
Influence of Sulfur-Curing Conditions on the Dynamics and Crosslinking of Rubber Networks: A Time-Domain NMR Study

 ,
,  , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Equilibrium Swelling, MDR, and DSC Experiments
2.3. NMR Experiments
2.4. Analysis of the DQ Build-Up Curves
2.5. Analysis of the 1H CPMG NMR Relaxation Curves
2.6. Analysis of the 1H FC NMRD Curves
3. Results and Discussion
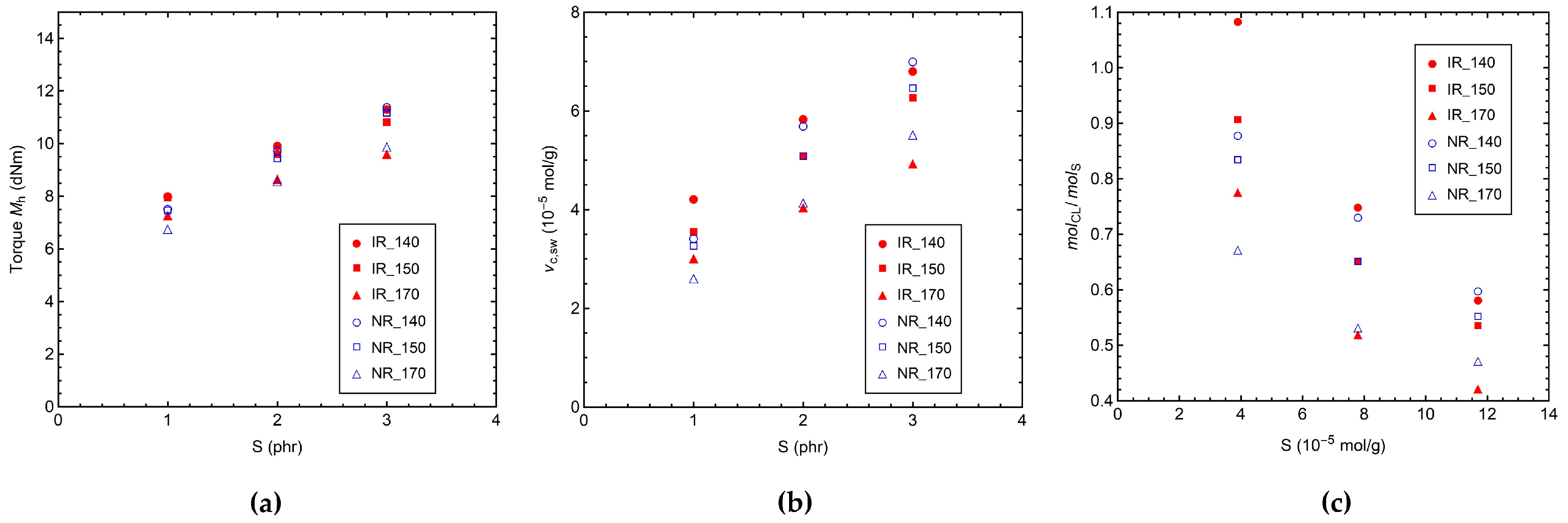
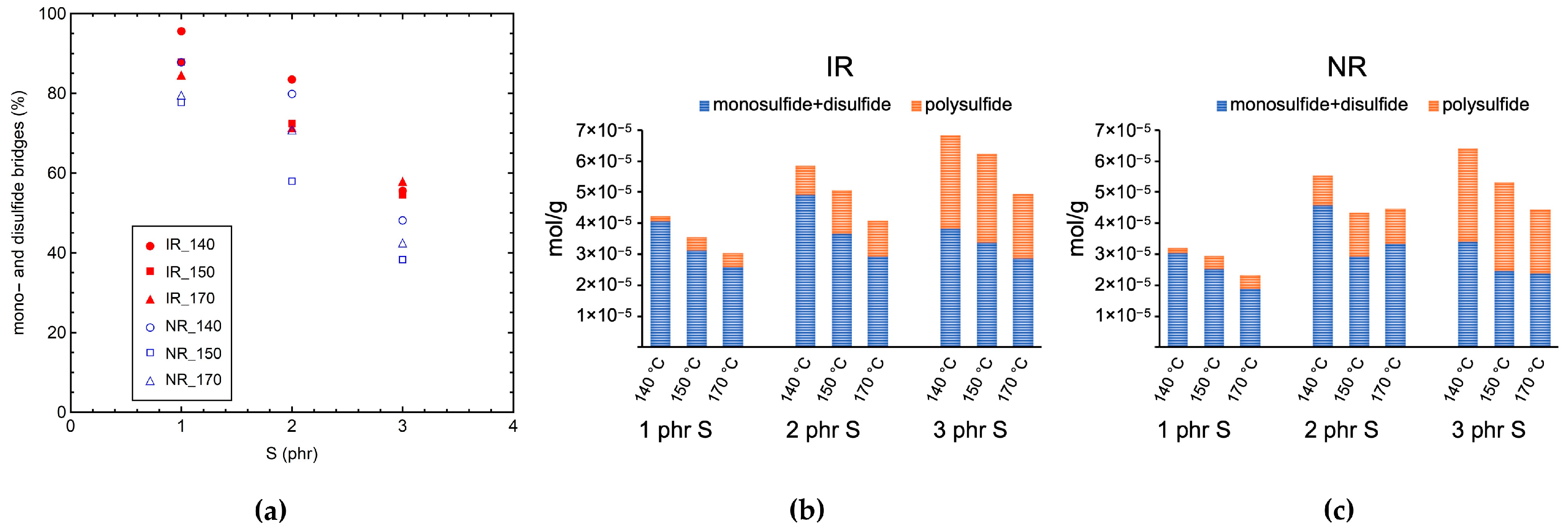
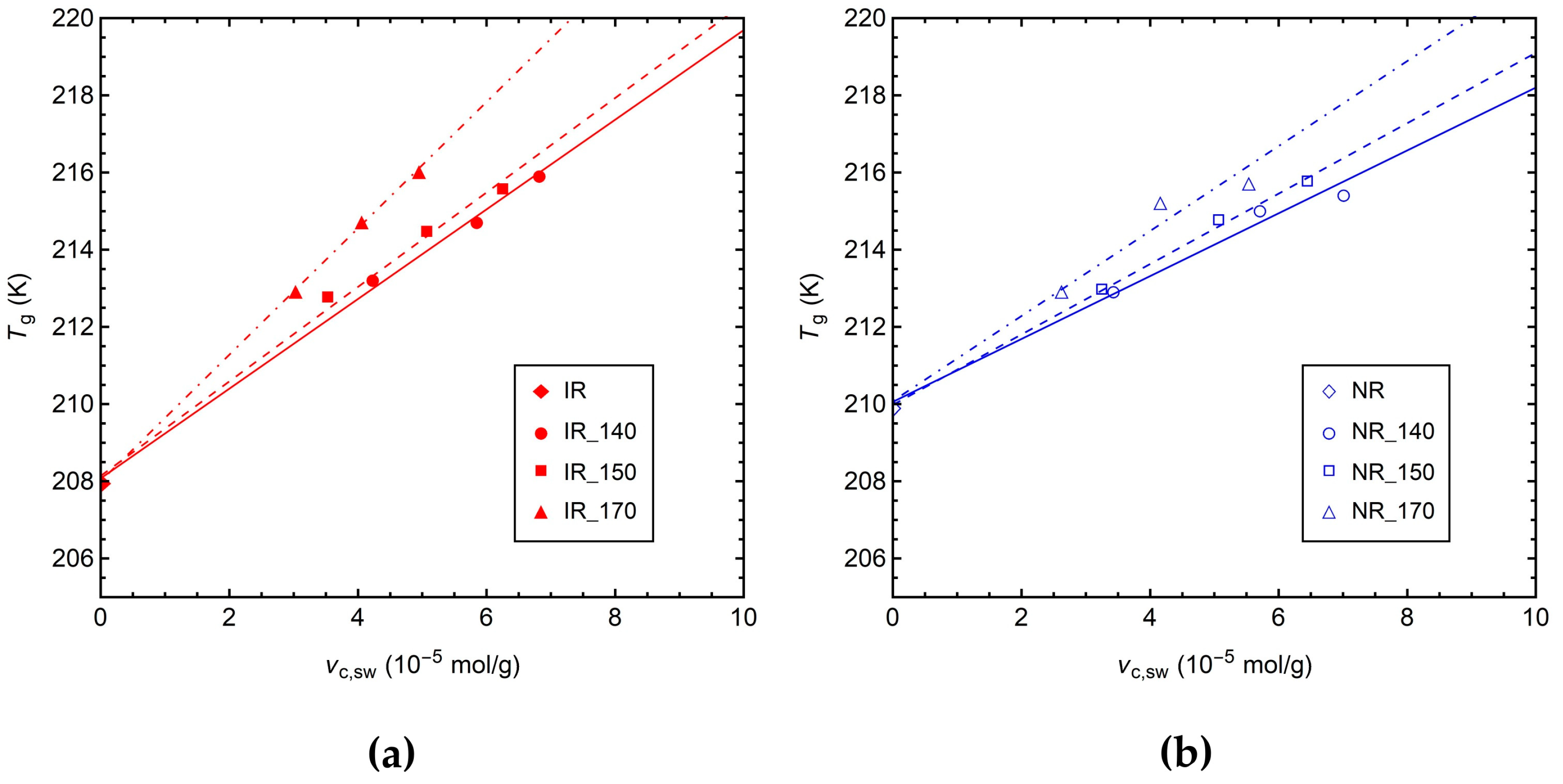
3.1. Characterization by Equilibrium Swelling, MDR, and DSC
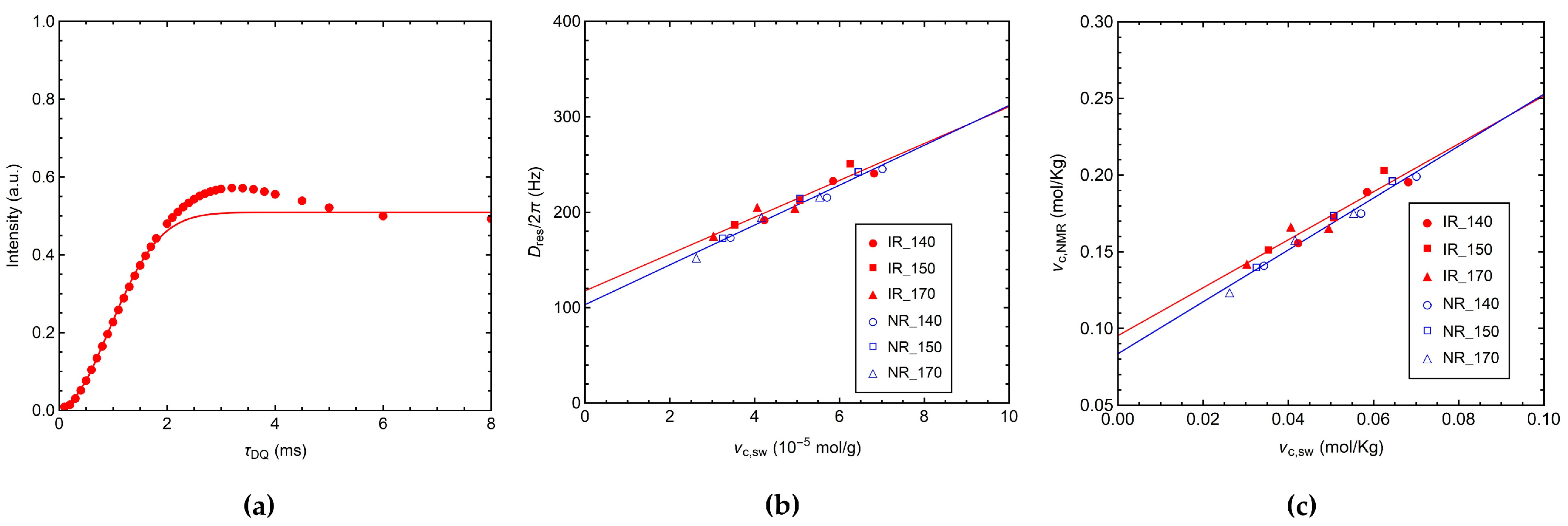
3.2. Characterization by 1H TD-NMR
3.2.1. 1H MQ NMR Experiments
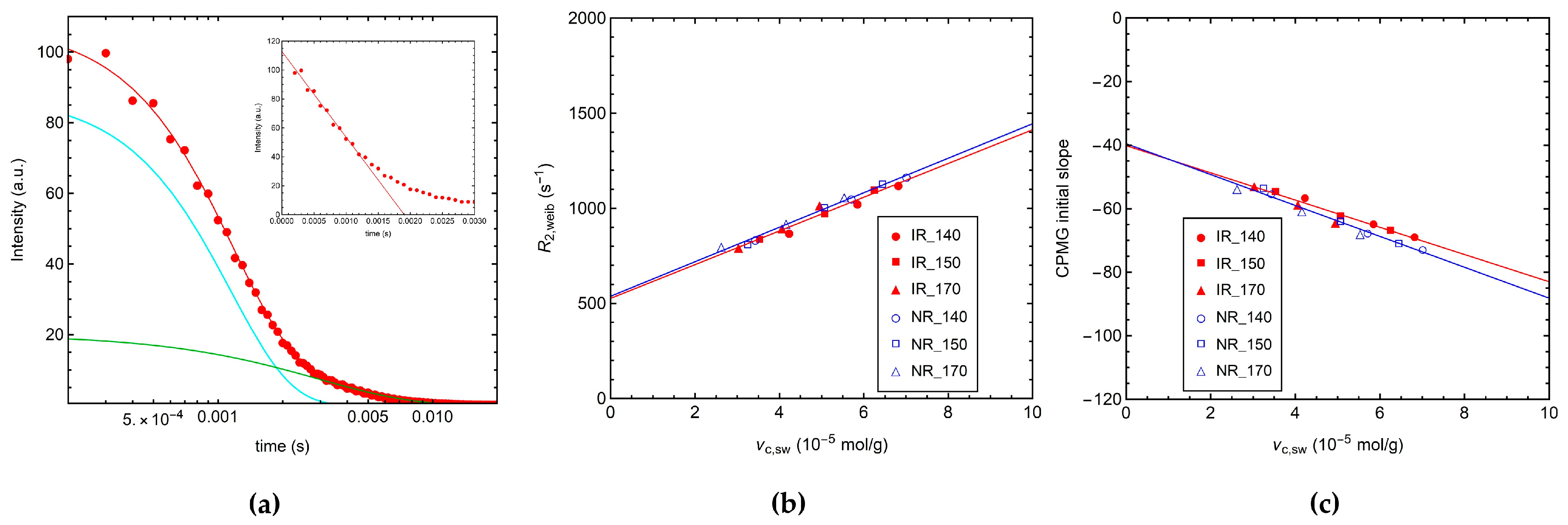
3.2.2. 1H Transverse Relaxation by CPMG Experiments
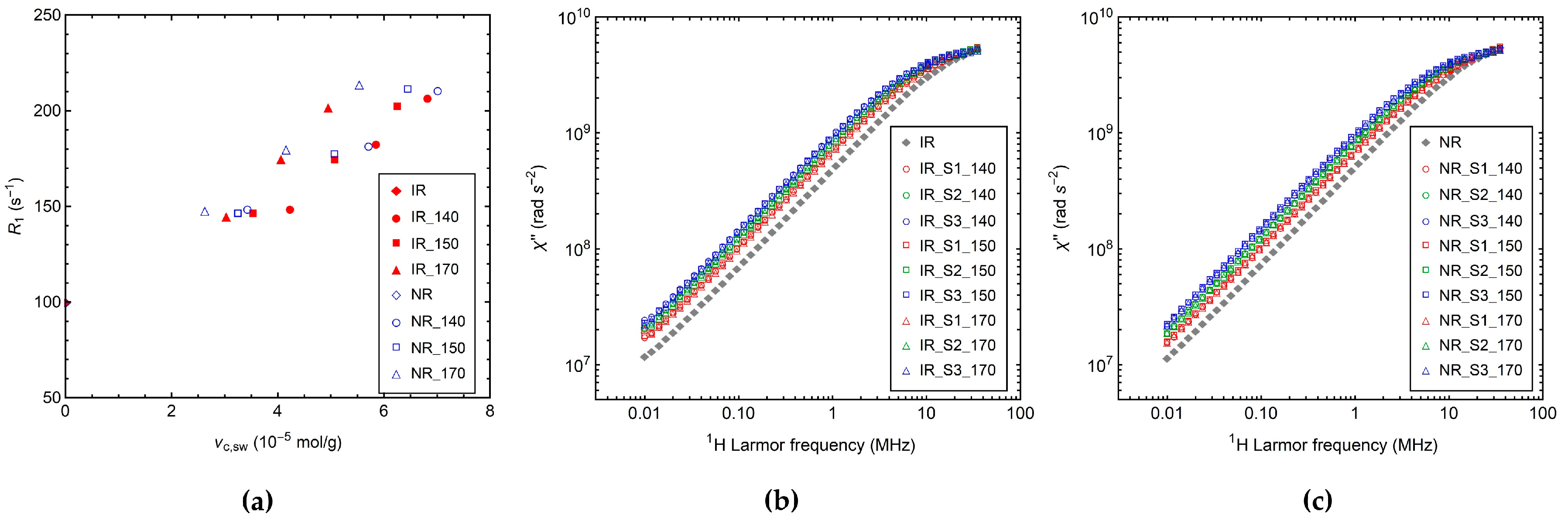
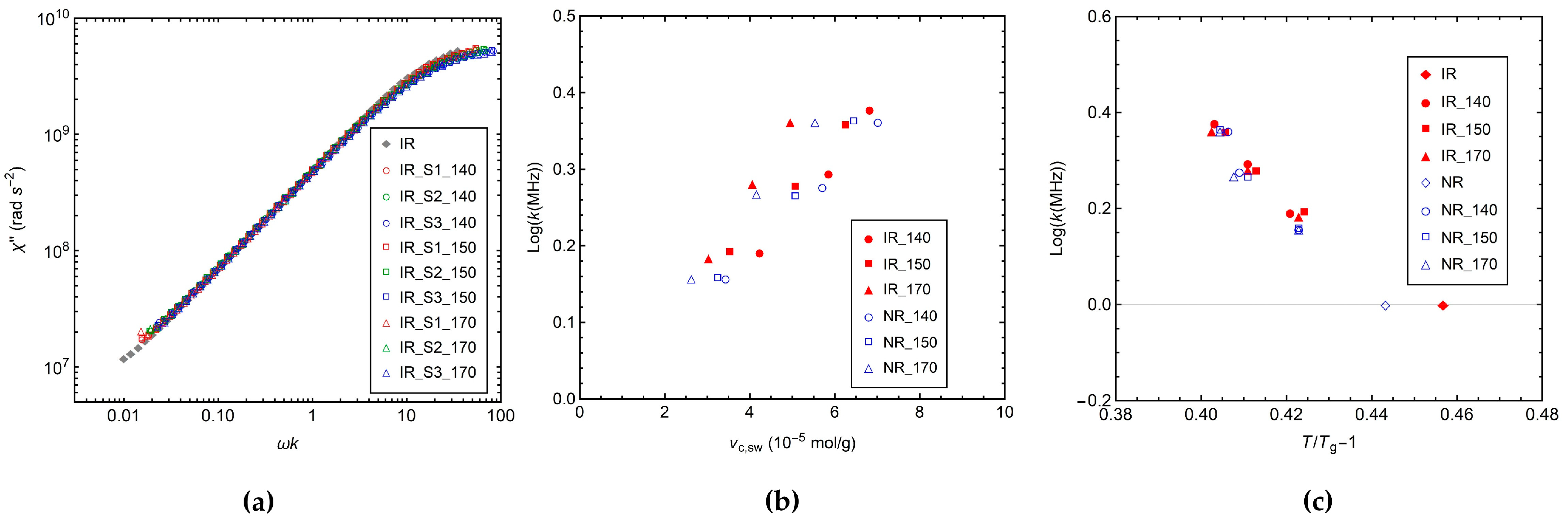
3.2.3. 1H Longitudinal Relaxation by FC NMR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krejsa, M.R.; Koenig, J.L. A Review of Sulfur Crosslinking Fundamentals for Accelerated and Unaccelerated Vulcanization. Rubber Chem. Technol. 1993, 66, 376–410. [Google Scholar] [CrossRef]
- Akiba, M.; Hashim, S. Vulcanization and crosslinking in elastomers. Prog. Polym. Sci. 1997, 22, 475–521. [Google Scholar] [CrossRef]
- Bhowmick, A.K.; Mukhopadhyay, R.; De, S.K. High Temperature Vulcanization of Elastomers. Rubber Chem. Technol. 1979, 52, 725–734. [Google Scholar] [CrossRef]
- Coran, A.Y. Vulcanization. In Science and Technology of Rubber, 3rd ed.; Mark, J.E., Erman, B., Eirich, F., Eds.; Academic Press: Burlington, MA, USA, 2005; Volume 7, pp. 339–385. [Google Scholar]
- Boonkerd, K.; Deeprasertkul, C.; Boonsomwong, K. Effect of sulfur to accelerator ratio on crosslink structure, reversion, and strength in natural rubber. Rubber Chem. Technol. 2016, 89, 450–464. [Google Scholar] [CrossRef]
- Zaper, A.M.; Koenig, J.L. Solid State Carbon-13 NMR Studies of Vulcanized Elastomers. III, Accelerated Sulfur Vulcanization of Natural Rubber. Rubber Chem. Technol. 1987, 60, 278–297. [Google Scholar] [CrossRef]
- Sombatsompop, N. Analysis of Cure Characteristics on Cross-Link Density and Type, and Viscoelastic Properties of Natural Rubber. Polym. Technol. Eng. 1998, 37, 333–349. [Google Scholar] [CrossRef]
