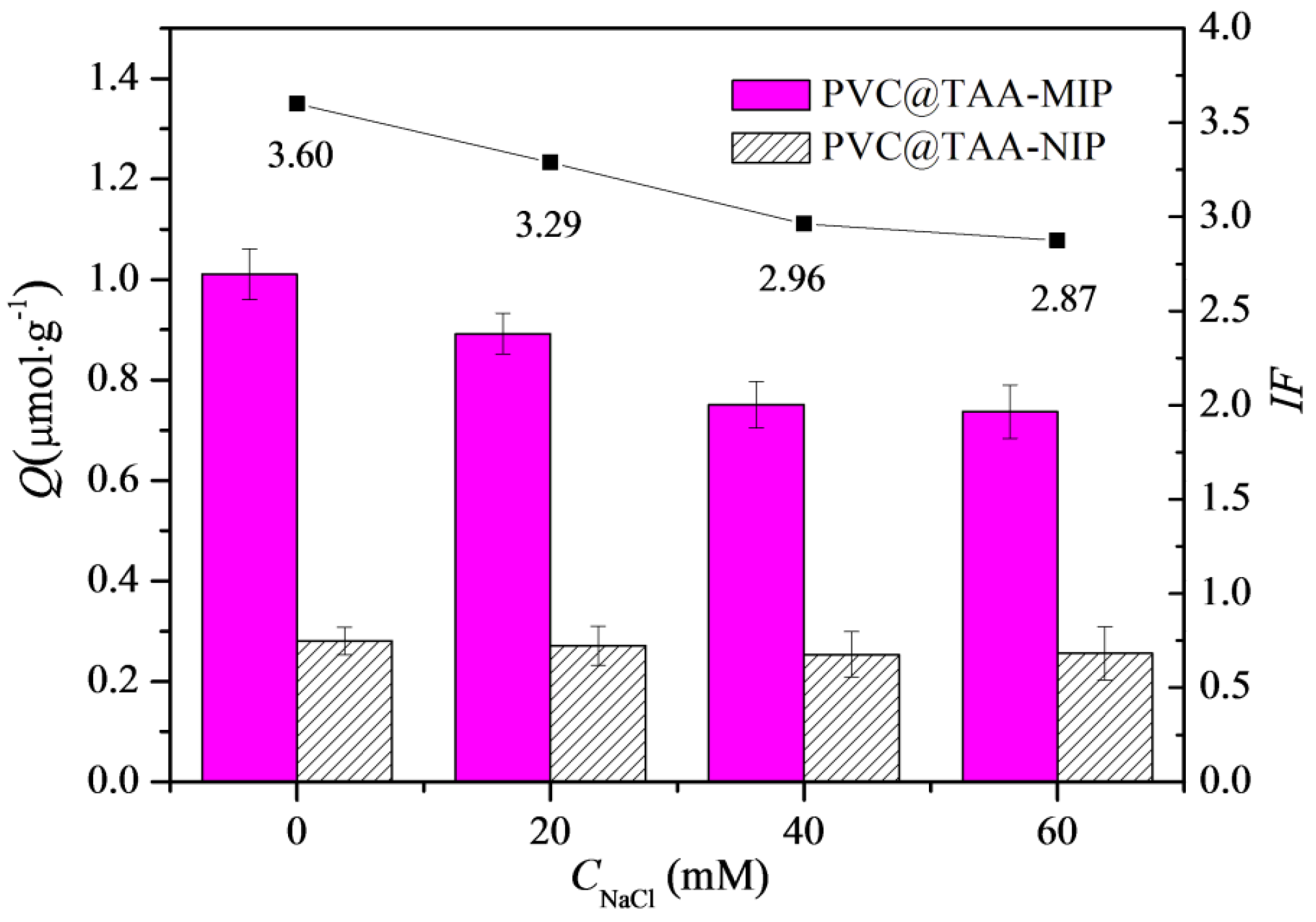
3.5. Influence of NaCl Concentration on Binding Properties
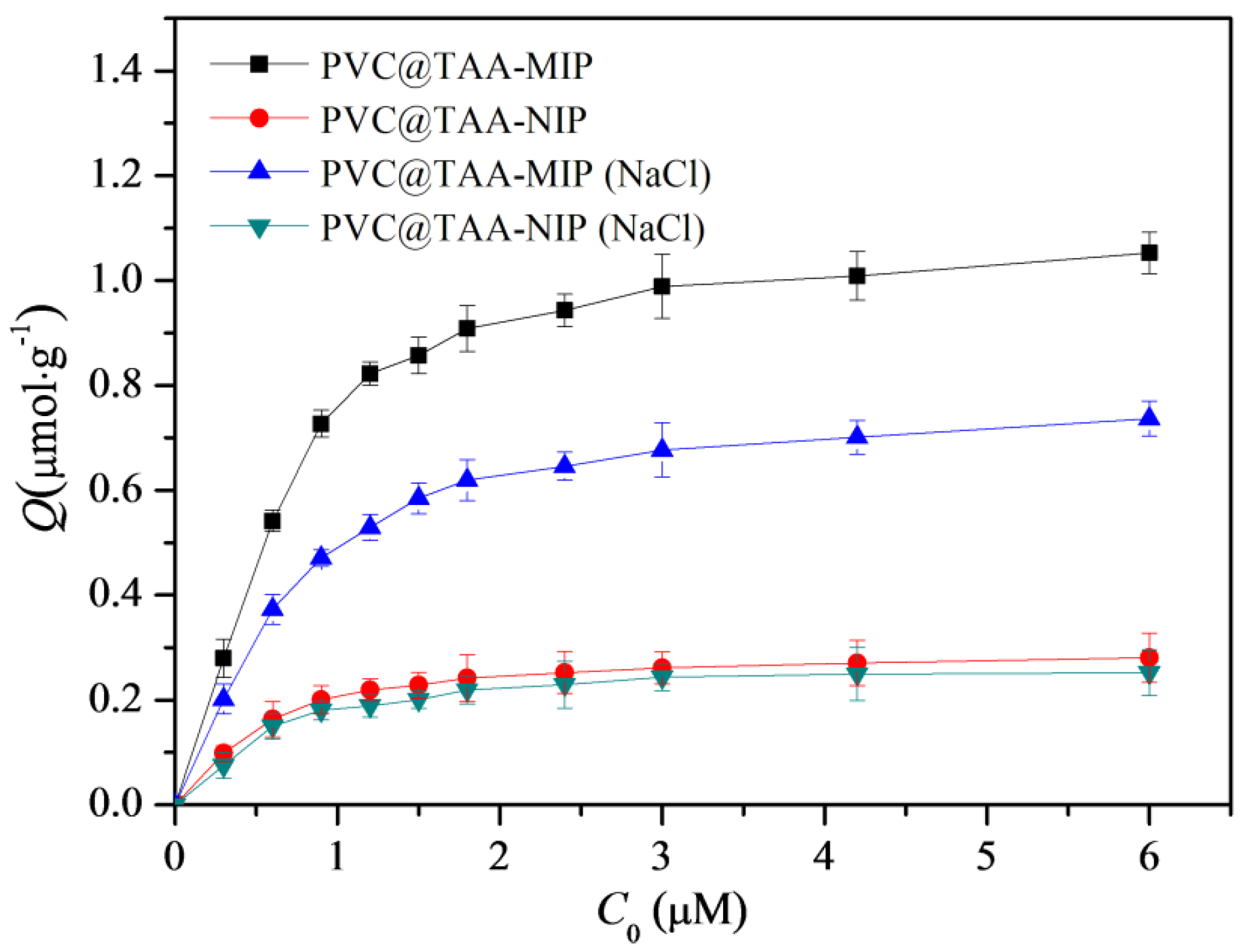
3.6. Binding Isotherms
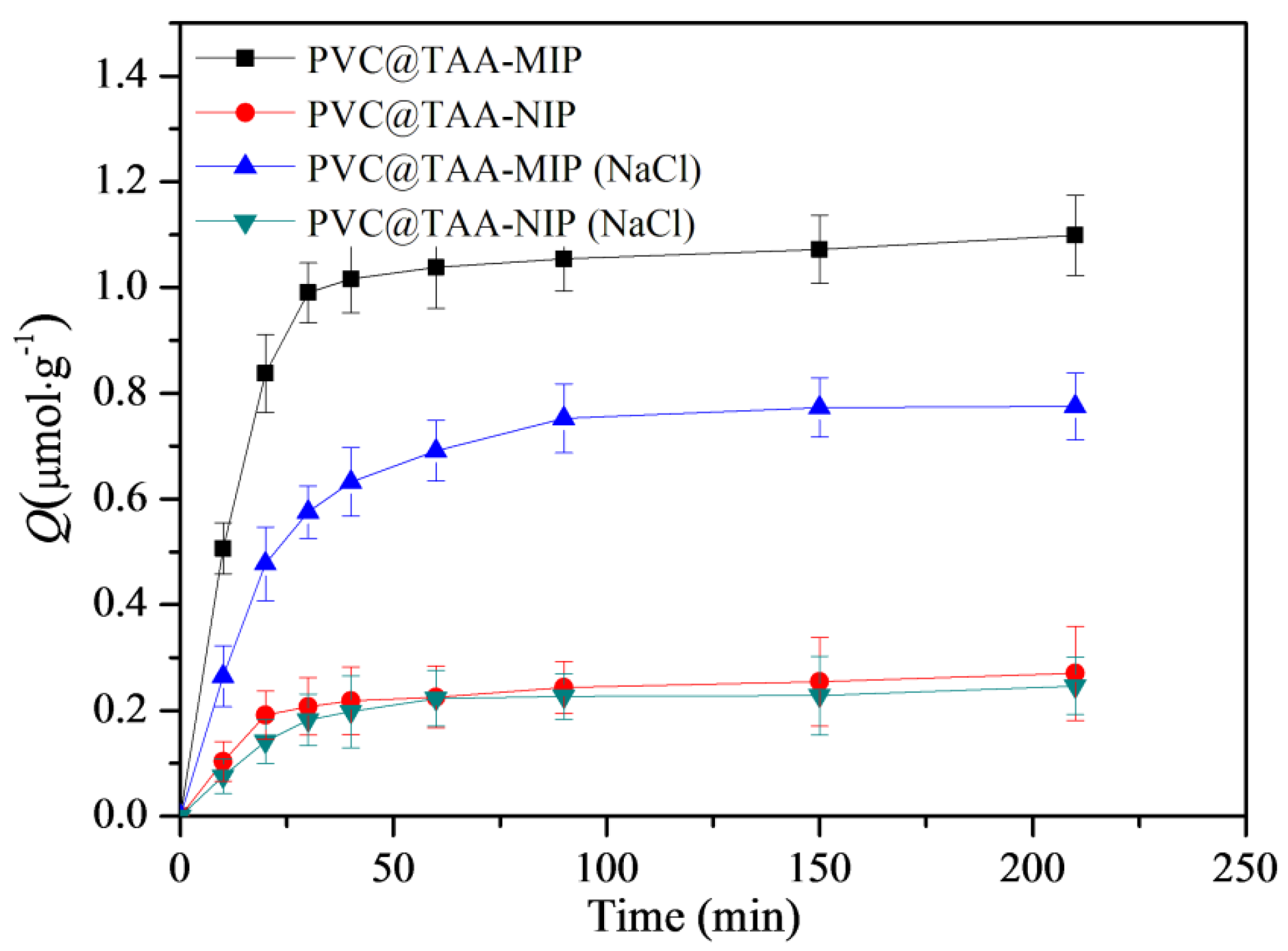
3.7. Binding Kinetics
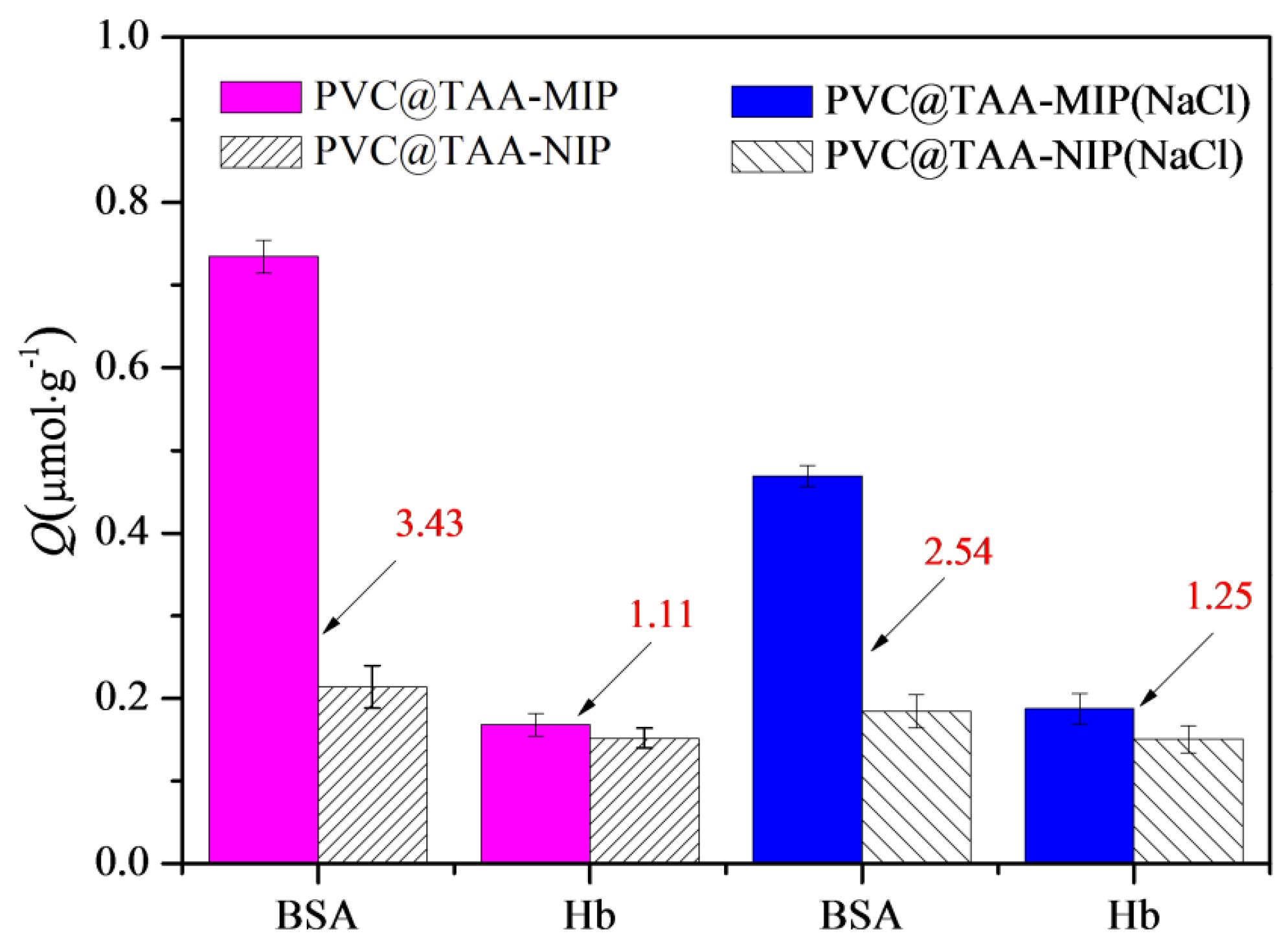
3.8. Binding Specificity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Babine, R.E.; Bender, S.L. Molecular recognition of protein-ligand complexes: Applications to drug design. Chem. Rev. 1997, 97, 1359–1472. [Google Scholar] [CrossRef] [PubMed]
- Rycke, E.D.; Leman, O.; Dubruel, P.; Hedström, M.; Völker, M.; Beloglazova, N.; Völker, M.; Beloglazova, N.; De Saeger, S. Novel multiplex capacitive sensor based on molecularly imprinted polymers: A promising tool for tracing specific amphetamine synthesis markers in sewage water. Biosens. Bioelectron. 2021, 178, 113006. [Google Scholar] [CrossRef] [PubMed]
- Mahani, M.; Mahmoudi, F.; Fassihi, J.; Hasani, Z.; Divsar, F. Carbon dots-embedded N-acetylneuraminic acid and glucuronic acid-imprinted polymers for targeting and imaging of cancer cells. Microchim. Acta 2021, 188, 224. [Google Scholar] [CrossRef]
- Li, F.; Li, X.; Su, J.; Li, Y.; Zhang, Y. Hydrophilic molecularly imprinted polymers functionalized magnetic carbon nanotubes for selective extraction of cyclic adenosine monophosphate from winter jujube. J. Sep. Sci. 2021, 44, 2131–2142. [Google Scholar] [CrossRef]
- Ma, J.; Yan, M.; Feng, G.; Ying, Y.; EI-Aty, A. An overview on molecular imprinted polymers combined with surface-enhanced Raman spectroscopy chemical sensors toward analytical applications. Talanta 2021, 225, 122031. [Google Scholar] [CrossRef]
