3.1. Synthesis and Morphology of ZnDOPs
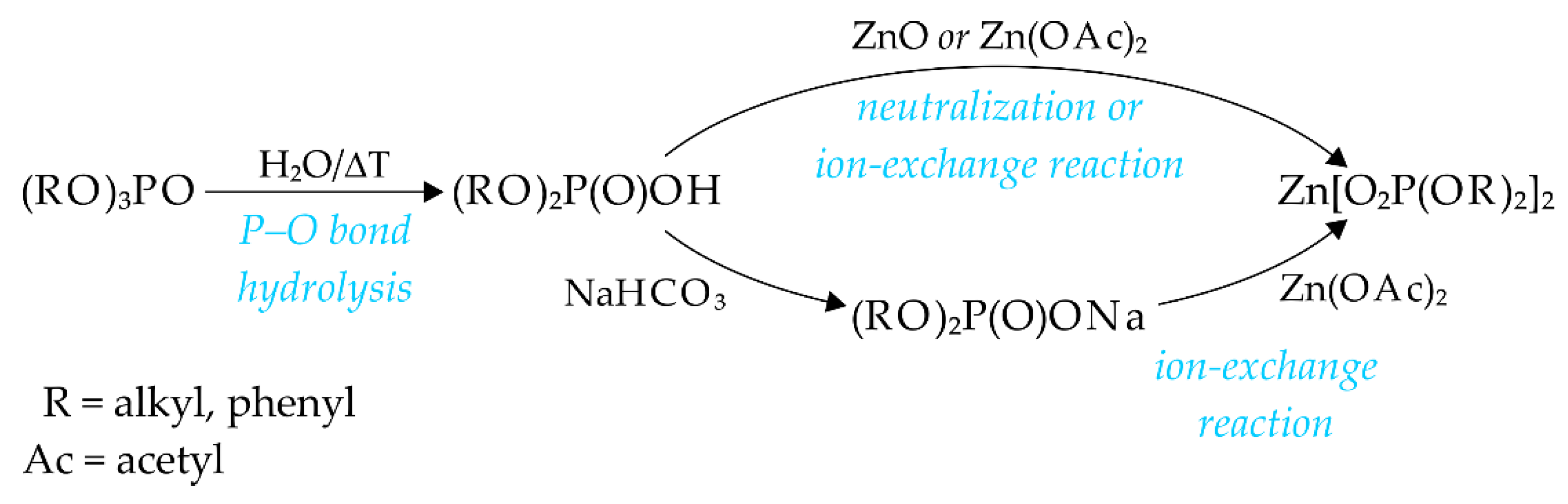
Two major synthetic routes were employed for the ZnDOPs synthesis, as shown on
Scheme 1. One of them was based on the one-pot, hydrothermal method previously described by Harrison and coworkers [
16,
18]; in our studies, we modified it by shortening the time necessary for its completion and applying milder conditions (e.g., simply refluxing ZnO with TMP or TEP in water). Under these conditions, one of the P–OR bonds in phosphoric acid triesters underwent hydrolysis yielding the respective dialkyl phosphates (strong acids), which subsequently reacted with ZnO resulting in the formation of the large amounts of ZnDOPs (58% for ZnDEP and 96% for ZnDMP) (see
Scheme 1). This hydrolytic strategy gave satisfactory results only for water-miscible triesters of H
3PO
4, albeit the yields of ZnDOPs quickly decreased when the size of organic group within the (RO)
3PO molecules increased (96% versus 58% in the case of ZnDMP and ZnDEP synthesis, respectively).
Hydrolytic reaction utilizing highly hydrophobic TBP required harsher reaction conditions (e.g., temperature of 150 °C applied for 48 h) and replacing ZnO with water-soluble ZnOAc·2H2O to produce ZnDBP. Nevertheless, the latter was obtained with a yield of ca. 34%; however, its purification from the unreacted TBP and n-butanol (liberated as a side product of hydrolysis) was cumbersome. An attempt to obtain ZnDnPP directly from TnPP using a hydrolytic method did not lead to the formation of the intended product. Thus, we had to first synthesize DnPP (via a controlled dealkylation of TnPP with CaCl2, and subsequent liberation of DnPP from its calcium salt under the action of H2SO4), and then react it with ZnO.
In the second method, ZnDOPs having long alkyl chains in their phosphate ligands, were obtained with good yields directly from sodium salt of the commercially available phosphoric acid diesters (DBP or BEHP) and zinc acetate. The chemistry behind this method is very simple and comprises two consecutive stages carried out in water at room temperature: neutralization of acidic dialkyl phosphate with sodium bicarbonate, followed by the ion-exchange reaction between the resulting water-soluble sodium diorganophosphate and zinc acetate (see
Scheme 1). Immediately after mixing the aqueous solutions of both substrates, the product precipitates and forms a fine dispersion of flocculent particles. From this dispersion ZnBEHP can be easily isolated almost quantitatively reaction yield of 95%). In comparison, the sedimentation of ZnDBP at room temperature is less efficient and the yield of isolated solid product typically does not exceed 30%; however, this can be easily changed by application of heating, since at temperatures around 55–60 °C the stability of ZnDBP dispersion is disrupted, resulting in a coalescence of the flocculated, small particles into a few, much larger agglomerates and an increase in the overall yield of ZnDBP up to 63%. It should be noted that the method described above is much more simple and environmentally benign than its other alternatives described in the literature for zinc-containing hybrid polymers, for example, the process based on a cation-exchange resin [
38]. Because of the properties of ZnDOPs, especially their solubility in different solvents (see a discussion in one of the following subsections of our manuscript), the latter method would require the use of a large amounts of polar, organic solvents, such as methanol or DMSO.
The carbon, hydrogen and zinc contents in ZnDOPs prepared by the hydrolytic or ion-exchange routes are in very good agreement with the values calculated for the polymers free of any auxiliary ligands (see
Table S1 in SM), thus clearly indicating that neither water nor any unreacted substrates are coordinated to the metallic centers. These results also confirm that the compounds investigated in the present study exhibit the same composition (e.g., the molar ratio of diorganophosphate ligands to zinc centers) and general formula {Zn[O
2P(OR)
2]
2}, as ZnDOPs already mentioned in the literature, containing either aliphatic [
4,
16,
18] or aromatic [
21,
33] substituents. Moreover, in the case of ZnDOPs obtained from triesters of H
3PO
4 via a hydrolytic route, they also exclude the possibility of an uncontrolled hydrolysis of phosphoester linkages leading to the formation of zinc salts containing either (ROPO
3)
2− or PO
43− ligands. A very similar conclusion can be made based on the analysis of the FTIR spectra of ZnDOPs (
Figure S1 in SM), which do not contain any absorption bands above 3000 cm
−1 or around 1650 cm
−1 resulting from the stretching or bending vibrations in the zinc-coordinated H
2O molecules, respectively [
33]. Instead, one can easily observe the bands ascribed to the C–H stretching (the 2800–3000 cm
−1 region), as well as bending and wagging modes (the 1360–1480 cm
−1 region) within the methyl, methylene or methanetriyl groups [
39,
40]. The four vibrational bands characteristic of the phosphate ligand occur in the wavenumber region of 720–1200 cm
−1: between 800–830 cm
−1 and 730–770 cm
−1 appear signals attributable to the P–O asymmetric and symmetric stretching modes within the phosphoester linkages, respectively, whereas their analogues from the Zn–O–P–O–Zn bridges are located around 1180–1195 cm
−1 and 1095–1100 cm
−1, respectively [
39,
40]. It is worth noting that the wavenumbers of the latter two vibrational bands in ZnDOPs are about 20–30 cm
−1 lower compared to their analogues characterizing AlDOPs [
15]. In addition to the absorption bands mentioned above, FTIR spectra of the aliphatic ZnDOPs also contain very strong signals ascribed to the C–O stretching modes located between 1000 and 1065 cm
−1 [
39,
40].
The as-synthesized samples of ZnDMP and ZnDEP consist of highly crystalline, large particles exhibiting a cuboid or fiber morphology, respectively (
Figure S7 in SM). Their PXRD patterns correspond very well to those simulated based on the diffraction data from single-crystal X-ray analyses carried out at room temperature and published by Harrison and coworkers [
16,
18] (
Figures S8 and S9 in SM). No evidence of any other crystalline phases can be found in these samples, indicating that our hydrolytic method of synthesis can be an interesting alternative to the hydrothermal one proposed in the literature. It should be noted, however, that some ZnDMP samples recrystallized from their aqueous solution during a slow evaporation at 70 °C also contain some additional reflections on their PXRD patterns, indicating the presence of another crystalline phase with an unknown structure. This observation prompted us to closely investigate the relationship between temperature and ZnDMP structure, the results of which will be discussed in a separate section of this manuscript.