- Marzocca, A.J.; Mansilla, M.A. Analysis of network structure formed in styrene–butadiene rubber cured with sulfur/TBBS system. J. Appl. Polym. Sci. 2007, 103, 1105–1112. [Google Scholar] [CrossRef]
- Bandzierz, K.S.; Reuvekamp, L.A.E.M.; Dryzek, J.; Dierkes, W.K.; Blume, A.; Bielinski, D. Influence of network structure on glass transition temperature of elastomers. Rubber Chem. Technol. 2019, 92, 69–89. [Google Scholar] [CrossRef] [Green Version]
- Bandzierz, K.; Reuvekamp, L.; Dryzek, J.; Dierkes, W.; Blume, A.; Bielinski, D. Effect of polymer chain modifications on elastomer properties. Materials 2016, 9, 607. [Google Scholar] [CrossRef] [Green Version]
- Morrison, N.J.; Porter, M. Temperature Effects on the Stability of Intermediates and Crosslinks in Sulfur Vulcanization. Rubber Chem. Technol. 1984, 57, 63–85. [Google Scholar] [CrossRef]
- Chueangchayaphan, N.; Nithi-Uthai, N.; Techakittiroj, K.; Manuspiya, H. In-situ dielectric cure monitoring as a method of measuring the influence of cure temperature on natural rubber vulcanization. Polym. Bull. 2021, 78, 3169–3182. [Google Scholar] [CrossRef]
- Kurian, T.; George, K.E.; Francis, D.J. Effect of vulcanization temperature on the cure characteristics and vulcanizate properties of natural rubber and styrene-butadiene rubber. Angewandte Makromol. Chem. 1988, 162, 123–134. [Google Scholar] [CrossRef]
- Kurian, T.; George, K.E. Effect of vulcanization temperature on the technical properties of NR, SBR, and BR. J. Appl. Polym. Sci. 1989, 37, 987–997. [Google Scholar] [CrossRef]
- Mukhopadhyay, R.; Bhowmick, A.K.; De, S.K. Effect of vulcanization temperature and synergism of accelerators on the network and technical properties of efficiently vulcanized natural rubber mixes. Polymer 1978, 19, 1176–1180. [Google Scholar] [CrossRef]
- Mukhopadhyay, R.; De, S.K.; Chakraborty, S. Effect of vulcanization temperature and vulcanization systems on the structure and properties of natural rubber vulcanizates. Polymer 1977, 18, 1243–1249. [Google Scholar] [CrossRef]
- Saville, B.R.; Watson, A.A. Structural Characterization of Sulfur-Vulcanized Rubber Networks. Rubber Chem. Technol. 1967, 40, 100–148. [Google Scholar] [CrossRef]
- Fox, T.G.; Loshaek, S. Influence of molecular weight and degree of crosslinking on the specific volume and glass temperature of polymers. J. Polym. Sci. 1955, 15, 371–390. [Google Scholar] [CrossRef]