- Alexander, C.; Davidson, L.; Hayes, W. Imprinted polymers: Artificial molecular recognition materials with applications in synthesis and catalysis. Tetrahedron 2003, 59, 2025–2057. [Google Scholar] [CrossRef]
- Zhao, X.; Mai, Y.; Chen, D.; Zhang, M.; Hu, H. Selective enrichment of clenbuterol onto molecularly imprinted polymer microspheres with tailormade structure and oxygen functionalities. Polymers 2019, 11, 1635. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, M.V.; Clay, O.; Moazami, M.P.; Watts, J.K.; Turner, N.W. Hybrid aptamer-molecularly imprinted polymer (aptaMIP) nanoparticles from protein recognition—A trypsin model. Macromol. Biosci. 2021, 21, 2100002. [Google Scholar] [CrossRef]
- Zhang, H.; Jiang, J.; Zhang, H.; Zhang, Y.; Sun, P. Efficient synthesis of molecularly imprinted polymers with enzyme inhibition potency by the controlled surface imprinting approach. ACS Macro Lett. 2013, 2, 566–570. [Google Scholar] [CrossRef]
- Nawaz, N.; Bakar, N.; Mahmud, H.; Jamaludin, N. Molecularly imprinted polymers-based DNA biosensors. Anal. Biochem. 2021, 630, 114328. [Google Scholar] [CrossRef]
- Varela-Garcia, A.; Gomez-Amoza, J.L.; Concheiro, A.; Alvarez-Lorenzo, C. Imprinted contact lenses for ocular administration of antiviral drugs. Polymers 2020, 12, 2026. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Kong, X.; Wang, X.; He, X.; Chen, L.; Zhang, Y. Preparation and characterization of uniformly sized molecularly imprinted polymers functionalized with core–shell magnetic nanoparticles for the recognition and enrichment of protein. J. Mater. Chem. 2011, 21, 17863–17871. [Google Scholar] [CrossRef]