ZnDnPP also exhibits a highly ordered crystalline structure (
Figure S10 in SM); however, the analysis of PXRD patterns obtained for ZnDBP and ZnBEHP (
Figures S11 and S12 in SM, respectively) points out that a further elongation of alkyl chains (above C3) within diorganophosphate ligand hinders the mutual ordering of ZnDOP particles into large domains having the regular internal structure: a low crystallinity of both these materials is clearly indicated by a substantial widening of their PXRD reflections, especially in the case of ZnBEHP, the X-ray diffractogram of which also shows the presence of a broad halo located at 2
θ angle of ca. 20°, which is characteristic of an amorphous phase (
Figure S12 in SM). This conclusion is further supported by SEM, since only a small fraction of fiber- or rod-like structures is observed at a high magnification for the ZnDBP sample obtained at room temperature (
Figure S13 in SM), whereas one cannot distinguish any discrete particles in the case of ZnDBP conditioned at 60 °C. The latter also applies to ZnBEHP and its samples exhibit a continuous morphology, without any distinct particles being visible (
Figure S14 in SM).
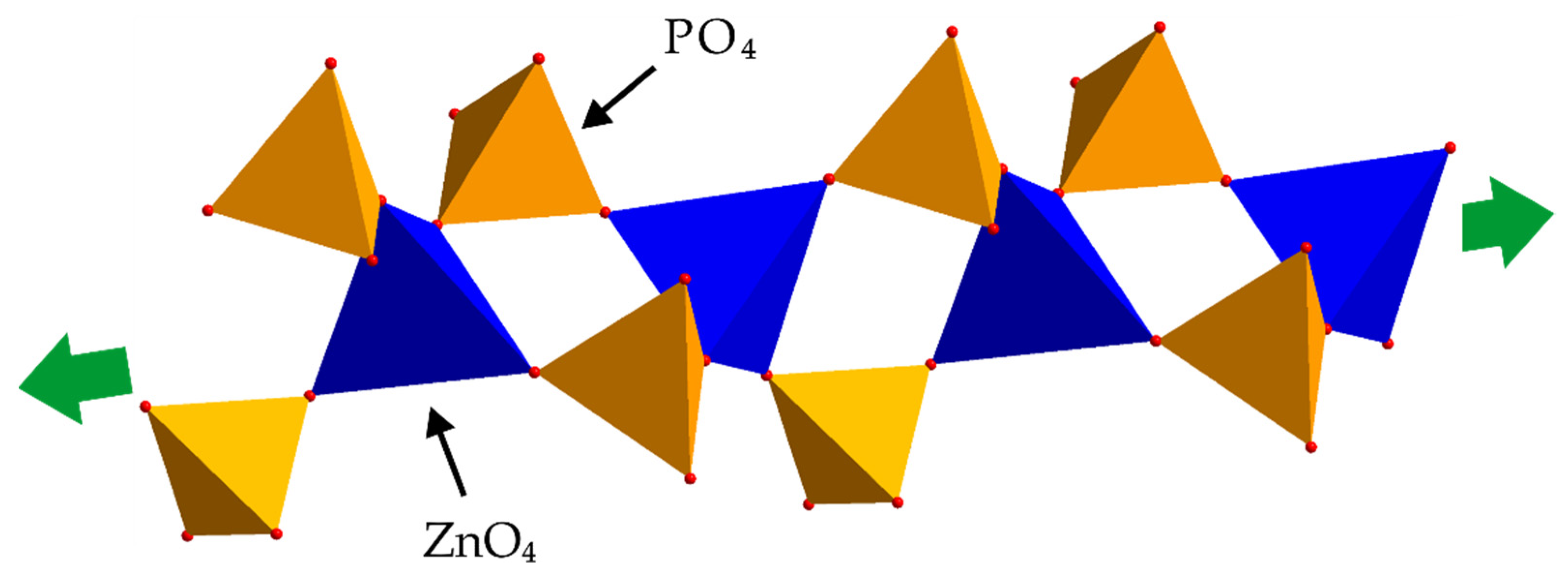
For the purpose of structural studies, we have prepared the needle-like crystals of ZnDBP by means of its recrystallization from a mixture of DMSO and methanol (1:5 vol/vol) carried out at −20 °C. This material was subjected to a single-crystal X-ray analysis (details of that procedure are included in SM). Unfortunately, the high flexibility of aliphatic chains within the (
n-BuO)
2PO
2 ligands resulting from rotations around the C–C bonds caused severe structural disorder outside the coordination polymer core. Although it did not permit full refinement and solution of that crystal structure, the connectivity pattern showing 2 + 2 bridging mode in the polymer chain is noted. Nevertheless, very good compliance between the theoretical PXRD pattern calculated based on this partially solved ZnDBP structure and the one recorded experimentally (see
Figure S11 in SM) indicates that the former may be considered a good approximation of the actual ZnDBP crystal structure, especially concerning its rigid core formed by ZnO
4 and PO
4 tetrahedra sharing their vertices. According to our proposal, the basic structural unit of ZnDBP is a polymeric chain in which tetrahedrally coordinated Zn centers are lined up on a single axis, and two bidentate diorganophosphate bridging ligands connect each pair of them. As a result, in a single ZnDBP chain, there are eight-membered Zn(O–P–O)
2Zn rings, the planes of which are arranged at an angle of ca. 90 (a spiro-type system) (
Figure 2). The average Zn···Zn distance in that structure (4.578–4.589 Å) is within the range observed for other aliphatic ZnDOPs exhibiting the same 2 + 2 metal center bridging mode (4.546 Å for ZnDEP [
16] or 4.665–4.697 Å for ZnDMP [
18]), but longer than in its aromatic analogues (4.329–4.425 Å [
21]). The PO
4 groups show distortions from the structure of a regular tetrahedron since the P–O bond lengths within the Zn(O–P–O)
2Zn rings are shorter than those responsible for bonding organic substituents, whereas the opposite effect occurs in the case of the angles between them. The O–P–O bond angles in the bridges between zinc centers (117.06–118.25°) are the largest ones and exceed the rest of them by about 5–20°. It should be noted that such features of the PO
4 tetrahedra seem to be typical for ZnDOPs regardless of the type of organic groups in their structure.
3.2. Thermogravimetric Analysis and Thermal Stability of Aliphatic ZnDOPs
The results of STA analysis show that when heated in an inert atmosphere, the samples of aliphatic ZnDOPs start to endothermally decompose at temperatures between 215 and 300 °C (as suggested by the values of the bend points on the respective TG curves,
Tb,
Table 1) and stop changing their weight at ca. 260–430 °C, depending on the type of organic substituents present within the (RO)
2PO
2 ligand (see the end points of the respective dTG peaks in
Figures S15–S19 in SM). As could be expected, the chemical bonds in most of the investigated aliphatic ZnDOPs are more prone to thermolysis than those present within aromatic ZnDOPs, although some thermally labile functional groups attached to the phenyl ring may alter that hierarchy [
21,
33]. Taking into account the values of
Tb, temperature corresponding to 2% or 5% of weight loss (
T98% or
T95%, respectively), as well as the extrapolated onset temperature of thermolysis (
Tonset), it is evident that ZnDOPs containing short (C1–C3) aliphatic chains are also less stable than their aluminum-based analogues having the same diorganophosphate ligands (AlDOPs) [
35]; however, this situation changes in the case of ZnDBP or ZnBEHP, whose pyrolysis is characterized by a higher energy barrier (initial temperature). Surprisingly, the latter compound is the most resistant to temperature among all of the investigated ZnDOPs, even when compared to ZnDMP—a derivative that can decompose only via more complex and energy-consuming processes involving homolytic cleavage of the P–OCH
3 or PO–CH
3 linkages and subsequent transformations of the liberated radicals [
35,
41]. The reason for such an unusually high thermal stability of ZnBEHP is still unclear and needs a more detailed, experimental investigation, which was out of the scope of the present study.
