
Detecting Micro- and Nanoplastics Released from Food Packaging: Challenges and Analytical Strategies

 , , , and
, , , and
Abstract
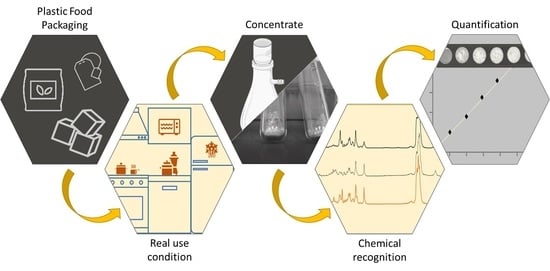
:
1. Introduction
2. Materials and Methods
2.1. Measures to Avoid Contamination
2.2. Sample Preparation
2.3. Identification Methods
2.4. Quantification Method
2.5. Statistical Analysis
2.6. Morpholigical Study
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Microplastics in Drinking-Water; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- O’Neill, S.M.; Lawler, J. Knowledge gaps on micro and nanoplastics and human health: A critical review. Case Stud. Chem. Environ. Eng. 2021, 3, 100091. [Google Scholar] [CrossRef]
- Burns, E.E.; Boxall, A.B.A. Microplastics in the aquatic environment: Evidence for or against adverse impacts and major knowledge gaps. Environ. Toxicol. Chem. 2018, 37, 2776–2796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.Q. Occurrence of microplastics and its pollution in the environment: A review. Sustain. Prod. Consum. 2018, 13, 16–23. [Google Scholar] [CrossRef]
- Ivleva, N.P.; Wiesheu, A.C.; Niessner, R. Microplastic in Aquatic Ecosystems. Angew. Chem. Int. Ed. 2017, 56, 1720–1739. [Google Scholar] [CrossRef]
- Boyle, K.; Örmeci, B. Microplastics and nanoplastics in the freshwater and terrestrial environment: A review. Water 2020, 12, 2633. [Google Scholar] [CrossRef]
- de Souza Machado, A.A.; Kloas, W.; Zarfl, C.; Hempel, S.; Rillig, M.C. Microplastics as an emerging threat to terrestrial ecosystems. Glob. Chang. Biol. 2018, 24, 1405–1416. [Google Scholar] [CrossRef] [Green Version]
- da Costa, J.P. Micro- and nanoplastics in the environment: Research and policymaking. Curr. Opin. Environ. Sci. Health 2018, 1, 12–16. [Google Scholar] [CrossRef]
- Pico, Y.; Alfarhan, A.; Barcelo, D. Nano- and microplastic analysis: Focus on their occurrence in freshwater ecosystems and remediation technologies. TrAC Trends Anal. Chem. 2019, 113, 409–425. [Google Scholar] [CrossRef]
- Ter Halle, A.; Jeanneau, L.; Martignac, M.; Jardé, E.; Pedrono, B.; Brach, L.; Gigault, J. Nanoplastic in the North Atlantic Subtropical Gyre. Environ. Sci. Technol. 2017, 51, 13689–13697. [Google Scholar] [CrossRef]
- Materić, D.; Kasper-Giebl, A.; Kau, D.; Anten, M.; Greilinger, M.; Ludewig, E.; Van Sebille, E.; Röckmann, T.; Holzinger, R. Micro-and Nanoplastics in Alpine Snow: A New Method for Chemical Identification and (Semi)Quantification in the Nanogram Range. Environ. Sci. Technol. 2020, 54, 2353–2359. [Google Scholar] [CrossRef]
- Wahl, A.; Le Juge, C.; Davranche, M.; El Hadri, H.; Grassl, B.; Reynaud, S.; Gigault, J. Nanoplastic occurrence in a soil amended with plastic debris. Chemosphere 2021, 262, 127784. [Google Scholar] [CrossRef] [PubMed]
- Valsesia, A.; Quarato, M.; Ponti, J.; Fumagalli, F.; Gilliland, D.; Colpo, P. Combining microcavity size selection with Raman microscopy for the characterization of Nanoplastics in complex matrices. Sci. Rep. 2021, 11, 362. [Google Scholar] [CrossRef] [PubMed]
- Alexy, P.; Anklam, E.; Emans, T.; Furfari, A.; Galgani, F.; Hanke, G.; Koelmans, A.; Pant, R.; Saveyn, H.; Sokull Kluettgen, B. Managing the analytical challenges related to micro- and nanoplastics in the environment and food: Filling the knowledge gaps. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2020, 37, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ECHA. Annex XV Restriction Report 2019; European Chemicals Agency: Helsinki, Finland, 2019. [Google Scholar]
- ECHA. Committee for Risk Assessment (RAC) Committee for Socio-Economic Analysis (SEAC) Background Document; European Chemicals Agency: Helsinki, Finland, 2020; Volume 1. [Google Scholar]
