
Effects of Alkyl Ester Chain Length on the Toughness of PolyAcrylate-Based Network Materials


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Polymer Networks
2.3. Characterization
3. Results and Discussion
3.1. Preparation of Network Materials
3.2. Thermal Properties of Polymer Materials
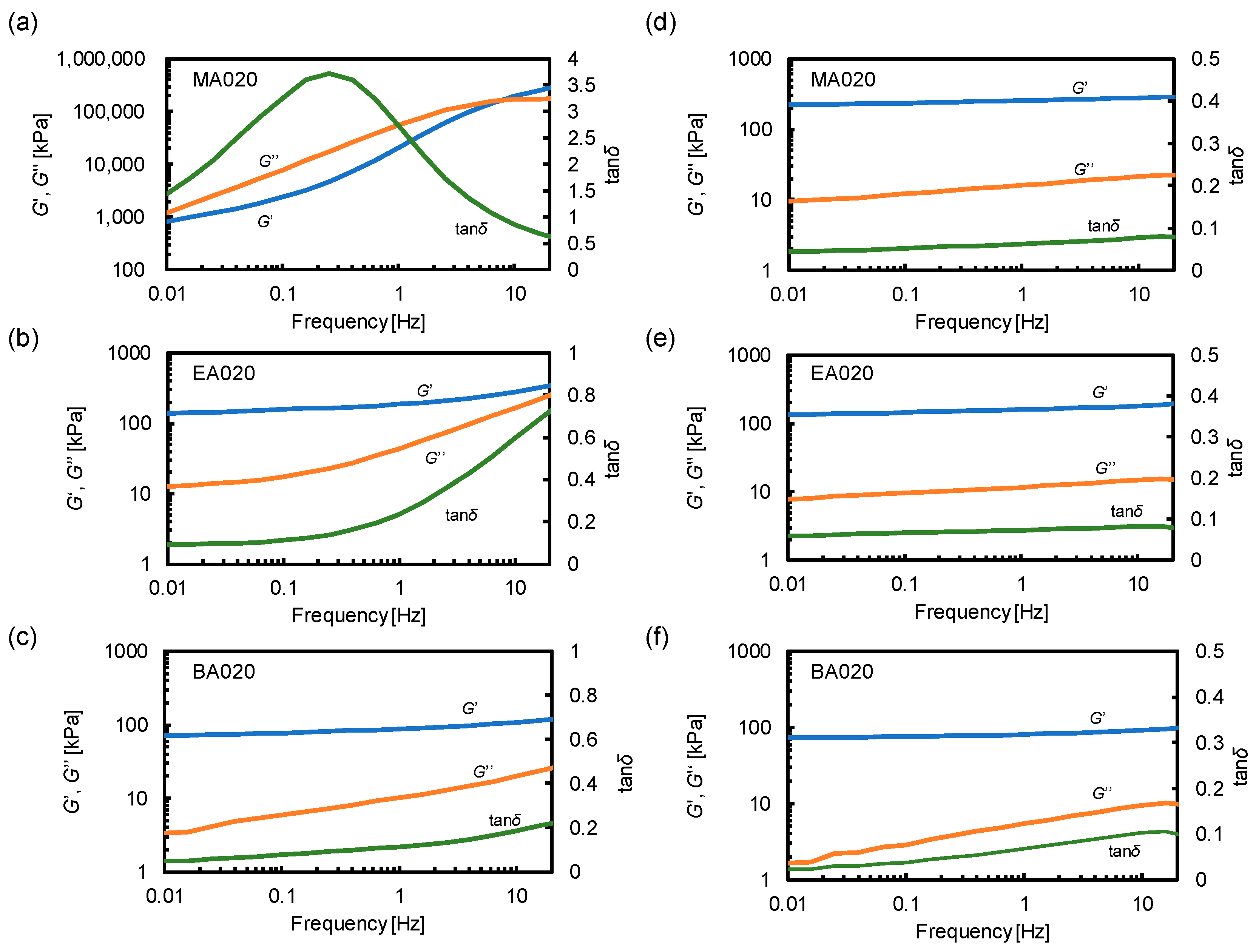
3.3. Viscoelastic Properties of Polymer Materials
3.4. Tensile Testing to Reveal the Toughness of the Network Materials
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zheng, K.; Zhang, Y.; Li, B.; Granick, S. Phosphorescent Extensophores Expose Elastic Nonuniformity in Polymer Networks. Nat. Commun. 2023, 14, 537. [Google Scholar] [CrossRef] [PubMed]
- Kannurpatti, A.R.; Anseth, J.W.; Bowman, C.N. A Study of the Evolution of Mechanical Properties and Structural Heterogeneity of Polymer Networks Formed by Photopolymerizations of Multifunctional (Meth)Acrylates. Polymer 1998, 39, 2507–2513. [Google Scholar] [CrossRef]
- Bandzierz, K.; Reuvekamp, L.; Dryzek, J.; Dierkes, W.; Blume, A.; Bielinski, D. Influence of Network Structure on Glass Transition Temperature of Elastomers. Materials 2016, 9, 607. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, W.; Liu, Z.; Xue, Y.; Lei, X.; Gong, G.; Zhang, Q. Towards a Tough Reprocessable and Self-Healable Acrylonitrile-Butadiene Rubber Based on Strong Hydrogen Bonding Interactions. J. Mater. Chem. C 2021, 9, 6241–6250. [Google Scholar] [CrossRef]
- Ferry, J.D.; Mancke, R.G.; Maekawa, E.; Oyanagi, Y.; Dickie, R.A. Dynamic Mechanical Properties of Cross-Linked Rubbers. I. Effects of Cross-Link Spacing in Natural Rubber. J. Phys. Chem. 1964, 68, 3414–3418. [Google Scholar] [CrossRef]
- Osaka, N.; Kato, M.; Saito, H. Mechanical Properties and Network Structure of Phenol Resin Crosslinked Hydrogenated Acrylonitrile-Butadiene Rubber. J. Appl. Polym. Sci. 2013, 129, 3396–3403. [Google Scholar] [CrossRef]
- Bendre, A.M.; Inamdar, S.R.; Parulekar, S.J. Multi-Equation Bifurcation Analysis of a Free Radical Polymerization in a CSTR. Mater. Today Proc. 2023, 72, 242–249. [Google Scholar] [CrossRef]
- Gao, H.; Li, W.; Matyjaszewski, K. Synthesis of Polyacrylate Networks by ATRP: Parameters Influencing Experimental Gel Points. Macromolecules 2008, 41, 2335–2340. [Google Scholar] [CrossRef]