QMS analysis of the evolved gases (
Figures S15b–S19b in SM) reveals that the chemical reactions occurring during pyrolysis of aliphatic ZnDOPs are limited only to the P–O–C bond system, whereas Zn–O–P linkages are resistant to any scission: no QMS peaks attributed to the typical volatile oxophosphorus species (such as PO, PO
2, HOPO, or HOPO
2;
m/
z = 47, 63, 64, or 80, respectively) [
42] could be detected. Moreover, a complete lack of any analytical signals above
m/
z = 60 (70 in the case of ZnBEHP) is strong evidence that, similar to the pyrolysis of AlDOPs [
35], the thermal degradation of aliphatic ZnDOPs in inert atmosphere proceeds essentially via the C–O bond splitting reactions involving the β-elimination of olefines (e.g., ethylene,
m/
z = 28, propene,
m/
z = 41 and 42, 1-butene,
m/
z = 41 and 56) [
43], as well as a subsequent rearrangement between the newly generated POH and the remaining phosphoester groups, leading to the OH-containing species [
44] (e.g., primary alcohols giving characteristic QMS signal of
+CH
2OH ions,
m/
z = 31 [
45]). The product that indirectly indicates the occurrence of the P–OR bond scission (e.g., ether resulting from the recombination of alkyl and alkoxy radicals [
41]) can be observed only in the case of ZnDMP pyrolysis—signals derived from dimethyl ether (
m/
z = 45 and 46) [
43], or formaldehyde (
m/
z = 29 and 30) [
43] are easily distinguishable on the QMS spectrum. It is worth noting that a common feature of the aliphatic ZnDOPs pyrolysis is a simultaneous condensation of the POH groups leading to the elimination of water molecules (
m/
z = 17 and 18) [
43].
3.3. Thermal Transitions in Aliphatic ZnDOPs below 200 °C
The DSC studies reveal that ZnDOPs undergo several endothermic phase transitions on heating from −100 to 200 °C. These processes proceed without any weight loss; additionally, based on the optical observations conducted on a Gallenkamp Melting Point Apparatus, they can be categorized into two groups: solid→liquid transitions and those occurring without any change in the physical state of the sample (solid→solid transitions). Some of them are reversible and are accompanied by the exothermic peaks on the DSC cooling curves, albeit the latter usually occurs at temperatures several degrees lower than the respective endotherms.
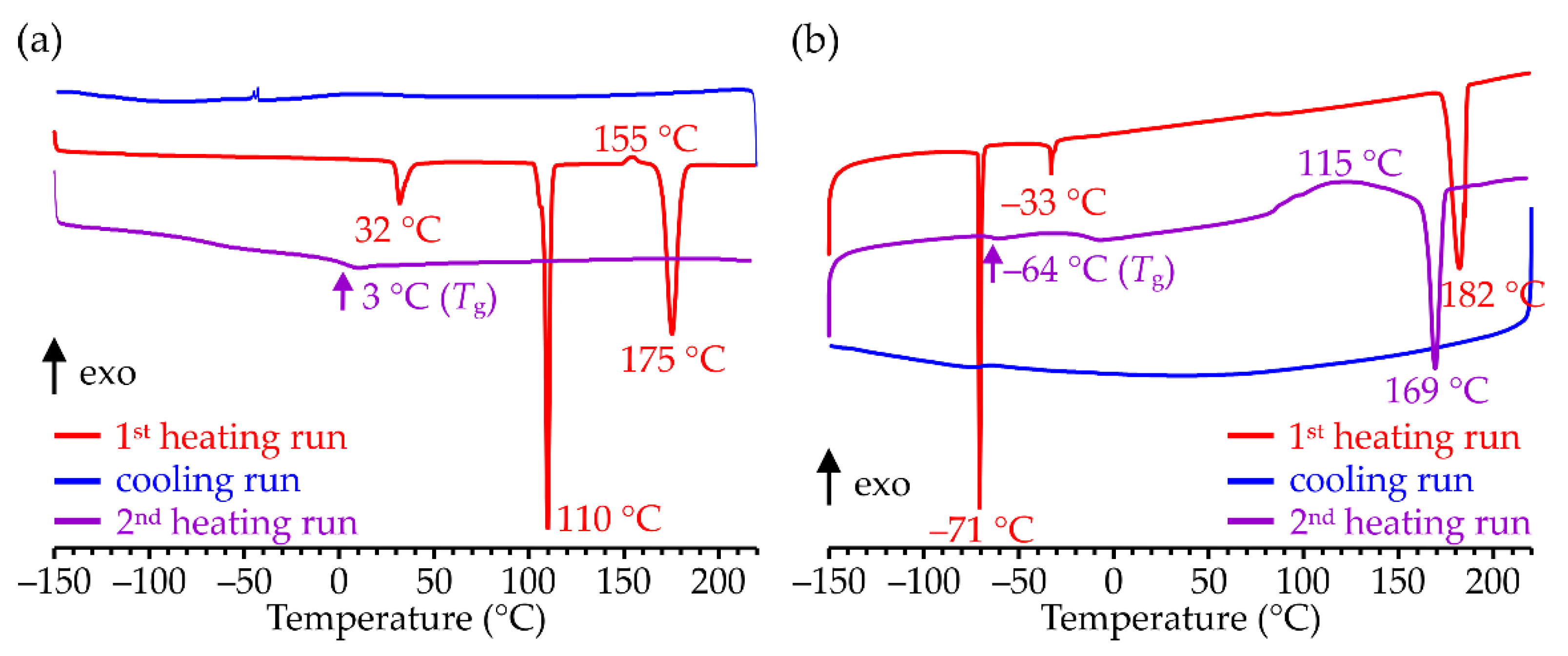
On heating, the pristine ZnDMP sample exhibits three endothermic transitions with maxima located at 32, 110 and 175 °C (
Figure 3a), whose enthalpies are equal to −2.4, −12.4 and −12.9 kJ/mol, respectively. The first two of them proceed in a solid state, while the one with the highest peak temperature arises simply from the melting of the sample. Interestingly, there exists also a small exothermic peak (Δ
H = 0.6 kJ/mol)) situated prior to the melting endotherm (its maximum at ca. 155 °C)—a very similar situation occurs in the case of several semi-crystalline polymers (e.g., polylactide) [
46] undergoing some structural rearrangement (for example, a disorder-to-order crystal phase transition) immediately before melting.
The systematic analysis of the X-ray diffractograms recorded at different temperatures by means of the VT–PXRD method gives evidence that the crystal structure of ZnDMP does not change significantly within the temperature range of 25–105 °C. As could be expected, within that area the increase in temperature resulted only in a gradual expansion of the ZnDMP unit cell volume due to its polymeric chains moving away from each other in a direction perpendicular to their axes. The results of our calculations (see
Table S2 in SM) show that within the margin of estimation error this process is anisotropic since it increases the value of the
c-axis unit length of ZnDMP, whereas the remaining lattice parameters
a and
b decrease their values (albeit with some fluctuations). It should be noted that the almost linear and continuous character of those changes is indicative of the absence of any polymorphic transitions during the heating of this low-temperature crystalline phase of ZnDMP (denoted as α-ZnDMP), which is structurally identical with the one determined by Harrison and coworkers for ZnDMP monocrystal [
18].
On heating above 105 °C (i.e., in the temperature region that corresponds to the first strong endothermic transition observed in the 1st heating DSC curve of the pristine ZnDMP), the PXRD reflections ascribed to α-ZnDMP start to disappear, while new ones show the indicating transformation of α-ZnDMP into a new crystalline phase (denoted as β-ZnDMP—during the VT-PXRD measurement, this process is completed at 165 °C (
Figure S20 in SM)). We tried to carry out a full profile refinement of the latter’s cell parameters, but we did not get any reliable results due to an insufficient number of observed reflections. Rapid cooling of the ZnDMP sample previously conditioned for 3 h at 130 °C does not reverse the α-ZnDMP → β-ZnDMP transition since the PXRD pattern of such a sample recorded at room temperature (
Figure S21a in SM) reveals a set of reflections attributable solely to β-ZnDMP—two of them exhibiting the highest intensity (2
θ angle of 9.626° and 13.277°) can be considered diagnostic PXRD signals for an identification of this crystalline phase. It is worth noting that β-ZnDMP is stable at room temperature and does not undergo any structural rearrangement while being stored for several days—nevertheless, after 7 days at that temperature, a development of the PXRD reflection characteristic for α-ZnDMP (2
θ = 11.263°) could be detected (
Figure S21b in SM). This process can be accelerated even further if the β-ZnDMP sample is conditioned at the temperature near that of the α-ZnDMP → β-ZnDMP transition (see
Figure S21a in SM).
The PXRD reflections of β-ZnDMP are clearly visible up to about 165 °C, but on further heating they gradually disappear, and above 180 °C, the sample of ZnDMP becomes an isotropic, viscous liquid (
Figure S20 in SM). Upon rapid cooling during a DSC test, it transforms into an amorphous solid that shows a second-order transition around 3 °C (see the 2nd heating DSC curve in
Figure S3a).