- Kentin, E.; Kaarto, H. An EU ban on microplastics in cosmetic products and the right to regulate. Rev. Eur. Comp. Int. Environ. Law 2018, 27, 254–266. [Google Scholar] [CrossRef] [Green Version]
- Hann, S.; Scholes, R.; Molteno, S.; Hilton, M.; Favoino, E.; Geest Jakobsen, L. Relevance of Biodegradable and Compostable Consumer Plastic Products and Packaging in a Circular Economy; Publications Office of the European Union: Luxembourg, 2020. [Google Scholar]
- Hann, S.; Fletcher, E.; Molteno, S.; Sherrington, C.; Elliott, L.; Kong, M.; Koite, A.; Sastre, S.; Martinez, V. Relevance of Conventional and Biodegradable Plastics in Agriculture; Publications Office of the European Union: Luxembourg, 2021. [Google Scholar]
- Dordevic, D.; Necasova, L.; Antonic, B.; Jancikova, S. Plastic Cutlery Alternative: Case Study with Biodegradable Spoons. Food 2021, 10, 1612. [Google Scholar] [CrossRef]
- EFSA Panel on Contaminants in the Food Chain Statement on the presence of microplastics and nanoplastics in food, with particular focus on seafood. EFSA J. 2016, 14, 4501–4530. [CrossRef] [Green Version]
- Hantoro, I.; Löhr, A.J.; Van Belleghem, F.G.A.J.; Widianarko, B.; Ragas, A.M.J. Microplastics in coastal areas and seafood: Implications for food safety. Food Addit. Contam.-Part A Chem. Anal. Control Expo. Risk Assess. 2019, 36, 674–711. [Google Scholar] [CrossRef]
- Toussaint, B.; Raffael, B.; Angers-Loustau, A.; Gilliland, D.; Kestens, V.; Petrillo, M.; Rio-Echevarria, I.M.; Van den Eede, G. Review of micro- and nanoplastic contamination in the food chain. Food Addit. Contam. Part A Chem. Anal. Control Expo. Risk Assess. 2019, 36, 639–673. [Google Scholar] [CrossRef]
- Van Raamsdonk, L.W.D.; Van Der Zande, M.; Koelmans, A.A.; Ron, L.A.P.; Hoogenboom, R.J.B.P.; Groot, M.J.; Peijnenburg, A.A.C.M.; Weesepoel, Y.J.A. Current Insights into Monitoring, Bioaccumulation and Potential Health Effects of Microplastics Present in the Food Chain. Foods 2020, 9, 72. [Google Scholar] [CrossRef] [Green Version]
- Shruti, V.C.; Pérez-Guevara, F.; Elizalde-Martínez, I.; Kutralam-Muniasamy, G. Toward a unified framework for investigating micro(nano)plastics in packaged beverages intended for human consumption. Environ. Pollut. 2021, 268, 115811. [Google Scholar] [CrossRef]
- Llorca, M.; Álvarez-Muñoz, D.; Ábalos, M.; Rodríguez-Mozaz, S.; Santos, L.H.M.L.M.; León, V.M.; Campillo, J.A.; Martínez-Gómez, C.; Abad, E.; Farré, M. Microplastics in Mediterranean coastal area: Toxicity and impact for the environment and human health. Trends Environ. Anal. Chem. 2020, 27, e00090. [Google Scholar] [CrossRef]
- Karbalaei, S.; Hanachi, P.; Walker, T.R.; Cole, M. Occurrence, sources, human health impacts and mitigation of microplastic pollution. Environ. Sci. Pollut. Res. 2018, 25, 36046–36063. [Google Scholar] [CrossRef] [PubMed]
- Mason, S.A.; Welch, V.G.; Neratko, J. Synthetic Polymer Contamination in Bottled Water. Front. Chem. 2018, 6, 407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oßmann, B.E.; Sarau, G.; Holtmannspötter, H.; Pischetsrieder, M.; Christiansen, S.H.; Dicke, W. Small-sized microplastics and pigmented particles in bottled mineral water. Water Res. 2018, 141, 307–316. [Google Scholar] [CrossRef]
- Schymanski, D.; Goldbeck, C.; Humpf, H.U.; Fürst, P. Analysis of microplastics in water by micro-Raman spectroscopy: Release of plastic particles from different packaging into mineral water. Water Res. 2018, 129, 154–162. [Google Scholar] [CrossRef]
- Zhang, Q.; Xu, E.G.; Li, J.; Chen, Q.; Ma, L.; Zeng, E.Y.; Shi, H. A Review of Microplastics in Table Salt, Drinking Water, and Air: Direct Human Exposure. Environ. Sci. Technol. 2020, 54, 3740–3751. [Google Scholar] [CrossRef]