- Braunecker, W.A.; Matyjaszewski, K. Controlled/Living Radical Polymerization: Features, Developments, and Perspectives. Prog. Polym. Sci. 2007, 32, 93–146. [Google Scholar] [CrossRef]
- Tan, M.; Hu, Z.; Dai, Y.; Peng, Y.; Zhou, Y.; Shi, Y.; Li, Y.; Chen, Y. A Simple Mechanochromic Mechanophore Based on Aminothiomaleimide. ACS Macro Lett. 2021, 10, 1423–1428. [Google Scholar] [CrossRef]
- Kury, M.; Ehrmann, K.; Gorsche, C.; Dorfinger, P.; Koch, T.; Stampfl, J.; Liska, R. Regulated Acrylate Networks as Tough Photocurable Materials for Additive Manufacturing. Polym. Int. 2022, 71, 897–905. [Google Scholar] [CrossRef]
- Ide, N.; Fukuda, T. Nitroxide-Controlled Free-Radical Copolymerization of Vinyl and Divinyl Monomers. 2. Gelation. Macromolecules 1999, 32, 95–99. [Google Scholar] [CrossRef]
- Russell, G.M.; Kaneko, T.; Ishino, S.; Masai, H.; Terao, J. Transient Photodegradability of Photostable Gel Induced by Simultaneous Treatment with Acid and UV–Light for Phototuning of Optically Functional Materials. Adv. Funct. Mater. 2022, 32, 2205855. [Google Scholar] [CrossRef]
- Corsaro, C.; Neri, G.; Santoro, A.; Fazio, E. Acrylate and Methacrylate Polymers’ Applications: Second Life with Inexpensive and Sustainable Recycling Approaches. Materials 2022, 15, 282. [Google Scholar] [CrossRef]
- Moretti, G.; Sarina, L.; Agostini, L.; Vertechy, R.; Berselli, G.; Fontana, M. Styrenic-Rubber Dielectric Elastomer Actuator with Inherent Stiffness Compensation. Actuators 2020, 9, 44. [Google Scholar] [CrossRef]
- Chen, Y.; Agostini, L.; Moretti, G.; Fontana, M.; Vertechy, R. Dielectric Elastomer Materials for Large-Strain Actuation and Energy Harvesting: A Comparison between Styrenic Rubber, Natural Rubber and Acrylic Elastomer. Smart Mater. Struct. 2019, 28, 114001. [Google Scholar] [CrossRef]
- Jin, F.L.; Lu, S.L.; Song, Z.B.; Pang, J.X.; Zhang, L.; De Sun, J.; Cai, X.P. Effect of Rubber Contents on Brittle-Tough Transition in Acrylonitrile-Butadiene-Styrene Blends. Mater. Sci. Eng. A 2010, 527, 3438–3441. [Google Scholar] [CrossRef]
- Beharaj, A.; Ekladious, I.; Grinstaff, M.W. Poly(Alkyl Glycidate Carbonate)s as Degradable Pressure-Sensitive Adhesives. Angew. Chem. Int. Ed. 2019, 58, 1407–1411. [Google Scholar] [CrossRef]