- Nishino, H.; Huang, C.S.; Shea, K.J. Selective protein capture by epitope imprinting. Angew. Chem. Int. Ed. 2006, 45, 2392–2396. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Z.; Liu, B.; Liu, J. Incorporation of boronic acid into aptamer-based molecularly imprinted hydrogels for highly specific recognition of adenosine. ACS Appl. Bio Mater. 2020, 3, 2568–2576. [Google Scholar] [CrossRef]
- Kempe, M.; Glad, M.; Mosbach, K. An approach towards surface imprinting using the enzyme ribonuclease. J. Mol. Recognit. 1995, 8, 35–39. [Google Scholar] [CrossRef]
- Gao, X.; Cheng, W.; Zhang, X.; Zhou, Z.; Ding, Z.; Zhou, X.; Lu, Q.; Kaplan, D.L. Nerve growth factor-laden anisotropic silk nanofiber hydrogels to regulate neuronal/astroglial differentiation for scarless spinal cord repair. ACS Appl. Mater. Interfaces 2022, 14, 3701–3715. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, J.; Liu, X. Fluorescent molecularly imprinted nanoparticles with boronate affinity for selective glycoprotein detection. J. Mater. Chem. B 2020, 8, 6469–6480. [Google Scholar] [CrossRef]
- Awsiuk, K.; Stetsyshyn, Y.; Raczkowska, J.; Lishchynskyi, O.; Dąbczyński, P.; Kostruba, A.; Ohar, H.; Shymborska, Y.; Nastyshyn, S.; Budkowski, A. Temperature-controlled orientation of proteins on temperature-responsive grafted polymer brushes: Poly(butyl methacrylate) vs. poly(butyl acrylate): Morphology, wetting, and protein adsorption. Biomacromolecules 2019, 20, 2185–2197. [Google Scholar] [CrossRef]