- Winkler, A.; Santo, N.; Ortenzi, M.A.; Bolzoni, E.; Bacchetta, R.; Tremolada, P. Does mechanical stress cause microplastic release from plastic water bottles? Water Res. 2019, 166, 115082. [Google Scholar] [CrossRef]
- Danopoulos, E.; Twiddy, M.; Rotchell, J.M. Microplastic contamination of drinking water: A systematic review. PLoS ONE 2020, 15, e0236838. [Google Scholar] [CrossRef]
- Mortensen, N.P.; Fennell, T.R.; Johnson, L.M. Unintended human ingestion of nanoplastics and small microplastics through drinking water, beverages, and food sources. NanoImpact 2021, 21, 100302. [Google Scholar] [CrossRef]
- Kedzierski, M.; Lechat, B.; Sire, O.; Le Maguer, G.; Le Tilly, V.; Bruzaud, S. Microplastic contamination of packaged meat: Occurrence and associated risks. Food Packag. Shelf Life 2020, 24, 100489. [Google Scholar] [CrossRef]
- Fadare, O.O.; Wan, B.; Guo, L.H.; Zhao, L. Microplastics from consumer plastic food containers: Are we consuming it? Chemosphere 2020, 253, 126787. [Google Scholar] [CrossRef]
- Du, F.; Cai, H.; Zhang, Q.; Chen, Q.; Shi, H. Microplastics in take-out food containers. J. Hazard. Mater. 2020, 399, 122969. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, L.M.; Xu, E.G.; Larsson, H.C.E.; Tahara, R.; Maisuria, V.B.; Tufenkji, N. Plastic Teabags Release Billions of Microparticles and Nanoparticles into Tea. Environ. Sci. Technol. 2019, 53, 12300–12310. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Shi, Y.; Yang, L.; Xiao, L.; Kehoe, D.K.; Gun, Y.K.; Boland, J.J.; Wang, J.J. Microplastic release from the degradation of polypropylene feeding bottles during infant formula preparation. Nat. Food 2020, 1, 746–754. [Google Scholar] [CrossRef]
- Dessì, C.; Okoffo, E.D.; O’Brien, J.W.; Gallen, M.; Samanipour, S.; Kaserzon, S.; Rauert, C.; Wang, X.; Thomas, K.V. Plastics contamination of store-bought rice. J. Hazard. Mater. 2021, 416, 125778. [Google Scholar] [CrossRef]
- Ranjan, V.P.; Joseph, A.; Goel, S. Microplastics and other harmful substances released from disposable paper cups into hot water. J. Hazard. Mater. 2021, 404, 124118. [Google Scholar] [CrossRef]
- Sobhani, Z.; Lei, Y.; Tang, Y.; Wu, L.; Zhang, X.; Naidu, R.; Megharaj, M.; Fang, C. Microplastics generated when opening plastic packaging. Sci. Rep. 2020, 10, 4841. [Google Scholar] [CrossRef] [Green Version]
- European Union. Council directive of 19 December 1985 laying down the list of simulants to be used for testing migration of constituents of plastic materials and articles intended to come into contact with foodstuffs. Off. J. Eur. Communities 1985, 372, 14–21. [Google Scholar]
- Jakubowska, N.; Beldi, G.; Robouch, P.; Hoekstra, E. Testing Conditions for Kitchenware Articles in Contact with Foodstuffs: Plastics and Metals; Technical Report JRC121622; European Commission: Ispra, Italy, 2020. [Google Scholar]
- Reed, C.R.; Loscombe, G.D. The Use of Microparticulate Guard Columns in Reverse-Phase High-Performance Liquid Chromatography. Chromatographia 1982, 15, 15–17. [Google Scholar] [CrossRef]
- Majors, R. Current Trends in HPLC Column Technology. LCGC N. Am. 2012, 30, 20–34. [Google Scholar]
- Seghers, J.; Stefaniak, E.A.; La Spina, R.; Cella, C.; Mehn, D.; Gilliland, D.; Held, A.; Jacobsson, U.; Emteborg, H. Preparation of a reference material for microplastics in water—evaluation of homogeneity. Anal. Bioanal. Chem. 2022, 414, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Schwaferts, C.; Niessner, R.; Elsner, M.; Ivleva, N.P. Methods for the analysis of submicrometer- and nanoplastic particles in the environment. TrAC Trends Anal. Chem. 2019, 112, 52–65. [Google Scholar] [CrossRef]
- Cowger, W.; Gray, A.; Hapich, H.; Rochman, C.; Lynch, J.; Primpke, S.; Munno, K.; De Frond, H.O.H. Open Specy. Available online: www.openspecy.org (accessed on 17 March 2022).