- Inui, T.; Sato, E.; Matsumoto, A. High-Molecular-Weight Polar Acrylate Block Copolymers as High-Performance Dismantlable Adhesive Materials in Response to Photoirradiation and Postbaking. RSC Adv. 2014, 4, 24719–24728. [Google Scholar] [CrossRef]
- Sakdapipanich, J.; Thananusont, N.; Pukkate, N. Synthesis of Acrylate Polymers by a Novel Emulsion Polymerization for Adhesive Applications. J. Appl. Polym. Sci. 2006, 100, 413–421. [Google Scholar] [CrossRef]
- Qin, X.; Li, X.; Wang, W.; Li, Q.; Wang, T.; Cao, L.; Piao, J.; Chen, S. Synthesis of Poly Ethyl Acrylate Interpenetrated Poly Methyl Acrylate Elastomer Coating and Its Corrosion Resistance Properties. Mater. Lett. 2022, 314, 131880. [Google Scholar] [CrossRef]
- Ducrot, E.; Creton, C. Characterizing Large Strain Elasticity of Brittle Elastomeric Networks by Embedding Them in a Soft Extensible Matrix. Adv. Funct. Mater. 2016, 26, 2482–2492. [Google Scholar] [CrossRef]
- Smith, K.E.; Parks, S.S.; Hyjek, M.A.; Downey, S.E.; Gall, K. The Effect of the Glass Transition Temperature on the Toughness of Photopolymerizable (Meth)Acrylate Networks under Physiological Conditions. Polymer 2009, 50, 5112–5123. [Google Scholar] [CrossRef] [PubMed]
- Pu, Z.; Mark, J.E.; Jethmalani, J.M.; Ford, W.T. Effects of Dispersion and Aggregation of Silica in the Reinforcement of Poly(Methyl Acrylate) Elastomers. Chem. Mater. 1997, 9, 2442–2447. [Google Scholar] [CrossRef]
- Huang, G.S.; Jiang, L.X.; Li, Q. Molecular Design of Damping Rubber Based on Polyacrylate-Containing Silicone. J. Appl. Polym. Sci. 2002, 85, 746–751. [Google Scholar] [CrossRef]
- Ducrot, E.; Chen, Y.; Bulters, M.; Sijbesma, R.P.; Creton, C. Toughening Elastomers with Sacrificial Bonds and Watching Them Break. Science 2014, 344, 186–189. [Google Scholar] [CrossRef]
- Kawai, Y.; Park, J.; Ishii, Y.; Urakawa, O.; Murayama, S.; Ikura, R.; Osaki, M.; Ikemoto, Y.; Yamaguchi, H.; Harada, A.; et al. Preparation of Dual-Cross Network Polymers by the Knitting Method and Evaluation of Their Mechanical Properties. NPG Asia Mater. 2022, 14, 32. [Google Scholar] [CrossRef]
- Wu, C.L.; Zhang, M.Q.; Rong, M.Z.; Friedrich, K. Tensile Performance Improvement of Low Nanoparticles Filled-Polypropylene Composites. Compos. Sci. Technol. 2002, 62, 1327–1340. [Google Scholar] [CrossRef]