- Danielsen, S.P.O.; Beech, H.K.; Wang, S.; El-Zaatari, B.M.; Wang, X.; Sapir, L.; Ouchi, T.; Wang, Z.; Johnson, P.N.; Hu, Y.; et al. Molecular Characterization of Polymer Networks. Chem. Rev. 2021, 121, 5042–5092. [Google Scholar] [CrossRef]
- Hagen, R.; Salmén, L.; Stenberg, B. Effects of the type of crosslink on viscoelastic properties of natural rubber. J. Polym. Sci. Part B Polym. Phys. 1996, 34, 1997–2006. [Google Scholar] [CrossRef]
- Nielsen, L.E. Cross-Linking–Effect on Physical Properties of Polymers. J. Macromol. Sci. Part C Polym. Rev. 1969, 3, 69–103. [Google Scholar] [CrossRef]
- Youssef, M. Influence of sulfur content and curing time on the glass transition parameters in SBR vulcanizates: An ultrasonic study. Polym. Test. 2003, 22, 235–242. [Google Scholar] [CrossRef]
- Grinberg, F.; Garbarczyk, M.; Kuhn, W. Influence of the cross-link density and the filler content on segment dynamics in dry and swollen natural rubber studied by the NMR dipolar-correlation effect. J. Chem. Phys. 1999, 111, 11222–11231. [Google Scholar] [CrossRef]
- Gronski, W.; Hoffmann, U.; Simon, G.; Wutzler, A.; Straube, E. Structure and Density of Crosslinks in Natural-Rubber Vulcanizates. A Combined Analysis by NMR Spectroscopy, Mechanical Measurements, and Rubber-Elastic Theory. Rubber Chem. Technol. 1992, 65, 63–77. [Google Scholar] [CrossRef]
- Saalwächter, K.; Herrero, B.; López-Manchado, M.A. Chain Order and Cross-Link Density of Elastomers as Investigated by Proton Multiple-Quantum NMR. Macromolecules 2005, 38, 9650–9660. [Google Scholar] [CrossRef]
- Klüppel, M.; Menge, H.; Schmidt, H.; Schneider, H.; Schuster, R.H. Influence of Preparation Conditions on Network Parameters of Sulfur-Cured Natural Rubber. Macromolecules 2001, 34, 8107–8116. [Google Scholar] [CrossRef]
- Valentín, J.L.; Carretero-González, J.C.; Mora-Barrantes, I.; Chassé, W.; Saalwächter, K. Uncertainties in the Determination of Cross-Link Density by Equilibrium Swelling Experiments in Natural Rubber. Macromolecules 2008, 41, 4717–4729. [Google Scholar] [CrossRef]
- Basterra-Beroiz, B.; Rommel, R.; Kayser, F.; Westermann, S.; Valentín, J.L.; Heinrich, G. New insights into rubber network structure by a combination of experimental techniques. Rubber Chem. Technol. 2017, 90, 347–366. [Google Scholar] [CrossRef]