- Stetsyshyn, Y.; Raczkowska, J.; Khrystyna, H.; Gajos, K.; Budkowski, A. Temperature-responsive and multi-responsive grafted polymer brushes with transitions based on critical solution temperature: Synthesis, properties, and applications. Colloid Polym. Sci. 2021, 299, 363–383. [Google Scholar] [CrossRef]
- Wei, D.; Cong, Z.; Zhao, B.; Chen, K.; Jia, P.; Yang, J.; Zhao, J. A negative correlation between water content and protein adsorption on polymer brushes. J. Mater. Chem. B 2019, 7, 2162–2168. [Google Scholar]
- Shiomi, T.; Matsui, M.; Mizukami, F.; Sakaguchi, K. A method for the molecular imprinting of hemoglobin on silica surfaces using silanes. Biomaterials 2005, 26, 5564–5571. [Google Scholar] [CrossRef] [PubMed]
- Bonini, F.; Piletsky, S.; Turner, A.P.F.; Speghini, A.; Bossi, A. Surface imprinted beads for the recognition of human serum albumin. Biosens. Bioelectron. 2007, 22, 2322–2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, G.; He, H.; Chai, Z.; Chen, H.; Kong, J.; Wang, Y.; Jiang, Y. Enhanced lysozyme imprinting over nanoparticles functionalized with carboxyl groups for noncovalent template sorption. Anal. Chem. 2011, 83, 1431–1436. [Google Scholar] [CrossRef] [PubMed]
- Franssen, M.C.R.; Steunenberg, P.; Scott, E.L.; Zuilhof, H.; Sanders, J.P.M. Immobilised enzymes in biorenewables production. Chem. Soc. Rev. 2013, 42, 6491–6533. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Moo-Young, M. The kinetic and mass transfer behavior of immobilized invertase on ion-exchange resin beads. Biotechnol. Bioeng. 1973, 15, 47–67. [Google Scholar] [CrossRef]