- Zhengcai, P.; James, E.M.; Jagdish, M.J.; Warren, T.F. Mechanical properties of a poly(methyl acrylate) nanocomposite containing regularly-arranged silica particles. Polym. Bull. 1996, 37, 545–551. [Google Scholar]
- Huang, X.; Nakagawa, S.; Houjou, H.; Yoshie, N. Insights into the Role of Hydrogen Bonds on the Mechanical Properties of Polymer Networks. Macromolecules 2021, 54, 4070–4080. [Google Scholar] [CrossRef]
- Kim, C.; Ejima, H.; Yoshie, N. Non-Swellable Self-Healing Polymer with Long-Term Stability under Seawater. RSC Adv. 2017, 7, 19288–19295. [Google Scholar] [CrossRef]
- Hayashi, M.; Noro, A.; Matsushita, Y. Highly Extensible Supramolecular Elastomers with Large Stress Generation Capability Originating from Multiple Hydrogen Bonds on the Long Soft Network Strands. Macromol. Rapid Commun. 2016, 37, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, T.; Hayashi, M. Potential of Graft Polymers Bearing Inner Molten Block and Outer Glassy Block at the Graft Chains for Thermoplastic Elastomers with Enhanced Properties. Macromol. Chem. Phys. 2022, 223, 2200073. [Google Scholar] [CrossRef]
- Urban, M.W.; Davydovich, D.; Yang, Y.; Demir, T.; Zhang, Y.; Casabianca, L. Key-and-lock commodity self-healing copolymers. Science 2018, 362, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Fytas, G.; Patkowski, A.; Meier, G.; Dorfmüller, T. A High Pressure Photon Correlation Study of Bulk Poly(Methylacrylate). Comparison with Relaxation Processes in Poly(Ethylacrylate) and Related Polymethacrylates. J. Chem. Phys. 1983, 80, 2214–2220. [Google Scholar] [CrossRef]
- Ribelles, J.L.G.; Duenas, J.M.M.; Pradas, M.M. Dielectric Relaxations in Poly(methyl Acrylate), Poly(ethyl Acrylate), and Poly(butyl Acrylate). J. Appl. Polym. Sci. 1989, 38, 1145–1157. [Google Scholar] [CrossRef]
- Coumoulos, G.D. The Electron Diffraction by Amorphous Polymers. Proc. R. Soc. London. Ser. A Math. Phys. Sci. 1943, 182, 166–179. [Google Scholar]
- Dahlquist, C.A.; Hendricks, J.O.; Taylor, N.W. Elasticity of Soft Polymers—Constant-Stress Elongation Tests. Ind. Eng. Chem. 1951, 43, 1404–1410. [Google Scholar] [CrossRef]