- Saalwächter, K. Proton multiple-quantum NMR for the study of chain dynamics and structural constraints in polymeric soft materials. Prog. Nucl. Magn. Reson. 2007, 51, 1–35. [Google Scholar] [CrossRef]
- Valentín, J.L.; Posadas, P.; Fernández-Torres, A.; Malmierca, M.A.; González, L.; Chassé, W.; Saalwächter, K. Inhomogeneities and Chain Dynamics in Diene Rubbers Vulcanized with Different Cure Systems. Macromolecules 2010, 43, 4210–4222. [Google Scholar] [CrossRef]
- Saalwächter, K. Microstructure and molecular dynamics of elastomers as studied by advanced low-resolution nuclear magnetic resonance methods. Rubber Chem. Technol. 2012, 85, 350–386. [Google Scholar] [CrossRef]
- Vieyres, A.; Pérez-Aparicio, R.; Albouy, P.-A.; Sanseau, O.; Saalwächter, K.; Long, D.R.; Sotta, P. Sulfur-Cured Natural Rubber Elastomer Networks: Correlating Cross-Link Density, Chain Orientation, and Mechanical Response by Combined Techniques. Macromolecules 2013, 46, 889–899. [Google Scholar] [CrossRef]
- Hernández, M.; Valentín, J.L.; López-Manchado, M.A.; Ezquerra, T.A. Influence of the vulcanization system on the dynamics and structure of natural rubber: Comparative study by means of broadband dielectric spectroscopy and solid-state NMR spectroscopy. Eur. Polym. J. 2015, 68, 90–103. [Google Scholar] [CrossRef]
- Schlögl, S.; Trutschel, M.-L.; Chassé, W.; Riess, G.; Saalwächter, K. Entanglement Effects in Elastomers: Macroscopic vs Microscopic Properties. Macromolecules 2014, 47, 2759–2773. [Google Scholar] [CrossRef]
- Hiranobe, C.T.; Ribeiroa, G.D.; Torres, G.B.; dos Reis, E.A.P.; Cabrera, F.C.; Job, A.E.; Paim, L.L.; dos Santos, R.J. Cross-Linked Density Determination of Natural Rubber Compounds by Different Analytical Techniques. Mater. Res. 2021, 24, 20210041. [Google Scholar] [CrossRef]
- Gabrielle, B.; Gomez, E.; Korb, J.-P. Probing Rubber Cross-Linking Generation of Industrial Polymer Networks at Nanometer Scale. J. Phys. Chem. B 2016, 120, 5581–5589. [Google Scholar] [CrossRef] [PubMed]
- Basterra-Beroiz, B.; Rommel, R.; Kayser, F.; Valentín, J.L.; Westermann, S.; Heinrich, G. Revisiting Segmental Order: A Simplified Approach for Sulfur-Cured Rubbers Considering Junction Fluctuations and Entanglements. Macromolecules 2018, 51, 2076–2088. [Google Scholar] [CrossRef]