- Otsu, T.; Yamashita, K.; Tsuda, K. Synthesis, reactivity, and role of 4-vinylbenzyl N,N-diethyldithiocarbamate as a monomer-iniferter in radical polymerization. Macromolecules 1986, 19, 287–290. [Google Scholar] [CrossRef]
- Pepper, K.W.; Paisley, H.M.; Young, M.A. Properties of ion-exchange resins in relation to their structure. J. Chem. Soc. 1953, 833, 4097–4105. [Google Scholar] [CrossRef]
- John, D.; Stanzel, M.; Andrieu-Runsen, A. Surface plasmons and visible light iniferter initiated polymerization for nanolocal functionalization of mesoporous separation layers. Adv. Funct. Mater. 2021, 31, 2009732. [Google Scholar] [CrossRef]
- Qian, L.; Hu, X.; Guan, P.; Gao, B.; Li, J.; Wang, C.; Tang, Y. Preparation of bovine serum albumin imprinting sensitive hydrogels using ionic liquid as co-monomer and stabilizer. Talanta 2014, 121, 56–64. [Google Scholar] [CrossRef]
- Bi, W.; Tian, M.; Row, K.H. Separation of phenolic acids from natural plant extracts using molecularly imprinted anion-exchange polymer confined ionic liquids. J. Chromatogr. A 2012, 1232, 37–42. [Google Scholar] [CrossRef]
- Bi, W.; Tian, M.; Row, K.H. Evaluation of molecularly imprinted anion-functionalized poly(ionic liquid)s by multi-phase dispersive extraction of flavonoids from plant. J. Chromatogr. B 2013, 913–914, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Tsou, H.S.; Graham, E.E. Prediction of adsorption and desorption of protein on dextran based ion-exchange resin. AICHE J. 1985, 31, 1959–1966. [Google Scholar] [CrossRef]
- Yu, Y.; Sun, Y. Macroporous poly(glycidyl methacrylate-triallyl isocyanurate-divinylbenzene) matrix as an anion-exchange resin for protein adsorption. J. Chromatogr. A 1999, 855, 129–136. [Google Scholar] [CrossRef]