- Safranski, D.L.; Gall, K. Effect of Chemical Structure and Crosslinking Density on the Thermo-Mechanical Properties and Toughness of (Meth)Acrylate Shape Memory Polymer Networks. Polymer 2008, 49, 4446–4455. [Google Scholar] [CrossRef]
- Li, T.; Li, H.; Wang, H.; Lu, W.; Osa, M.; Wang, Y.; Mays, J.; Hong, K. Chain Flexibility and Glass Transition Temperatures of Poly(n-Alkyl (Meth)Acrylate)s: Implications of Tacticity and Chain Dynamics. Polymer 2021, 213, 123207. [Google Scholar] [CrossRef]
- Jakubowski, W.; Juhari, A.; Best, A.; Koynov, K.; Pakula, T.; Matyjaszewski, K. Comparison of Thermomechanical Properties of Statistical, Gradient and Block Copolymers of Isobornyl Acrylate and n-Butyl Acrylate with Various Acrylate Homopolymers. Polymer 2008, 49, 1567–1578. [Google Scholar] [CrossRef]
- Ueberreiter, K.; Kanig, G. Second-Order Transitions and Mesh Distribution Functions of Cross-Linked Polystyrenes. J. Chem. Phys. 1950, 18, 399–406. [Google Scholar] [CrossRef]
- Loshaek, S. Crosslinked Polymers. II. Glass Temperatures of Copolymers of Methyl Methacrylate and Glycol Dimethacrylates. J. Polym. Sci. 1955, 15, 391–404. [Google Scholar] [CrossRef]
- Qu, L.; Nie, Y.; Huang, G.; Weng, G.; Wu, J. Dynamic Fatigue Behavior of Natural Rubber Reinforced with Nanoclay and Carbon Black. J. Macromol. Sci. Part B Phys. 2011, 50, 1646–1657. [Google Scholar] [CrossRef]
- Gong, J.P.; Katsuyama, Y.; Kurokawa, T.; Osada, Y. Double-Network Hydrogels with Extremely High Mechanical Strength. Adv. Mater. 2003, 15, 1155–1158. [Google Scholar] [CrossRef]
- Gong, J.P. Why Are Double Network Hydrogels so Tough? Soft Matter 2010, 6, 2583–2590. [Google Scholar] [CrossRef]
- Nakajima, T.; Ozaki, Y.; Namba, R.; Ota, K.; Maida, Y.; Matsuda, T.; Kurokawa, T.; Gong, J.P. Tough Double-Network Gels and Elastomers from the Nonprestretched First Network. ACS Macro Lett. 2019, 8, 1407–1412. [Google Scholar] [CrossRef]
- Kang, B.; Lang, Q.; Tu, J.; Bu, J.; Ren, J.; Lyu, B.; Gao, D. Preparation and Properties of Double Network Hydrogel with High Compressive Strength. Polymers 2022, 14, 966. [Google Scholar] [CrossRef]
- Lee, H.; Hong, H.J.; Ahn, S.; Kim, D.; Kang, S.H.; Cho, K.; Koh, W.G. One-Pot Synthesis of Double-Network PEG/Collagen Hydrogel for Enhanced Adipogenic Differentiation and Retrieval of Adipose-Derived Stem Cells. Polymers 2023, 15, 1777. [Google Scholar] [CrossRef]