The DSC heating curve of pristine ZnDEP (
Figure S3b) contains 3 endotherms with maxima at approximately −71 °C (Δ
H = −7.5 kJ/mol), −33 °C (Δ
H = −0.8 kJ/mol) and 182 °C (Δ
H = −21 kJ/mol). Similar to ZnDMP, when heated above 180 °C ZnDEP becomes a viscous liquid, the cooling of which leads to the formation of an amorphous material exhibiting a second-order transition at −64 °C and no DSC peaks below room temperature. In contrast to ZnDMP, the amorphized ZnDEP sample is prone to a slow cold crystallization during the 2nd heating run—a process that begins above 80 °C, ends around 150 °C and turns immediately into melting. Interestingly, its enthalpy closely matches that of the melting, and they both constitute approximately 80% of the value of enthalpy of melting detected during the 1st heating cycle. Moreover, the crystalline domains formed during cold crystallization are characterized by a lower melting temperature (169 °C) than the pristine ZnDEP.
In order to investigate the first two endothermic processes occurring during the heating of ZnDEP, we carried out the DSC and VT-PXRD measurements between −100 and room temperature. The obtained calorimetric results (
Figure S22 in SM) indicate that both those transitions are fully reversible since their endotherms are accompanied on the DSC cooling curves by the respective exothermic peaks characterized by almost the same absolute values of enthalpy: between cooling and heating mode, the maxima of the related DSC peaks are shifted by only ca. 5−8 °C relative to each other. Previously, a very similar situation was observed on the DSC trace of the aluminum-based analogue of ZnDEP, aluminum tris(diethylphosphate), and the overcoming of energy barriers related to the rotations around C–C and C–O bonds within the (RO)
2PO
2 ligand was proposed as the source of that low-temperature DSC endotherms [
35]—the same explanation can be given in the case of ZnDEP. In the latter’s crystal structure determined by Harrison and coworkers at room temperature [
16], all ethyl groups around phosphorus center occur in the
gauche-
anticlinal (G
−A
−) conformation (the O–P–O–C torsion angles of −75(1)° and −104(2)°), which has a higher potential energy than the most stable
gauche-
gauche conformer (or the
trans-
gauche-
gauche-
trans one, if both the carbon-oxygen and phosphorus-oxygen torsion angles are considered) [
39,
40]. One can expect that with a decreasing temperature this structure might be inclined to adopt a spatial configuration with less potential energy, thus giving rise to exotherms observed on the DSC cooling curve. The results of the VT-PXRD measurements (
Figure S23 in SM) support this hypothesis, since the temperature-dependent structural changes below 0 °C impact mainly the reflections located at the high 2
θ angle region of the ZnDEP X-ray diffractogram, which originate from diffractions occurring on the planes at least partially created by carbon atoms—for example, the (401) plane (see
Figure S24 in SM) disappears between −70 and −80 °C. The two main reflections ascribed to the (200) and (110) planes, which are parallel to the axis of the ZnDEP polymeric chain and pass through the positions of zinc atoms, undergo only the typical changes resulting from a temperature-driven variation of the unit cell dimensions (e.g., they shift to higher 2
θ values with lowering temperature due to a contraction of the ZnDEP unit cell and its polymeric chains getting closer to each other). Based on this conception, the lack of any structural transitions in ZnDMP below 100 °C is entirely understandable, since at room temperature the methyl groups in α-ZnDMP are already in the most stable
gauche-
gauche arrangement, according to the crystallographic data published in the literature [
18]. Still, the presented explanation is only a hypothesis that needs further, experimental verification (e.g., ZnDEP single crystal XRD measurements below 0 °C), since other structural factors related to the thermal shrinkage of the ZnDEP unit cell may also play an important role in those processes.
Thermal effects of the subzero temperature structural transitions are also observed for several other homologues of ZnDEP containing longer alkyl chains, such as:
n-propyl (between ca. −14 and −27 °C),
n-butyl (between −88 and −95 °C) and 2-ethylhexyl (between −25 and −42 °C) (see
Figure S25 in SM). Based on the VT-PXRD measurements of the cooled samples of ZnDnPP (
Figure S26 in SM) or ZnBEHP (
Figure S27 in SM), one can conclude that with the lowering of temperature the crystal structures of these compounds not only undergo a standard thermal contraction resulting in a shift of PXRD reflections to higher values of 2
θ angle, but also some larger reorganization of their atoms giving rise to new X-ray diffraction planes. In both these derivatives, the low-temperature processes are related mainly to the X-ray reflections with the highest intensities and the lowest 2
θ values, which most probably have the same origin as in ZnDEP (e.g., zinc atoms in the parallelly located ZnDOP polymeric chains). This means that the proposed structural reorganization must involve not only the conformational changes within organic substituents, but also a non-continuous relocation of the Zn(OPO)
2Zn rigid cores. However, due to the lack of structural data, the exact nature of the abovementioned changes remains unknown and needs further investigation. Interestingly, a careful analysis of the DSC traces (see
SM, Figure S25a for ZnDnPP and
Figure S25c for ZnBEHP) and VT-PXRD patterns recorded during cooling and heating steps indicates that the occurring structural transformations seem to be completely reversible, since the original crystal structure of the previously cooled samples of ZnDnPP or ZnBEHP is restored upon their reheating to room temperature. This restoring is slower than the opposite process, since the characteristic reflections of the low-temperature phases are still observed on the PXRD diffractograms at the temperatures higher by 10–20 °C than those during the earlier cooling step—for example, α- and β-ZnBEHP phases coexist at room temperature within the sample at the end of the cooling-heating procedure, while only the former was present at the beginning of it (see
Figure S27 in SM). It should be emphasized that the temperature window in which all these low-temperature structural transformations proceed corresponds very well to the one defined by the positions of the endo- and exotherms observed on the DSC heating and cooling curves below room temperature. It is also worth noting that, apart from the transition detected at the subzero temperature region, ZnDOPs with long alkyl (C3–C8) substituents also undergo another low-temperature structural reorganization around room temperature (ZnBEHP), or close to it (ZnDnPP and ZnDBP), the nature of which strongly depends on the type of organic group: ZnDnPP becomes a very viscous liquid, while the samples of the
n-butyl and 2-ethylhexyl derivatives remain crystalline solids (although with a changed structure). Upon cooling from a temperature higher than that of the second structural transition, the melted
n-propyl derivative solidifies into a material which do not show any transformations at the subzero temperature region, and whose melting temperature (55.8 °C) and enthalpy of fusion (Δ
H = −6.6 kJ/mol) differ from those exhibited by the pristine ZnDnPP sample (62.1 °C and Δ
H = −18.2 kJ/mol,
Figure S25a in SM). At room temperature, ZnDBP preheated earlier to 90 °C exhibits the PXRD pattern, which differs from that of the initial sample (see
Figure S11 in SM) indicative of its temperature-impeded structural reorganization, and similarly to an analogous sample of ZnDnPP is structurally stable at the subzero temperatures (no thermal effects below room temperature during the DSC 2nd cooling run, see
Figure S25b in SM).
