
Polyethylene Glycol-b-poly(trialkylsilyl methacrylate-co-methyl methacrylate) Hydrolyzable Block Copolymers for Eco-Friendly Self-Polishing Marine Coatings

 ,
,  , , and
, , and 
Abstract
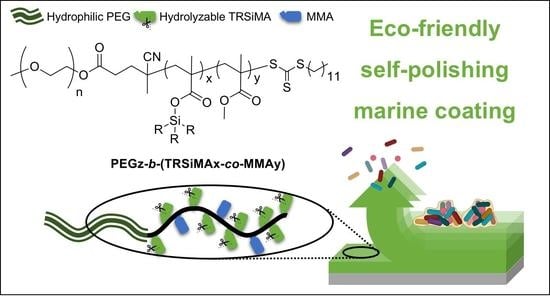
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterizations
2.3. Synthesis
2.3.1. Synthesis of the Macroinitiators PEGz-CTA
2.3.2. Synthesis of PEGz-b-(TRSiMAx-co-MMAy) Copolymers
2.4. Film Preparation
2.5. Biological Assay
3. Results and Discussion
3.1. Synthesis of Copolymers
3.2. Hydrolysis Kinetics of Copolymers
3.3. Mass Erosion of Copolymer Films in Artificial Seawater
3.4. Dynamic Contact Angle Analysis
3.5. Atomic Force Microscopy
3.6. Biological Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qiu, H.; Feng, K.; Gapeeva, A.; Meurisch, K.; Kaps, S.; Li, X.; Yu, L.; Mishra, Y.K.; Adelung, R.; Baum, M. Functional Polymer Materials for Modern Marine Biofouling Control. Prog. Polym. Sci. 2022, 127, 101516. [Google Scholar] [CrossRef]
- Pourhashem, S.; Seif, A.; Saba, F.; Nezhad, E.G.; Ji, X.; Zhou, Z.; Zhai, X.; Mirzaee, M.; Duan, J.; Rashidi, A.; et al. Antifouling Nanocomposite Polymer Coatings for Marine Applications: A Review on Experiments, Mechanisms, and Theoretical Studies. J. Mater. Sci. Technol. 2022, 118, 73–113. [Google Scholar] [CrossRef]
- Tian, L.; Yin, Y.; Bing, W.; Jin, E. Antifouling Technology Trends in Marine Environmental Protection. J. Bionic Eng. 2021, 18, 239–263. [Google Scholar] [CrossRef] [PubMed]
- Maan, A.M.C.; Hofman, A.H.; Vos, W.M.; Kamperman, M. Recent Developments and Practical Feasibility of Polymer-Based Antifouling Coatings. Adv. Funct. Mater. 2020, 30, 2000936. [Google Scholar] [CrossRef]
- Pradhan, S.; Kumar, S.; Mohanty, S.; Nayak, S.K. Environmentally Benign Fouling-Resistant Marine Coatings: A Review. Polym.-Plast. Technol. Mater. 2019, 58, 498–518. [Google Scholar] [CrossRef]
- Leonardi, A.K.; Ober, C.K. Polymer-Based Marine Antifouling and Fouling Release Surfaces: Strategies for Synthesis and Modification. Annu. Rev. Chem. Biomol. Eng. 2019, 10, 241–264. [Google Scholar] [CrossRef]
- Guazzelli, E.; Perondi, F.; Criscitiello, F.; Pretti, C.; Oliva, M.; Casu, V.; Maniero, F.; Gazzera, L.; Galli, G.; Martinelli, E. New Amphiphilic Copolymers for PDMS-Based Nanocomposite Films with Long-Term Marine Antifouling Performance. J. Mater. Chem. B 2020, 8, 9764–9776. [Google Scholar] [CrossRef]
- Martinelli, E.; Guazzelli, E.; Glisenti, A.; Galli, G. Surface Segregation of Amphiphilic PDMS-Based Films Containing Terpolymers with Siloxane, Fluorinated and Ethoxylated Side Chains. Coatings 2019, 9, 153. [Google Scholar] [CrossRef] [Green Version]
- Hu, P.; Xie, Q.; Ma, C.; Zhang, G. Silicone-Based Fouling-Release Coatings for Marine Antifouling. Langmuir 2020, 36, 2170–2183. [Google Scholar] [CrossRef] [Green Version]
- Leigh, B.L.; Cheng, E.; Xu, L.; Derk, A.; Hansen, M.R.; Guymon, C.A. Antifouling Photograftable Zwitterionic Coatings on PDMS Substrates. Langmuir 2019, 35, 1100–1110. [Google Scholar] [CrossRef]