- Hernández, M.; Carretero-González, J.; Verdejo, R.; Ezquerra, T.A.; López-Manchado, M.A. Molecular Dynamics of Natural Rubber/Layered Silicate Nanocomposites as Studied by Dielectric Relaxation Spectroscopy. Macromolecules 2010, 43, 643–651. [Google Scholar] [CrossRef] [Green Version]
- Kariyo, S.; Stapf, S. Influence of Cross-Link Density and Deformation on the NMR Relaxation Dispersion of Natural Rubber. Macromolecules 2002, 35, 9253–9255. [Google Scholar] [CrossRef]
- Kariyo, S.; Stapf, S. NMR relaxation dispersion of vulcanized natural rubber. Solid State Nucl. Magn. Reson. 2004, 25, 64–71. [Google Scholar] [CrossRef]
- Kariyo, S.; Stapf, S. Restricted Molecular Dynamics of Polymer Chains by Means of NMR Field Cycling Relaxometry. Macromol. Chem. Phys. 2005, 206, 1300–1310. [Google Scholar] [CrossRef]
- Martini, F.; Carignani, E.; Nardelli, F.; Rossi, E.; Borsacchi, S.; Cettolin, M.; Susanna, A.; Geppi, M.; Calucci, L. Glassy and Polymer Dynamics of Elastomers by 1H Field-Cycling NMR Relaxometry: Effects of Cross-Linking. Macromolecules 2020, 53, 10028–10039. [Google Scholar] [CrossRef] [PubMed]
- Nardelli, F.; Martini, F.; Carignani, E.; Rossi, E.; Borsacchi, S.; Cettolin, M.; Susanna, A.; Arimondi, M.; Giannini, L.; Geppi, M.; et al. Glassy and Polymer Dynamics of Elastomers by 1H-Field-Cycling NMR Relaxometry: Effects of Fillers. J. Phys. Chem. B 2021, 125, 4546–4554. [Google Scholar] [CrossRef]
- Martini, F.; Borsacchi, S.; Geppi, M.; Ruggeri, G.; Pucci, A. Understanding the aggregation of bis(benzoxazolyl)stilbene in PLA/PBS blends: A combined spectrofluorimetric, calorimetric and solid state NMR approach. Polym. Chem. 2014, 5, 828–835. [Google Scholar] [CrossRef]
- Borsacchi, S.; Sudhakaran, U.P.; Calucci, L.; Martini, F.; Carignani, E.; Messori, M.; Geppi, M. Rubber-Filler Interactions in Polyisoprene Filled with In Situ Generated Silica: A Solid State NMR Study. Polymers 2018, 10, 822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobani, E.; Tagliaro, I.; Geppi, M.; Giannini, L.; Leclère, P.; Martini, F.; Nguyen, T.C.; Lazzaroni, R.; Scotti, R.; Tadiello, L.; et al. Hybrid Interface in Sepiolite Rubber Nanocomposites: Role of Self-Assembled Nanostructure in Controlling Dissipative Phenomena. Nanomaterials 2019, 9, 486. [Google Scholar] [CrossRef] [Green Version]
- Redaelli, M.; D’Arienzo, M.; Brus, J.; Di Credico, B.; Geppi, M.; Giannini, L.; Matejka, L.; Martini, F.; Panattoni, F.; Spirkova, M.; et al. On the key role of SiO2@POSS hybrid filler in tailoring networking and interfaces in rubber nanocomposites. Polym. Test. 2018, 65, 429–439. [Google Scholar] [CrossRef]