3.4. Aliphatic ZnDOPs in Solution
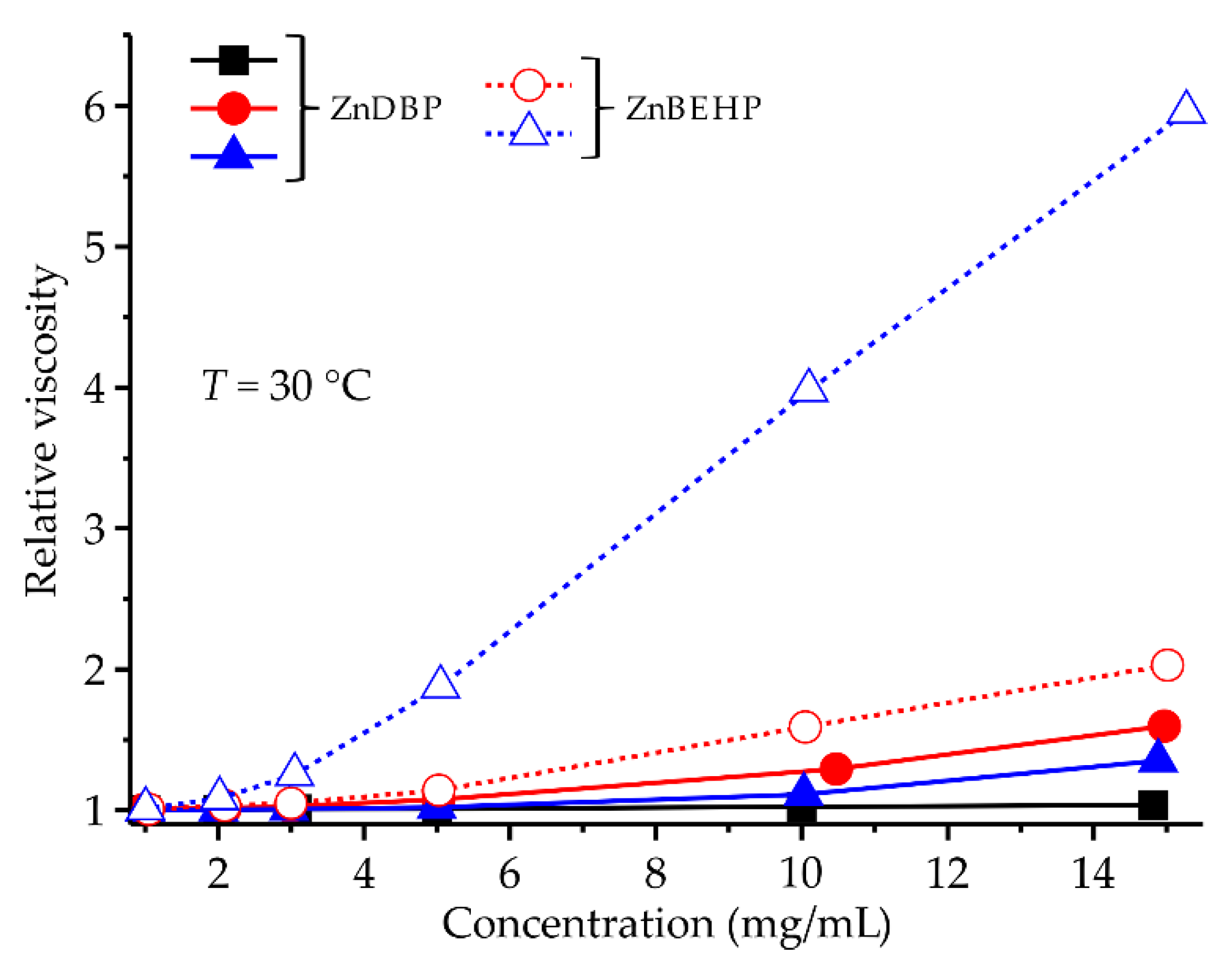
The solubility of aliphatic ZnDOPs depends significantly on the type of the alkyl substituent in their molecule. The butyl and 2-ethylhexyl derivatives are soluble in chloroform and aprotic, nonpolar solvents such as cyclohexane, benzene, or toluene (ZnDBP has also good solubility in methanol), and we have investigated the variation in the relative viscosity of the resulting solutions with their concentration (1–15 mg/mL) at 30 °C—the results of these studies are shown in
Figure 4.
In diluted solutions (sample concentration up to 3 mg/mL), only a slight linear increase in the relative viscosity values is observed, which can be attributed mainly to the interactions between solvent and the individual molecules of ZnDOPs. On the other hand, at higher sample concentrations the rheological behavior of the system depends on the type of the applied solvent: in methanol, the viscosity of solutions is almost constant, while in the case of systems based on less polar chloroform or toluene it increases significantly. One can assume that in the last two solvents ZnDBP and ZnBEHP exist in the form of large (polymeric) particles and above a certain critical concentration, hydrodynamic interactions between them give rise to the additional viscosity increase.
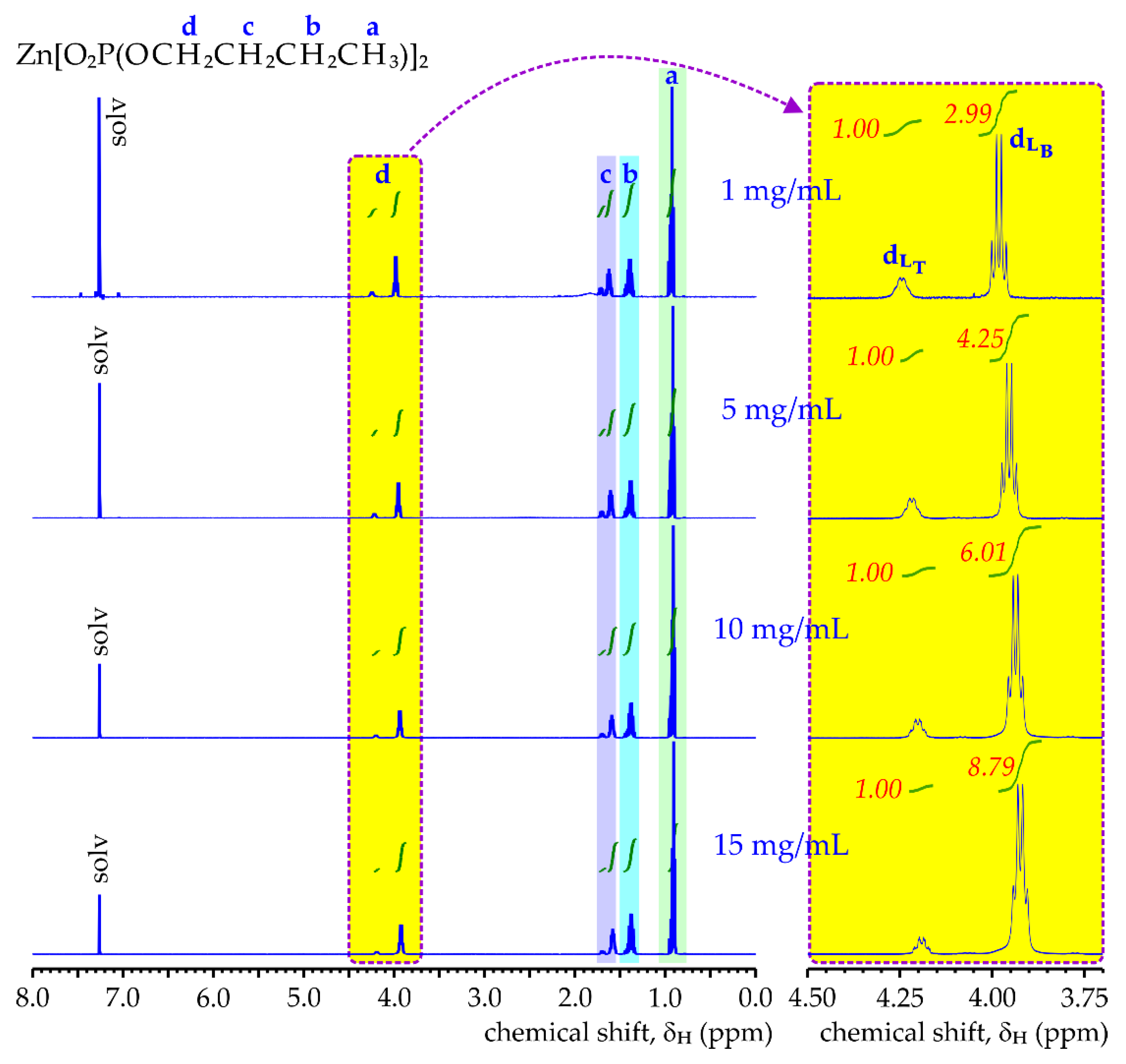
Figure 5 shows the
1H NMR spectra of ZnDBP dissolved in CDCl
3. These spectra contain signals characteristic for protons within the
n-butyl chain constituting: methyl group (
δH,a ≈ 0.9 ppm,), two inner methylene groups (
δH,b ≈ 1.4 ppm
δH,c ≈ 1.6–1.7 ppm) and methylene moiety connected to oxygen (
δH,d at 4.0–4.2 ppm). As can be seen, a common feature of these spectra is the fact that each peak of the chemically equivalent group of protons is divided into two components having very similar multiplicity and chemical shifts (the difference in their
δH values not larger than 0.3 ppm), but significantly differing in integrals—it is especially evident in the case of the nuclear resonance of the most deshielded protons from the –OCH
2– group. Interestingly, the relative peak integral of the main component increases when increasing the concentration of the ZnDBP solution. A plausible explanation for that phenomenon is that within each NMR signal the main multiplet comes from diorganophosphate ligands in the polymer bridging groups (L
B), whereas the second one with a smaller integral can be associated with (RO)
2PO
2 groups at the ends of the ZnDBP polymeric chain (terminal ligands, L
T)—exactly the same “duplication” of
1H NMR signals is reported in the literature for ZnDMP [
18] and ZnDEP [
16] solutions in D
2O.