- Cowger, W.; Steinmetz, Z.; Gray, A.; Munno, K.; Lynch, J.; Hapich, H.; Primpke, S.; De Frond, H.; Rochman, C.; Herodotou, O. Microplastic Spectral Classification Needs an Open Source Community: Open Specy to the Rescue! Anal. Chem. 2021, 93, 7543–7548. [Google Scholar] [CrossRef] [PubMed]
- Pigments Checker, v.5. Available online: https://chsopensource.org/pigments-checker/ (accessed on 17 March 2022).
- Shard, A.G. Practical guides for X-ray photoelectron spectroscopy: Quantitative XPS. J. Vac. Sci. Technol. A 2020, 38, 041201. [Google Scholar] [CrossRef]
- Baer, D.R.; Artyushkova, K.; Cohen, H.; Easton, C.D.; Engelhard, M.; Gengenbach, T.R.; Greczynski, G.; Mack, P.; Morgan, D.J.; Roberts, A. XPS guide: Charge neutralization and binding energy referencing for insulating samples. J. Vac. Sci. Technol. A 2020, 38, 031204. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J. Statistical Power Analysis for the Behavioral Sciences, 2nd ed.; Lawrence Erlbaum Associate: Mahwah, NJ, USA, 1988; ISBN 0-8058-0283-5. [Google Scholar]
- Lenth, R.V. Some Practical Guidelines for Effective Sample Size Determination. Am. Stat. 2001, 55, 187–193. [Google Scholar] [CrossRef]
- Sawilowsky, S.S. New Effect Size Rules of Thumb. J. Mod. Appl. Stat. Methods 2009, 8, 26. [Google Scholar] [CrossRef]
- Valsesia, A.; Desmet, C.; Ojea-Jiménez, I.; Oddo, A.; Capomaccio, R.; Rossi, F.; Colpo, P. Direct quantification of nanoparticle surface hydrophobicity. Commun. Chem. 2018, 1, 53. [Google Scholar] [CrossRef]
- Desmet, C.; Valsesia, A.; Oddo, A.; Ceccone, G.; Spampinato, V.; Rossi, F.; Colpo, P. Characterisation of nanomaterial hydrophobicity using engineered surfaces. J. Nanopart. Res. 2017, 19, 117. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Basantes, M.F.; Conesa, J.A.; Fullana, A. Microplastics in honey, beer, milk and refreshments in Ecuador as emerging contaminants. Sustainability 2020, 12, 5514. [Google Scholar] [CrossRef]
- Magrì, D.; Sánchez-Moreno, P.; Caputo, G.; Gatto, F.; Veronesi, M.; Bardi, G.; Catelani, T.; Guarnieri, D.; Athanassiou, A.; Pompa, P.P.; et al. Laser ablation as a versatile tool to mimic polyethylene terephthalate nanoplastic pollutants: Characterization and toxicology assessment. ACS Nano 2018, 12, 7690–7700. [Google Scholar] [CrossRef] [PubMed]
- Electronic Code of Federal Regulations PART 178—Indirect Food Additives: Adjuvants, Production Aids, and Sanitizers. Available online: https://www.ecfr.gov/cgi-bin/text-idx?SID=a1ef4942d858446924d35877fa61effc&mc=true&node=pt21.3.178&rgn=div5 (accessed on 17 March 2022).