- Nakayama, Y.; Matsuda, T. Surface macromolecular architectural designs using photo-graft copolymerization based on photochemistry of benzyl N,N-diethyldithiocarbamate. Macromolecules 1996, 29, 8622–8630. [Google Scholar] [CrossRef]
- Ali, A.; Mayes, A.G. Preparation of polymeric core shell and multilayer nanoparticles: Surface-initiated polymerization using in situ synthesized photoiniferters. Macromolecules 2010, 43, 837–844. [Google Scholar] [CrossRef]
- Fan, L.; Wu, P.; Zhang, J.; Gao, S.; Wang, L.; Li, M.; Sha, M.; Xie, W.; Nie, M. Synthesis and anticoagulant activity of the quaternary ammonium chitosan sulfates. Int. J. Biol. Macromol. 2012, 50, 31–37. [Google Scholar] [CrossRef]
- Olivier, G.K.; Shin, D.; Gilbert, J.B.; Monzon, L.M.A.; Frechette, J. Supramolecular ion-pair interactions to control monolayer assembly. Langmuir 2009, 25, 2159–2165. [Google Scholar] [CrossRef]
- Cheng, Z.; Zhu, X.; Shi, Z.L.; Neoh, K.G.; Kang, E.T. Polymer microspheres with permanent antibacterial surface from surface-initiated atom transfer radical polymerization. Ind. Eng. Chem. Res. 2005, 44, 7098–7104. [Google Scholar] [CrossRef]
- Hirashima, Y.; Sato, H.; Suzuki, A. ATR-FTIR spectroscopic study on hydrogen bonding of poly(N-isopropylacrylamide-co-sodium acrylate) gel. Macromolecules 2005, 38, 9280–9286. [Google Scholar] [CrossRef]
- Ataka, K.; Richter, B.; Heberle, J. Orientational control of the physiological reaction of cytochrome c oxidase tethered to a gold electrode. J. Phys. Chem. B 2006, 110, 9339–9347. [Google Scholar] [CrossRef] [Green Version]