Taking into account the zero electric charge of the ZnDBP chain, individual charges of its components {Zn
2+ and [(RO)
2PO
2]
−1 ions} and the fact of its formation via Zn(OPO)
2Zn bridging units, one can expect that every ZnDBP (or generally ZnDOP) chain should have one diorganophosphate group at each of its ends, meaning two L
T ligands per single polymeric chain. Based on this, the formula of ZnDBP can be rewritten as L
T[Zn(L
B)
2]
(n − 1)ZnL
T. The L
B/L
T NMR peak integral ratio allows one to calculate the average number of repeating units (degree of polymerization,
n) in a ZnDBP molecule (L
B/L
T =
n − 1), and its value depends on the concentration of the sample. As can be seen from data presented in
Table 2, for the investigated range of the concentration of ZnDBP solution in CDCl
3, the
n value changes from ca. 4 (ZnDBP tetramer) up to 10 (ZnDBP dekamer), which corresponds to the number average molecular weight between 1900 and 4800 g/mol, respectively.
Conclusions very similar to the ones described above can be made based on the analysis of the
31P NMR spectra of ZnDBP solutions in CDCl
3 (
Figure S28 in SM). They contain two peaks located around −2 ppm (main signal) and −13 ppm—a relative integral ratio of the former to the latter increases when increasing the mass concentration of the respective solution. The estimated degrees of polymerization and number average molecular weights are very close to the values derived from
1H NMR—the observed small differences can result from lower signal-to-noise ratio and problems with peak integration occurring in the case of phosphorus NMR.
It should be noted that
1H and
31P NMR spectra of ZnDBP in either non-polar (benzene-d
6,
Figure S29 in SM), or strongly polar (DMSO-d
6, methanol-d
4,
Figures S5 and S30 in SM, respectively) solvents show no differentiation between the inner and terminal diorganophosphate ligands: only one set of the proton resonance peaks ascribed to the
n-C
4H
9 group and one population of phosphorus nuclei are visible, respectively, suggesting that in these solvents ZnDBP molecules are either in the form of long polymeric chains (in which the concentration of the chain-ends is below the detection point of the NMR method), or they are completely depolymerized to monomeric species or free ions. The same applies to other ZnDOPs, which, in the
31P NMR spectra recorded in a polar solvent, give one average signal at about 0–−2 ppm (see
Figures S1–S4 in SM). In order to further investigate the abovementioned phenomena, we have measured the diffusion coefficients of ZnDBP particles (
DZnDBP) in diluted solutions (mass concentration of 1 mg/mL) in perdeuterated methanol, chloroform and benzene at 25 °C by means of the
1H DOSY NMR method. The obtained values of
DZnDBP are summarized in
Table 3, together with those of the ZnDBP hydrodynamic radii (
RH,ZnDBP) estimated based on the Stokes–Einstein equation [
46,
47]:
where
kB is the Boltzmann constant,
T is the absolute temperature,
f is friction factor (
f = 1 for a hard sphere), and
η is the solution viscosity.
Although the presented data should be considered a gross approximation of the real values of
RH,ZnDBP (for example, they do not take into account the deviation of the shape of ZnDBP particles from sphericity), one can conclude that the size of ZnDBP particles strongly depends on the polarity of the solvent. The largest particles are found in nonpolar C
6D
6, while the smallest ones are found in CD
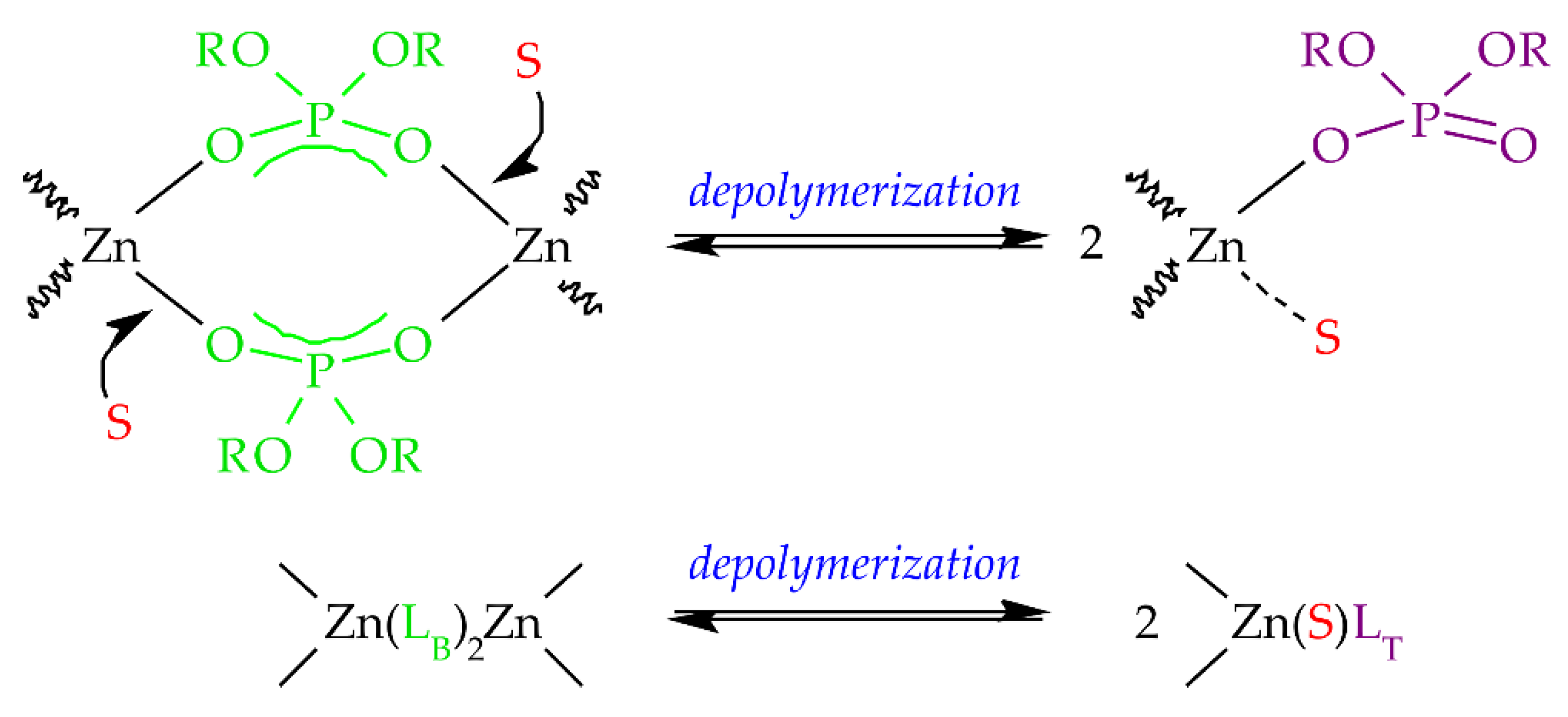
3OD solution. Probably polar solvents can complex zinc atoms in ZnDBP (or ZnDOPs in general) and break down the bridging bonds into small oligomers, as shown in
Scheme 2, or mononuclear complexes.