- Guazzelli, E.; Lusiani, N.; Monni, G.; Oliva, M.; Pelosi, C.; Wurm, F.R.; Pretti, C.; Martinelli, E. Amphiphilic Polyphosphonate Copolymers as New Additives for PDMS-Based Antifouling Coatings. Polymers 2021, 13, 3414. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, E.; Pretti, C.; Oliva, M.; Glisenti, A.; Galli, G. Sol-Gel Polysiloxane Films Containing Different Surface-Active Trialkoxysilanes for the Release of the Marine Foulant Ficopomatus Enigmaticus. Polymer 2018, 145, 426–433. [Google Scholar] [CrossRef]
- Pretti, C.; Oliva, M.; Mennillo, E.; Barbaglia, M.; Funel, M.; Reddy Yasani, B.; Martinelli, E.; Galli, G. An Ecotoxicological Study on Tin- and Bismuth-Catalysed PDMS Based Coatings Containing a Surface-Active Polymer. Ecotoxicol. Environ. Saf. 2013, 98, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Yang, J.; Guo, H.; Xu, T.; Li, Q.; Wen, C.; Sui, X.; Lin, C.; Zhang, J.; Zhang, L. Slime-Resistant Marine Anti-Biofouling Coating with PVP-Based Copolymer in PDMS Matrix. Chem. Eng. Sci. 2019, 207, 790–798. [Google Scholar] [CrossRef]
- Kuliasha, C.A.; Finlay, J.A.; Franco, S.C.; Clare, A.S.; Stafslien, S.J.; Brennan, A.B. Marine Anti-Biofouling Efficacy of Amphiphilic Poly(Coacrylate) Grafted PDMSe: Effect of Graft Molecular Weight. Biofouling 2017, 33, 252–267. [Google Scholar] [CrossRef]
- Wang, D.; Xu, J.; Tan, J.; Yang, J.; Zhou, S. In Situ Generation of Amphiphilic Coatings Based on a Self-Catalytic Zwitterionic Precursor and Their Antifouling Performance. Chem. Eng. J. 2021, 422, 130115. [Google Scholar] [CrossRef]
- Galli, G.; Barsi, D.; Martinelli, E.; Glisenti, A.; Finlay, J.A.; Callow, M.E.; Callow, J.A. Copolymer Films Containing Amphiphilic Side Chains of Well-Defined Fluoroalkyl-Segment Length with Biofouling-Release Potential. RSC Adv. 2016, 6, 67127–67135. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, C.; Wang, J.; Feng, X.; He, C. Dual-Mode Antifouling Ability of Thiol–Ene Amphiphilic Conetworks: Minimally Adhesive Coatings via the Surface Zwitterionization. ACS Sustain. Chem. Eng. 2016, 4, 3803–3811. [Google Scholar] [CrossRef]
- Galli, G.; Martinelli, E. Amphiphilic Polymer Platforms: Surface Engineering of Films for Marine Antibiofouling. Macromol. Rapid Commun. 2017, 38, 1600704. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, J.; Liu, Z.; Jiang, X.; Yu, L. Antifouling Property of Cu2O-Free Self-Polishing Antifouling Coatings Based on Amide Derivatives Inspired by Capsaicin. Langmuir 2022, 38, 10244–10255. [Google Scholar] [CrossRef]
- Wang, X.; Yang, J.; Jiang, X.; Yu, L. Preparation and Properties of Environmentally Friendly Marine Antifouling Coatings Based on a Collaborative Strategy. Langmuir 2022, 38, 6676–6689. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Cao, M.; Li, S.; Yao, J.; Wu, B.; Wang, Y.; Wang, H.; Dong, J.; Yi, J. A Novel Marine Antifouling Coating Based on a Self-Polishing Zinc-Polyurethane Copolymer. J. Coat. Technol. Res. 2021, 18, 1333–1343. [Google Scholar] [CrossRef]
- Ni, C.; Feng, K.; Li, X.; Zhao, H.; Yu, L. Study on the Preparation and Properties of New Environmentally Friendly Antifouling Acrylic Metal Salt Resins Containing Indole Derivative Group. Prog. Org. Coat. 2020, 148, 105824. [Google Scholar] [CrossRef]