- European Union. Commission Regulation (EU) No 10/2011 of 14 January 2011 on plastic materials and articles intended to come into contact with food (Text with EEA relevance). Off. J. Eur. Union 2011, 45, 1–89. [Google Scholar]
- Osticioli, I.; Mendes, N.F.C.; Nevin, A.; Gil, F.P.S.C.; Becucci, M.; Castellucci, E. Analysis of natural and artificial ultramarine blue pigments using laser induced breakdown and pulsed Raman spectroscopy, statistical analysis and light microscopy. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2009, 73, 525–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, D.; Chen, S.; Liu, Q. Review of Fluorescence Suppression Techniques in Raman Spectroscopy. Appl. Spectrosc. Rev. 2015, 50, 387–406. [Google Scholar] [CrossRef]
- Yakubovskaya, E.; Zaliznyak, T.; Martínez Martínez, J.; Taylor, G.T. Tear Down the Fluorescent Curtain: A New Fluorescence Suppression Method for Raman Microspectroscopic Analyses. Sci. Rep. 2019, 9, 15785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valsesia, A.; Parot, J.; Ponti, J.; Mehn, D.; Marino, R.; Melillo, D.; Muramoto, S.; Verkouteren, M.; Hackley, V.A.; Colpo, P. Detection, counting and characterization of nanoplastics in marine bioindicators: A proof of principle study. Microplast. Nanoplast. 2021, 1, 5. [Google Scholar] [CrossRef]
- Ivleva, N.P. Chemical Analysis of Microplastics and Nanoplastics: Challenges, Advanced Methods, and Perspectives. Chem. Rev. 2021, 121, 11886–11936. [Google Scholar] [CrossRef]
- Li, J.; Liu, H.; Paul Chen, J. Microplastics in freshwater systems: A review on occurrence, environmental effects, and methods for microplastics detection. Water Res. 2018, 137, 362–374. [Google Scholar] [CrossRef]
- Shim, W.J.; Hong, S.H.; Eo, S.E. Identification methods in microplastic analysis: A review. Anal. Methods 2017, 9, 1384–1391. [Google Scholar] [CrossRef]
- Busse, K.; Ebner, I.; Humpf, H.U.; Ivleva, N.; Kaeppler, A.; Oßmann, B.E.; Schymanski, D. Comment on “plastic Teabags Release Billions of Microparticles and Nanoparticles into Tea”. Environ. Sci. Technol. 2020, 54, 14134–14135. [Google Scholar] [CrossRef]
- Schuck, P.; Perugini, M.A.; Gonzales, N.R.; Hewlett, G.J.; Schubert, D. Size-distribution analysis of proteins by analytical ultracentrifugation: Strategies and application to model systems. Biophys. J. 2002, 82, 1096–1111. [Google Scholar] [CrossRef] [Green Version]
- Mehn, D.; Capomaccio, R.; Gioria, S.; Gilliland, D.; Calzolai, L. Analytical ultracentrifugation for measuring drug distribution of doxorubicin loaded liposomes in human serum. J. Nanopart. Res. 2020, 22, 158. [Google Scholar] [CrossRef]
- Mehn, D.; Iavicoli, P.; Cabaleiro, N.; Borgos, S.E.; Caputo, F.; Geiss, O.; Calzolai, L.; Rossi, F.; Gilliland, D. Analytical ultracentrifugation for analysis of doxorubicin loaded liposomes. Int. J. Pharm. 2017, 523, 320–326. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Filter Pore Size µm | mg of Nylon 6 in Hot-Filtered Leachate | mg of Nylon 6 in Cold-Filtered Leachate | Cohen’s d | p Value (Two Sample t-Test) |
|---|---|---|---|---|
| 0.02 | 1.13 ± 0.05 | 0.71 ± 0.10 | 5.3 | 0.003 |
| 0.2 | 0.60 ± 0.10 | 0.84 ± 0.14 | 1.9 | 0.080 |
| 0.4 | 0.91 ± 0.14 | 0.93 ± 0.03 | 0.2 | NA 1 |
| 2.0 | 0.92 ± 0.08 | 1.03 ± 0.10 | 1.2 | 0.271 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cella, C.; La Spina, R.; Mehn, D.; Fumagalli, F.; Ceccone, G.; Valsesia, A.; Gilliland, D. Detecting Micro- and Nanoplastics Released from Food Packaging: Challenges and Analytical Strategies. Polymers 2022, 14, 1238. https://doi.org/10.3390/polym14061238
Cella C, La Spina R, Mehn D, Fumagalli F, Ceccone G, Valsesia A, Gilliland D. Detecting Micro- and Nanoplastics Released from Food Packaging: Challenges and Analytical Strategies. Polymers. 2022; 14(6):1238. https://doi.org/10.3390/polym14061238
Chicago/Turabian StyleCella, Claudia, Rita La Spina, Dora Mehn, Francesco Fumagalli, Giacomo Ceccone, Andrea Valsesia, and Douglas Gilliland. 2022. "Detecting Micro- and Nanoplastics Released from Food Packaging: Challenges and Analytical Strategies" Polymers 14, no. 6: 1238. https://doi.org/10.3390/polym14061238
APA StyleCella, C., La Spina, R., Mehn, D., Fumagalli, F., Ceccone, G., Valsesia, A., & Gilliland, D. (2022). Detecting Micro- and Nanoplastics Released from Food Packaging: Challenges and Analytical Strategies. Polymers, 14(6), 1238. https://doi.org/10.3390/polym14061238