3.5. Synthesis and Characterization of Hybrid Copolymers
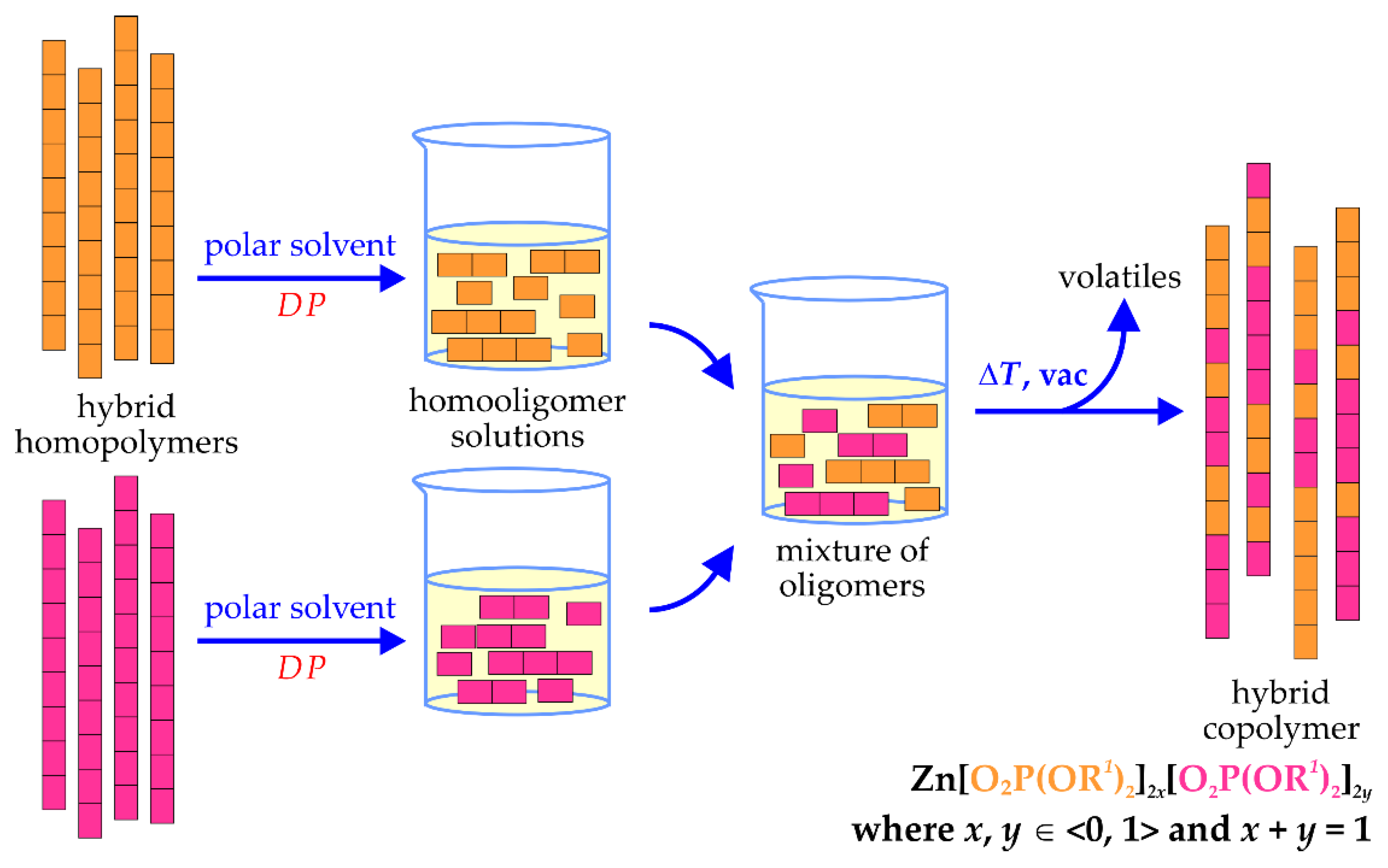
To confirm the reversible dissociation of aliphatic ZnDOPs in polar solvents, we have made attempts to obtain hybrid copolymers by dissolving two homopolymers in methanol and then evaporating the volatiles (see
Scheme 3). For that purpose, we have used both the aliphatic ZnDOPs described in this manuscript, as well as their simplest aromatic analogue, namely, ZnDPhP, which was fully characterized in our previous work [
33].
We have found that the abovementioned procedure leads to fully amorphous or semicrystalline materials, which are well-soluble in non-polar solvents.
Figures S31–S33 (SM) contain
1H DOSY NMR spectra recorded at 25 °C for several hybrid copolymers dissolved in C
6D
6 (a mass concentration of each system was 5 mg/mL), whereas
Table 4 shows the values of their diffusion coefficients (
Dcop) and hydrodynamic radii (
Rcop) approximated based on the Stokes–Einstein formula (see Equation (1), where
DZnDBP and
RH,ZnDBP are replaced with
Dcop and
RH,cop, respectively).
It should be noted that the Dcop values for the various groups of protons in the respective hybrid copolymer are the same within the experimental error. Thus, it can be assumed that the different organic substituents (e.g., n-butyl and phenyl groups in the case of ZnDBP-co-ZnDPhP copolymer) present in its structure constitute the same macromolecule. The mobility and RH,cop of the copolymer molecules depends on the type of diorganophosphate ligands. For example, a copolymer containing methyl and ethyl groups (ZnDMP-co-ZnDEP) forms smaller molecules with a higher diffusion coefficient than its analogue bearing longer alkyl chains—n-butyl and 2-ethylhexyl groups (ZnDBP-co-ZnBEHP).
More detailed studies carried out for the copolymer containing phenyl and
n-butyl groups (ZnDBP-
co-ZnDPhP) show that the distribution of ligands in its chains is not fully uniform and, by using an appropriate solvent, it can be separated into fractions with different compositions. For example,
1H NMR analysis (
Figure S34 in SM) indicates that a sample of ZnDBP-
co-ZnDPhP, in which the overall molar ratio of the aromatic and aliphatic monomeric units is 1:1, contains a fraction of polymeric chains enriched with aromatic ligands (the molar ratio of (PhO)
2PO
2/(
nBuO)
2PO
2 is 4:1) and is insoluble in cyclohexane—just like pure ZnDPhP homopolymer. At the same time, the cyclohexane-soluble part of that sample retains the overall equimolar composition. The
31P NMR spectrum of this copolymer in C
6D
6 (
Figure S35 in SM) contains two signals located at −1.18 and −12.94 ppm, which can be attributed to phosphorus nuclei in the bridging diorganophosphate ligands containing aliphatic and aromatic groups, respectively. Taking into account that ZnDPhP homopolymer is insoluble in non-polar media, the presence of its
1H and
31P NMR peaks in the spectrum of ZnDBP-
co-ZnDPhP suggests that zinc diphenylphosphate monomeric units or blocks “gain” solubility in C
6D
6 due to their incorporation into the macromolecule containing blocks of ZnDBP.
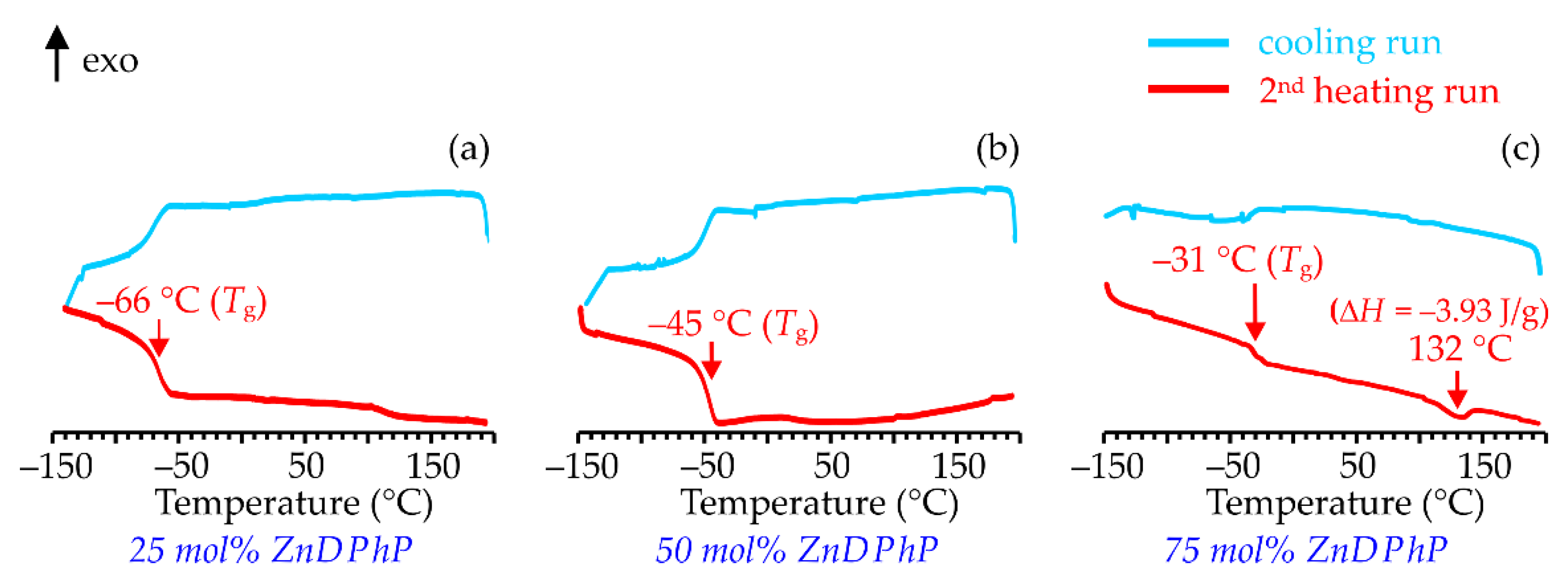
The DSC thermograms of the ZnDBP-
co-ZnDPhP copolymers indicate the presence of a homogeneous amorphous phase, whose glass transition temperature (
Tg) depends on the molar ratio of the alkyl and aromatic (RO)
2PO
2 ligands—for copolymers containing 25, 50 and 75 mol% of diphenylphosphate units the
Tg values are −66, −45 and −31 °C, respectively (
Figure 6). Interestingly, in the ZnDBP-
co-ZnDPhP copolymers with an excess of monomeric units of a given type, a small fraction of the crystalline domains may also occur—they give rise to the PXRD reflections (e.g., the one with the highest intensity located at 2
θ = 7.87°,
Figure S36 in SM) closely matching those previously reported for the samples of a pure ZnDPhP [
33]. It is possible that in these systems a high concentration of oligomers of one type favors a quick formation of larger particles of the respective homopolymer, thus changing the overall molar ratio of the remaining alkyl and aryl building blocks from which a copolymer molecule can be synthesized.