- Kavouras, P.; Trompeta, A.F.; Larroze, S.; Maranhão, M.; Teixeira, T.; Beltri, M.; Koumoulos, E.P.; Charitidis, C.A. Correlation of Mechanical Properties with Antifouling Efficacy of Coatings Containing Loaded Microcapsules. Prog. Org. Coat. 2019, 136, 105249. [Google Scholar] [CrossRef]
- Chen, R.; Li, Y.; Tang, L.; Yang, H.; Lu, Z.; Wang, J.; Liu, L.; Takahashi, K. Synthesis of Zinc-Based Acrylate Copolymers and Their Marine Antifouling Application. RSC Adv. 2017, 7, 40020–40027. [Google Scholar] [CrossRef] [Green Version]
- Song, F.; Wang, J.; Zhang, L.; Chen, R.; Liu, Q.; Liu, J.; Yu, J.; Liu, P.; Duan, J. Synergistically Improved Antifouling Efficiency of a Bioinspired Self-Renewing Interface via a Borneol/Boron Acrylate Polymer. J. Colloid Interface Sci. 2022, 612, 459–466. [Google Scholar] [CrossRef]
- Sha, J.; Yu, J.; Chen, R.; Liu, Q.; Liu, J.; Zhu, J.; Liu, P.; Li, R.; Wang, J. Eco-Friendly Self-Polishing Antifouling Coating via Eugenol Ester Hydrolysis. Prog. Org. Coat. 2022, 172, 107077. [Google Scholar] [CrossRef]
- Pan, J.; Ai, X.; Ma, C.; Zhang, G. Degradable Vinyl Polymers for Combating Marine Biofouling. Acc. Chem. Res. 2022, 55, 1586–1598. [Google Scholar] [CrossRef]
- Zhao, X.; Hao, H.; Duan, Y. A Self-Polishing Polyacrylate-g-Polysiloxane Paint for Marine Antifouling Application. Polym. Sci. Ser. A 2020, 62, 184–195. [Google Scholar] [CrossRef]
- Dai, G.; Xie, Q.; Ai, X.; Ma, C.; Zhang, G. Self-Generating and Self-Renewing Zwitterionic Polymer Surfaces for Marine Anti-Biofouling. ACS Appl. Mater. Interfaces 2019, 11, 41750–41757. [Google Scholar] [CrossRef]
- Xie, Q.; Ma, C.; Zhang, G.; Bressy, C. Poly(ester)–Poly(silyl methacrylate) Copolymers: Synthesis and Hydrolytic Degradation Kinetics. Polym. Chem. 2018, 9, 1448–1454. [Google Scholar] [CrossRef]
- Nguyen, M.N.; Pham, Q.T.; Le, V.D.; Nguyen, T.B.V.; Bressy, C.; Margaillan, A. Synthesis and Characterization of Random and Block-Random Diblock Silylated Terpolymers via RAFT Polymerization. Asian J. Chem. 2018, 30, 1125–1130. [Google Scholar] [CrossRef]
- Pavlović, D.; Lafond, S.; Margaillan, A.; Bressy, C. Facile Synthesis of Graft Copolymers of Controlled Architecture. Copolymerization of Fluorinated and Non-Fluorinated Poly(dimethylsiloxane) Macromonomers with Trialkylsilyl Methacrylates Using RAFT Polymerization. Polym. Chem. 2016, 7, 2652–2664. [Google Scholar] [CrossRef]
- Duong, T.H.; Briand, J.-F.; Margaillan, A.; Bressy, C. Polysiloxane-Based Block Copolymers with Marine Bacterial Anti-Adhesion Properties. ACS Appl. Mater. Interfaces 2015, 7, 15578–15586. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Xie, L.; He, C.; Liu, J.; Zhang, G.; Wu, C. Effects of Hydrolyzable Comonomer and Cross-Linking on Anti-Biofouling Terpolymer Coatings. Polymer 2013, 54, 2966–2972. [Google Scholar] [CrossRef]
- Wang, P.; He, B.; Wang, B.; Wang, L.; Yu, H.; Liu, S.; Ye, Q.; Zhou, F. Durable Self-Polishing Antifouling Coating Based on Fluorine-Containing Pyrrolidone Amphiphilic Copolymer-Functionalized Nanosilica. Prog. Org. Coat. 2022, 165, 106706. [Google Scholar] [CrossRef]
- Xie, Q.; Pan, J.; Ma, C.; Zhang, G. Dynamic Surface Antifouling: Mechanism and Systems. Soft Matter 2019, 15, 1087–1107. [Google Scholar] [CrossRef]