- Martini, F.; Guazzelli, E.; Martinelli, E.; Borsacchi, S.; Geppi, M.; Galli, G. Molecular Dynamics of Amphiphilic Random Copolymers in the Bulk: A 1H and 19F NMR Relaxometry Study. Macromol. Chem. Phys. 2019, 220, 1900177. [Google Scholar] [CrossRef]
- Brereton, M.G. NMR transverse relaxation function calculated for constrained polymer chains: Application to entanglements and networks. Macromolecules 1990, 23, 1119–1131. [Google Scholar] [CrossRef]
- Knörgen, M.; Menge, H.; Hempel, G.; Schneider, H.; Ries, M.E. Relationship between the transverse NMR decay and the dipolar interaction in elastomers: A comparison of two models. Polymers 2002, 43, 4091–4096. [Google Scholar] [CrossRef]
- Simon, G.; Baumann, K.; Gronski, W. Mc determination and molecular dynamics in crosslinked 1,4-cis-polybutadiene: A comparison of transversal proton and deuterium NMR relaxation. Macromolecules 1992, 25, 3624–3628. [Google Scholar] [CrossRef]
- Heuert, U.; Knörgen, M.; Menge, H.; Scheler, G.; Schneider, H. New aspects of transversal 1H-NMR relaxation in natural rubber vulcanizates. Polym. Bull. 1996, 37, 489–496. [Google Scholar] [CrossRef]
- Saalwächter, K. Artifacts in Transverse Proton NMR Relaxation Studies of Elastomers. Macromolecules 2005, 38, 1508–1512. [Google Scholar] [CrossRef]
- Saalwächter, K.; Ziegler, P.; Spyckerelle, O.; Haidar, B.; Vidal, A.; Sommer, J.-U. 1H multiple-quantum nuclear magnetic resonance investigations of molecular order distributions in poly(dimethylsiloxane) networks: Evidence for a linear mixing law in bimodal systems. J. Chem. Phys. 2003, 119, 3468–3482. [Google Scholar] [CrossRef] [Green Version]
- Suh, B.; Borsa, F.; Torgeson, D. Use of an Alternating-Phase CPMG Sequence to Avoid Spin-Locking Effects in T2 Measurements in Solids. J. Magn. Reson. Ser. A 1994, 110, 58–61. [Google Scholar] [CrossRef]
- Anoardo, E.; Galli, G.; Ferrante, G. Fast-field-cycling NMR: Applications and instrumentation. Appl. Magn. Reson. 2001, 20, 365–404. [Google Scholar] [CrossRef]
- Kimmich, R.; Anoardo, E. Field-cycling NMR relaxometry. Prog. Nucl. Magn. Reson. 2004, 44, 257–320. [Google Scholar] [CrossRef]