- Wright, A.K.; Thompson, M.R. Hydrodynamic structure of bovine serum albumin determined by transient electric birefringence. Biophys. J. 1975, 15, 137–141. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Shi, W.; Zhang, W.; Zhang, X. Force spectroscopy study on poly(acrylamide) derivatives: Effects of substitutes and buffers on single-chain elasticity. Nano Lett. 2002, 2, 1169–1172. [Google Scholar] [CrossRef]
- Harmer, S.L.; Nesbitt, H.W. Stabilization of pyrite (FeS2), marcasite (FeS2), arsenopyrite (FeAsS) and loellingite (FeAs2) surfaces by polymerization and auto-redox reactions. Sur. Sci. 2004, 564, 38–52. [Google Scholar] [CrossRef]
- Shaikha, H.; Memona, N.; Bhangera, M.I.; Nizamania, S.M.; Denizlib, A. Core-shell molecularly imprinted polymer-based solid-phase microextraction fiber for ultra trace analysis of endosulfan I and II in real aqueous matrix through gas chromatography-micro electron capture detector. J. Chromatogr. A 2014, 1337, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Fu, J.; Fu, Z.; Li, Y.; Su, X.; Zou, L.; He, L.; Liu, S.; Ao, X.; Yang, Y. Preparation and characterization of magnetic molecular imprinted polymers with ionic liquid for the extraction of carbaryl in food. Anal. Bioanal. Chem. 2020, 412, 1049–1062. [Google Scholar] [CrossRef]
- Sunayama, H.; Takeuchi, T. Protein-imprinted polymer films prepared via cavity-selective multi-step post-imprinting modifications for highly selective protein recognition. Anal. Bioanal. Chem. 2021, 413, 6183–6189. [Google Scholar] [CrossRef]