- Wang, S.; Liu, X.; Yu, L.; Zhao, Y.; Sun, M. Low Surface Energy Self-polishing Polymer Grafted MWNTs for Antibacterial Coating and Controlled-release Property of Cu2O. J. Appl. Polym. Sci. 2021, 138, 50267. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.; Wang, X.; Zhang, C.; Liu, H.; Yang, W.; Cai, M.; Pei, X.; Zhou, F. Self-Polishing Emulsion Platforms: Eco-Friendly Surface Engineering of Coatings toward Water Borne Marine Antifouling. Prog. Org. Coat. 2020, 149, 105945. [Google Scholar] [CrossRef]
- Xie, C.; Guo, H.; Zhao, W.; Zhang, L. Environmentally Friendly Marine Antifouling Coating Based on a Synergistic Strategy. Langmuir 2020, 36, 2396–2402. [Google Scholar] [CrossRef]
- Wang, D.; Xu, J.; Yang, J.; Zhou, S. Preparation and Synergistic Antifouling Effect of Self-Renewable Coatings Containing Quaternary Ammonium-Functionalized SiO2 Nanoparticles. J. Colloid Interface Sci. 2020, 563, 261–271. [Google Scholar] [CrossRef]
- Zhou, X.; Xie, Q.; Ma, C.; Chen, Z.; Zhang, G. Inhibition of Marine Biofouling by Use of Degradable and Hydrolyzable Silyl Acrylate Copolymer. Ind. Eng. Chem. Res. 2015, 54, 9559–9565. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Z.; Han, S.; Han, J.; Jiang, D. Poly(propylene carbonate) Polyurethane Self-Polishing Coating for Marine Antifouling Application. J. Appl. Polym. Sci. 2016, 133, 43667. [Google Scholar] [CrossRef]
- Guazzelli, E.; Martinelli, E.; Pelloquet, L.; Briand, J.-F.; Margaillan, A.; Bunet, R.; Galli, G.; Bressy, C. Amphiphilic Hydrolyzable Polydimethylsiloxane-b-Poly(ethyleneglycol methacrylate-co-trialkylsilyl methacrylate) Block Copolymers for Marine Coatings. II. Antifouling Laboratory Tests and Field Trials. Biofouling 2020, 36, 378–388. [Google Scholar] [CrossRef]
- Guazzelli, E.; Galli, G.; Martinelli, E.; Margaillan, A.; Bressy, C. Amphiphilic Hydrolyzable Polydimethylsiloxane-b-Poly(ethyleneglycol methacrylate-co-trialkylsilyl methacrylate) Block Copolymers for Marine Coatings. I. Synthesis, Hydrolysis and Surface Wettability. Polymer 2020, 186, 121954. [Google Scholar] [CrossRef]
- Holland, R.; Dugdale, T.M.; Wetherbee, R.; Brennan, A.B.; Finlay, J.A.; Callow, J.A.; Callow, M.E. Adhesion and Motility of Fouling Diatoms on a Silicone Elastomer. Biofouling 2004, 20, 323–329. [Google Scholar] [CrossRef]
- Sader, J.E.; Sanelli, J.A.; Adamson, B.D.; Monty, J.P.; Wei, X.; Crawford, S.A.; Friend, J.R.; Marusic, I.; Mulvaney, P.; Bieske, E.J. Spring Constant Calibration of Atomic Force Microscope Cantilevers of Arbitrary Shape. Rev. Sci. Instrum. 2012, 83, 103705. [Google Scholar] [CrossRef] [Green Version]
- Derjaguin, B.V.; Muller, V.M.; Toporov, Y.P. Effect of Contact Deformations on the Adhesion of Particles. J. Colloid Interface Sci. 1975, 53, 314–326. [Google Scholar] [CrossRef]
- Lejars, M.; Margaillan, A.; Bressy, C. Well-defined graft copolymers of tert-butyldimethylsilyl methacrylate and poly(dimethylsiloxane) macromonomers synthesized by raft polymerization. Polym. Chem. 2013, 4, 3282–3292. [Google Scholar] [CrossRef]