- Saalwächter, K.; Klüppel, M.; Luo, H.; Schneider, H. Chain order in filled SBR elastomers: A proton multiple-quantum NMR study. Appl. Magn. Reson. 2004, 27, 401–417. [Google Scholar] [CrossRef]
- Sun, X.; Isayev, A.I.; Joshi, T.R.; Von Meerwall, E. Molecular Mobility of Unfilled and Carbon-Black-Filled Isoprene Rubber: Proton NMR Transverse Relaxation and Diffusion. Rubber Chem. Technol. 2007, 80, 854–872. [Google Scholar] [CrossRef]
- Asano, A. NMR Relaxation Studies of Elastomers. In Annual Reports on NMR Spectroscopy Vol. 86; Graham, A.W., Ed.; Academic Press (Elsevier): Waltham, MA, USA, 2015; Chapter 1; pp. 1–72. [Google Scholar] [CrossRef]
- Massey, J.L.; Parr, J.C.; Wagler, A.T.; von Meerwall, E.; Hong, C.K.; Isayev, I.A. Ultrasound devulcanization of unfilled natural rubber networks, studied via component molecular mobility. Polym. Int. 2007, 56, 860–869. [Google Scholar] [CrossRef]
- Rössler, E.A.; Hofmann, M.; Fatkullin, N. Application of Field- Cycling 1H NMR Relaxometry to the Study of Translational and Rotational Dynamics in Liquids and Polymers. In Field-Cycling NMR Relaxometry: Instrumentation, Model Theories and Applications; Kimmich, R., Ed.; The Royal Society of Chemistry: Cambridge, UK, 2019; Chapter 8; pp. 181–206. [Google Scholar] [CrossRef]
- Ferry, J.D. Viscoelasticity Properties of Polymers; John Wiley & Sons Ltd.: New York, NY, USA, 1980. [Google Scholar]
- Ding, Y.; Sokolov, A.P. Breakdown of Time−Temperature Superposition Principle and Universality of Chain Dynamics in Polymers. Macromolecules 2006, 39, 3322–3326. [Google Scholar] [CrossRef]
- Blochowicz, T.; Gainaru, C.; Medick, P.; Tschirwitz, C.; Rössler, E.A. The dynamic susceptibility in glass forming molecular liquids: The search for universal relaxation patterns II. J. Chem. Phys. 2006, 124, 134503. [Google Scholar] [CrossRef] [PubMed]
- Loo, C. High temperature vulcanization of elastomers: 3. Network structure of efficiently vulcanized natural rubber mixes. Polymer 1974, 15, 729–737. [Google Scholar] [CrossRef]
- Ueberreiter, K.; Kanig, G. Second-Order Transitions and Mesh Distribution Functions of Cross-Linked Polystyrenes. J. Chem. Phys. 1950, 18, 399–406. [Google Scholar] [CrossRef]
- Chang, S.-S. Effect of curing history on ultimate glass transition temperature and network structure of crosslinking polymers. Polymer 1992, 33, 4768–4778. [Google Scholar] [CrossRef]