- Mashita, K.; Hirooka, M. Alternating copolymers of isobutylene and acrylic ester by complexed copolymerization. Polymers 1995, 36, 2983–2988. [Google Scholar] [CrossRef]




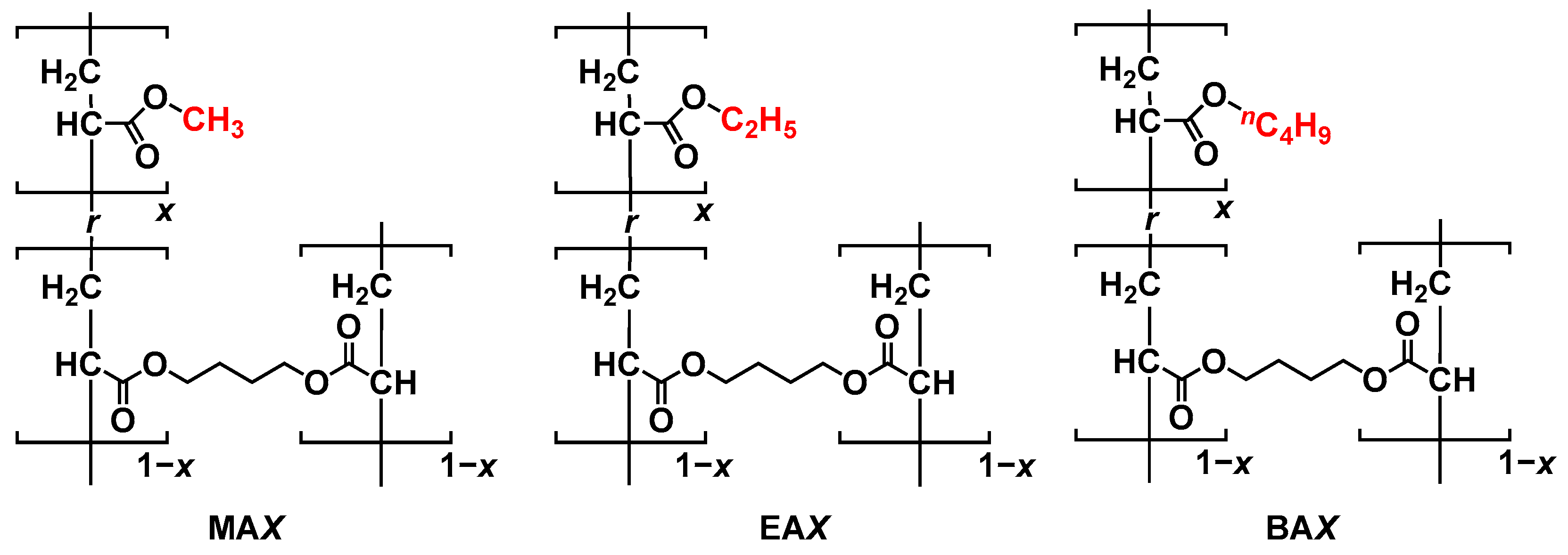{kind=link}
{kind=link}
{kind=link}
| Network Material | Monomer (MA, EA, or BA) | Crosslinker (1,4-Butanediol Diacrylate) | Initiator (ADVN) | Solvent (DMF) | |||
|---|---|---|---|---|---|---|---|
| [mL] | [mmol] | [μL] | [mmol] | [mg] | [mmol] | [mL] | |
| MA005 | 1.00 | 11 | 1.05 | 0.0055 | 2.8 | 0.011 | 0.20 |
| MA020 | 1.00 | 11 | 4.21 | 0.022 | 2.8 | 0.011 | 0.20 |
| MA100 | 1.00 | 11 | 21.0 | 0.11 | 2.8 | 0.011 | 0.20 |
| EA005 | 1.19 | 11 | 1.05 | 0.0055 | 2.8 | 0.011 | 0.20 |
| EA020 | 1.19 | 11 | 4.21 | 0.022 | 2.8 | 0.011 | 0.20 |
| EA100 | 1.19 | 11 | 21.0 | 0.11 | 2.8 | 0.011 | 0.20 |
| BA005 | 1.59 | 11 | 1.05 | 0.0055 | 2.8 | 0.011 | 0.20 |
| BA020 | 1.59 | 11 | 4.21 | 0.022 | 2.8 | 0.011 | 0.20 |
| BA100 | 1.59 | 11 | 21.0 | 0.11 | 2.8 | 0.011 | 0.20 |
| X | Tg of Network Materials [°C] | ||
|---|---|---|---|
| MAX | EAX | BAX | |
| 0 (homopolymer) | 10.041 | −24.041 | −54.041 |
| 005 | 16.5 | −15.1 | −49.0 |
| 020 | 16.1 | −14.7 | −50.2 |
| 100 | 17.6 | −12.1 | −46.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kawano, Y.; Masai, H.; Nakagawa, S.; Yoshie, N.; Terao, J. Effects of Alkyl Ester Chain Length on the Toughness of PolyAcrylate-Based Network Materials. Polymers 2023, 15, 2389. https://doi.org/10.3390/polym15102389
Kawano Y, Masai H, Nakagawa S, Yoshie N, Terao J. Effects of Alkyl Ester Chain Length on the Toughness of PolyAcrylate-Based Network Materials. Polymers. 2023; 15(10):2389. https://doi.org/10.3390/polym15102389
Chicago/Turabian StyleKawano, Yutaro, Hiroshi Masai, Shintaro Nakagawa, Naoko Yoshie, and Jun Terao. 2023. "Effects of Alkyl Ester Chain Length on the Toughness of PolyAcrylate-Based Network Materials" Polymers 15, no. 10: 2389. https://doi.org/10.3390/polym15102389
APA StyleKawano, Y., Masai, H., Nakagawa, S., Yoshie, N., & Terao, J. (2023). Effects of Alkyl Ester Chain Length on the Toughness of PolyAcrylate-Based Network Materials. Polymers, 15(10), 2389. https://doi.org/10.3390/polym15102389