- Lejars, M.; Margaillan, A.; Bressy, C. Synthesis and Characterization of Diblock and Statistical Copolymers Based on Hydrolyzable Siloxy Silylester Methacrylate Monomers. Polym. Chem. 2014, 5, 2109–2117. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Copolymer | Mon:CTA a | p b | PEG/TRSiMA/MMA c (mole%) | Mn SEC d (g/mol) | Ð d | TRSiMA e DPn | MMA e DPn | |
|---|---|---|---|---|---|---|---|---|
| Feed | Copolymer | |||||||
| PEG2-b-(TPSiMA28-co-MMA38) | 117:1 | 80 | 30/30/40 | 34/28/38 | 14,700 | 1.1 | 41 | 56 |
| PEG2-b-(TPSiMA30-co-MMA50) | 450:1 | 57 | 10/30/60 | 20/30/50 | 42,300 | 1.1 | 74 | 125 |
| PEG1-b-(TPSiMA34-co-MMA39) | 40:1 | 89 | 30/30/40 | 27/34/39 | 10,400 | 1.1 | 21 | 24 |
| PEG1-b-(TPSiMA29-co-MMA60) | 153:1 | 71 | 10/30/60 | 11/29/60 | 24,400 | 1.1 | 42 | 88 |
| PEG2-b-(TBSiMA26-co-MMA41) | 117:1 | 89 | 30/30/40 | 33/26/41 | 12,700 | 1.1 | 40 | 63 |
| PEG2-b-(TBSiMA30-co-MMA54) | 450:1 | 70 | 10/30/60 | 16/30/54 | 25,400 | 1.2 | 96 | 172 |
| PEG1-b-(TBSiMA35-co-MMA37) | 40:1 | 96 | 30/30/40 | 28/35/37 | 7100 | 1.2 | 22 | 23 |
| PEG1-b-(TBSiMA24-co-MMA49) | 153:1 | 92 | 10/30/60 | 27/24/49 | 5900 | 1.4 | 46 | 95 |
| TPSiMA45-co-MMA55 | 58:1 | 86 | 0/40/60 | 0/45/55 | 7400 | 1.1 | 40 | 49 |
| TBSiMA40-co-MMA60 | 64:1 | 96 | 0/40/60 | 0/40/60 | 6300 | 1.1 | 31 | 45 |
| Film | Days of Immersion | ||||
|---|---|---|---|---|---|
| 0 | 21 | 30 | 75 | ||
| PEG2-b-(TPSiMA30-co-MMA50) | θadv (°) | 110 ± 5 | 72 ± 6 | 64 ± 1 | 95 ± 4 |
| θrec (°) | 30 ± 2 | 22 ± 2 | 23 ± 2 | 26 ± 2 | |
| Δ (°) | 80 ± 3 | 50 ± 5 | 41 ± 3 | 69 ± 2 | |
| PEG2-b-(TPSiMA28-co-MMA38) | θadv (°) | 71 ± 3 | 66 ± 2 | 56 ± 1 | 57 ± 2 |
| θrec (°) | 21 ± 2 | 19 ± 4 | 18 ± 1 | 19 ± 1 | |
| Δ (°) | 50 ± 3 | 47 ± 6 | 38 ± 1 | 38 ± 1 | |
| PEG2-b-(TBSiMA26-co-MMA41) | θadv (°) | 82 ± 3 | 77 ± 6 | 68 ± 2 | 66 ± 5 |
| θrec (°) | 28 ± 4 | 18 ± 2 | 18 ± 1 | 17 ± 4 | |
| Δ (°) | 54 ± 3 | 59 ± 4 | 50 ± 3 | 49 ± 2 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guazzelli, E.; Oliva, M.; Pretti, C.; Monni, G.; Fahs, A.; Bressy, C.; Martinelli, E. Polyethylene Glycol-b-poly(trialkylsilyl methacrylate-co-methyl methacrylate) Hydrolyzable Block Copolymers for Eco-Friendly Self-Polishing Marine Coatings. Polymers 2022, 14, 4589. https://doi.org/10.3390/polym14214589
Guazzelli E, Oliva M, Pretti C, Monni G, Fahs A, Bressy C, Martinelli E. Polyethylene Glycol-b-poly(trialkylsilyl methacrylate-co-methyl methacrylate) Hydrolyzable Block Copolymers for Eco-Friendly Self-Polishing Marine Coatings. Polymers. 2022; 14(21):4589. https://doi.org/10.3390/polym14214589
Chicago/Turabian StyleGuazzelli, Elisa, Matteo Oliva, Carlo Pretti, Gianfranca Monni, Armand Fahs, Christine Bressy, and Elisa Martinelli. 2022. "Polyethylene Glycol-b-poly(trialkylsilyl methacrylate-co-methyl methacrylate) Hydrolyzable Block Copolymers for Eco-Friendly Self-Polishing Marine Coatings" Polymers 14, no. 21: 4589. https://doi.org/10.3390/polym14214589
APA StyleGuazzelli, E., Oliva, M., Pretti, C., Monni, G., Fahs, A., Bressy, C., & Martinelli, E. (2022). Polyethylene Glycol-b-poly(trialkylsilyl methacrylate-co-methyl methacrylate) Hydrolyzable Block Copolymers for Eco-Friendly Self-Polishing Marine Coatings. Polymers, 14(21), 4589. https://doi.org/10.3390/polym14214589