- Chassé, W.; Lang, M.; Sommer, J.-U.; Saalwächter, K. Cross-Link Density Estimation of PDMS Networks with Precise Consideration of Networks Defects. Macromolecules 2012, 45, 899–912. [Google Scholar] [CrossRef]
- Lang, M.; Sommer, J.-U. Analysis of Entanglement Length and Segmental Order Parameter in Polymer Networks. Phys. Rev. Lett. 2010, 104, 177801. [Google Scholar] [CrossRef]
- Kimmich, R.; Fatkullin, N. Polymer Chain Dynamics and NMR. In NMR-3D Analysis—Photopolymerization. Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 2004; Volume 170, pp. 1–113. [Google Scholar] [CrossRef]
- Stapf, S.; Lozovoi, A. Field-Cycling Relaxometry of Polymers. In Field-Cycling NMR Relaxometry: Instrumentation, Model Theories and Applications; Kimmich, R., Ed.; The Royal Society of Chemistry: Cambridge, UK, 2019; Chapter 13; pp. 322–357. [Google Scholar]
- Kimmich, R.; Gille, K.; Fatkullin, N.; Seitter, R.; Hafner, S.; Müller, M. Field-cycling nuclear magnetic resonance relaxometry of thermoreversible polybutadiene networks. J. Chem. Phys. 1997, 107, 5973–5978. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nardelli, F.; Calucci, L.; Carignani, E.; Borsacchi, S.; Cettolin, M.; Arimondi, M.; Giannini, L.; Geppi, M.; Martini, F. Influence of Sulfur-Curing Conditions on the Dynamics and Crosslinking of Rubber Networks: A Time-Domain NMR Study. Polymers 2022, 14, 767. https://doi.org/10.3390/polym14040767
Nardelli F, Calucci L, Carignani E, Borsacchi S, Cettolin M, Arimondi M, Giannini L, Geppi M, Martini F. Influence of Sulfur-Curing Conditions on the Dynamics and Crosslinking of Rubber Networks: A Time-Domain NMR Study. Polymers. 2022; 14(4):767. https://doi.org/10.3390/polym14040767
Chicago/Turabian StyleNardelli, Francesca, Lucia Calucci, Elisa Carignani, Silvia Borsacchi, Mattia Cettolin, Marco Arimondi, Luca Giannini, Marco Geppi, and Francesca Martini. 2022. "Influence of Sulfur-Curing Conditions on the Dynamics and Crosslinking of Rubber Networks: A Time-Domain NMR Study" Polymers 14, no. 4: 767. https://doi.org/10.3390/polym14040767
APA StyleNardelli, F., Calucci, L., Carignani, E., Borsacchi, S., Cettolin, M., Arimondi, M., Giannini, L., Geppi, M., & Martini, F. (2022). Influence of Sulfur-Curing Conditions on the Dynamics and Crosslinking of Rubber Networks: A Time-Domain NMR Study. Polymers, 14(4), 767. https://doi.org/10.3390/polym14040767