3.6. Curing of Epoxy Resins in the Presence of ZnDOPs
The results presented above suggest that ZnDOPs are Lewis acids and can form complexes with organic compounds of nucleophilic properties. This feature prompted us to conduct some preliminary studies on the potential catalytic activity of ZnDOPs in the curing of epoxy resin—for that purpose, a thermosetting epoxy monomer, bisphenol A diglycidyl ether (BADGE), was chosen. Initially, we observed that by heating above 160 °C, one can solidify BADGE compositions containing a few wt% of ZnDMP, ZnDEP, ZnDBP or ZnDPhP (namely, ZnDOP/BADGE compositions), whereas its pure sample remains liquid under similar treatment. This finding led us to have a closer look into the changes of viscoelastic properties occurring during the heating of ZnDOP/BADGE by utilizing the method of oscillatory rheology. In these experiments, the changes in the complex viscosity (|
η*|), storage modulus (
G′) and loss modulus (
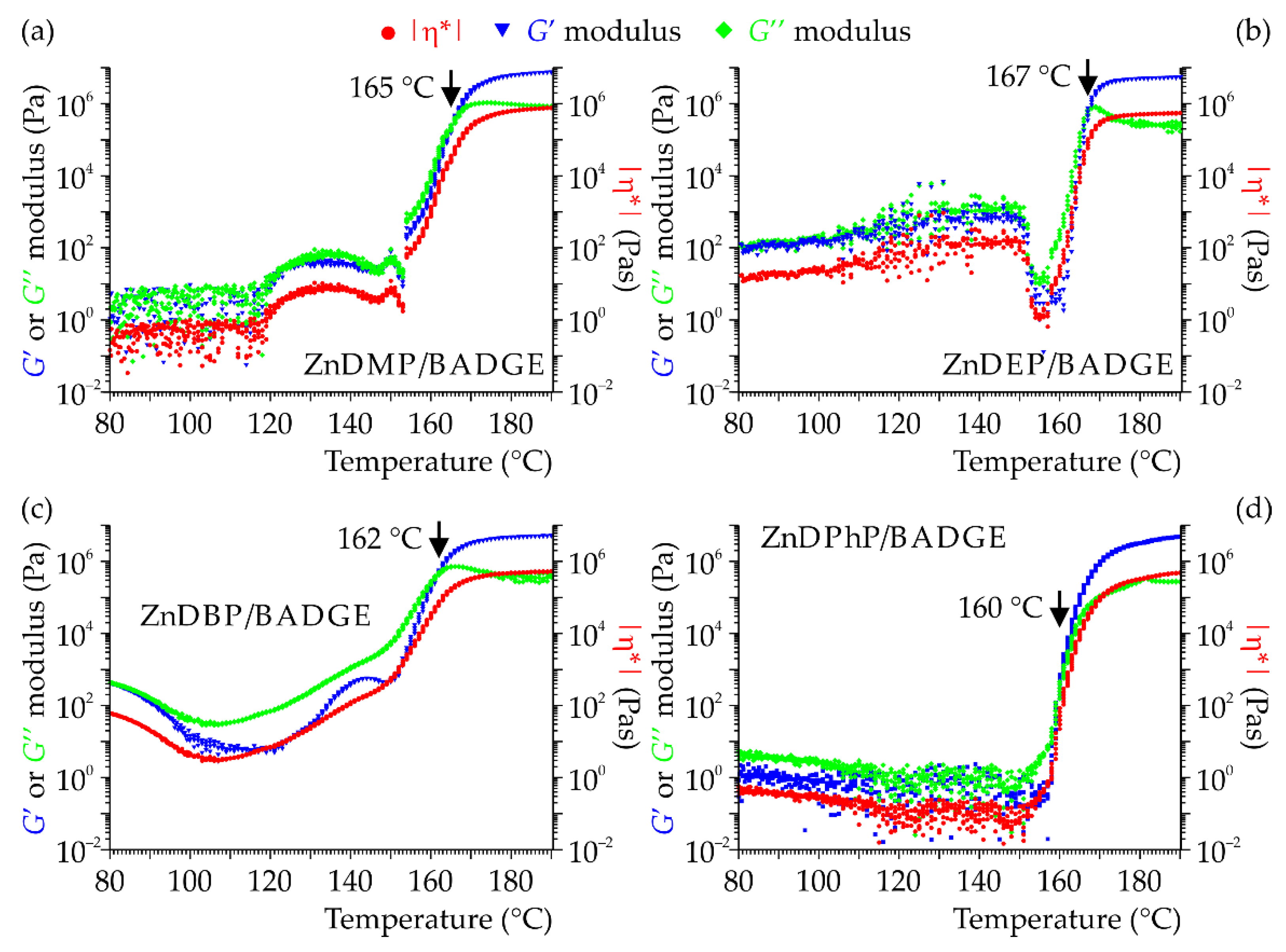
G″) were recorded while heating the samples at a constant rate from 80 to 190 °C. The results from these measurements carried out for the compositions filled with 20 wt% of the selected ZnDOPs are presented in
Figure 7.
As can be seen, upon heating to ca. 150 °C, the viscous properties prevail in all systems since G′ has lower values than G″: within this temperature region, the compositions behave like viscous liquids. Around 140–150 °C, a transition region starts, in which both moduli and viscosity rapidly increase their values. Subsequently, between 160 and 170 °C the storage modulus reaches values higher than G″, indicating that the material’s elastic properties start prevailing and it becomes solid. The whole process ends at around 180 °C and all of the studied rheological parameters stabilize their values at levels that are 5–7 orders of magnitude higher than those at the beginning of the curing process.
The changes in rheological parameters in the temperature range of 80–140 °C are much smaller and depend on the type of the curing agent. In the case of ZnDPhP/BADGE, the values of these parameters decrease slightly, whereas in compositions based on ZnDMP, ZnDEP or ZnDBP, some fluctuation in |η*|, G′ and G″ are observed, which may be related to changes in the structure of the respective ZnDOP and initiation of the curing process.
The thermal effects during the isothermal curing of BADGE in the presence of 20 wt% of ZnDEP were investigated within a 30 min time span at the temperatures of 130, 140, 150 and 160 °C, using the DSC method (
Figure S37 in SM). The results of these tests show that at 130 °C a possible exothermic transition (polymerization of the epoxy groups) requires at least 26 min to begin. At higher temperatures, the induction period is getting shorter (starting from about 10 min at 140 °C to virtually immediate polymerization at 160 °C), and the maximum curing rate is reached after 3.5–23 min. Based on these results, we think that ZnDOPs can be used as very effective, latent curing agents in the so-called one-component epoxy resin composition. An important advantage is that such compositions are stable when stored at ambient conditions, and will cure quickly when heated to a moderately high temperature.
The curing process often leads to a change in the structure of ZnDOPs. For example, the PXRD measurements (
Figure S38 in SM) clearly show that the filler/catalyst particles in the ZnDMP/BADGE composition (a 20 or 50 wt% filler load) hardened at 160 °C have an amorphous structure, while in the case of the same materials cured at lower temperature (130 °C) one can also observe the reflections attributable to β-ZnDMP, with only a small trace of those characteristic for the structure of ZnDMP before curing (its α phase). On the other hand, the crystalline domains of ZnDPhP immobilized in the epoxy resin are detectable at polyepoxide hardened at 160 °C (
Figure S39 in SM), albeit only for the more concentrated system—their structure corresponds very well to the one exhibited at the same temperature by the high temperature ZnDPhP crystal phase forming at temperatures exceeding 145 °C [
33]. Therefore, it can be suspected that in the solidified polyepoxide matrix the particles of ZnDOPs at least partially (their amorphization always occurs) retain the ordered structure they adopt at the temperature applied during curing. Moreover, it is likely that, beside the catalytic activity, some of these particles can also react with either epoxide or the newly formed polyepoxide; however, this hypothesis needs more detailed studies since the structure of the resulting products, as well as the mechanism of the epoxide curing, has not been fully elucidated yet.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}