- Chen, T.; Shao, M.; Xu, H.; Zhuo, S.; Liu, S.S.; Lee, T. Molecularly imprinted polymer-coated silicon nanowires for protein specific recognition and fast separation. J. Mater. Chem. 2012, 22, 3990–3996. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Q (µmol·g−1) | α | β | |
|---|---|---|---|---|
| PVC@TAA-MIP | PVC@TAA-NIP | |||
| BSA | 1.068 ± 0.0235 | 0.2830 ± 0.0184 | 3.77 | — |
| OVA | 0.3636 ± 0.0166 | 0.2010 ± 0.0143 | 1.81 | 2.08 |
| Hb | 0.2039 ± 0.0189 | 0.2295 ± 0.0125 | 0.89 | 4.24 |
| Lys | 0.0776 ± 0.0138 | 0.0919 ± 0.0116 | 0.84 | 4.49 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, R.; Yu, X.; Liu, M.; Hu, X.; Zhu, S. Anion Exchange Affinity-Based Controllable Surface Imprinting Synthesis of Ultrathin Imprinted Films for Protein Recognition. Polymers 2022, 14, 2011. https://doi.org/10.3390/polym14102011
Song R, Yu X, Liu M, Hu X, Zhu S. Anion Exchange Affinity-Based Controllable Surface Imprinting Synthesis of Ultrathin Imprinted Films for Protein Recognition. Polymers. 2022; 14(10):2011. https://doi.org/10.3390/polym14102011
Chicago/Turabian StyleSong, Renyuan, Xiaofeng Yu, Muxin Liu, Xiaoling Hu, and Shengqing Zhu. 2022. "Anion Exchange Affinity-Based Controllable Surface Imprinting Synthesis of Ultrathin Imprinted Films for Protein Recognition" Polymers 14, no. 10: 2011. https://doi.org/10.3390/polym14102011
APA StyleSong, R., Yu, X., Liu, M., Hu, X., & Zhu, S. (2022). Anion Exchange Affinity-Based Controllable Surface Imprinting Synthesis of Ultrathin Imprinted Films for Protein Recognition. Polymers, 14(10), 2011. https://doi.org/10.3390/polym14102011