
Dynamic Light Scattering Based Microrheology of End-Functionalised Triblock Copolymer Solutions
 , , , , and
, , , , and
Abstract
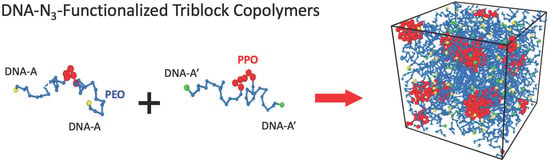
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.1.1. Functionalisation of F108 with Azide Groups (N-PEG-PPO-PEG-N)
2.1.2. Dibenzylcyclooctane (DBCO)-DNA Preparation
2.1.3. DNA Functionalised F108 Preparation
2.2. F108 Phase Diagram Study
2.3. Dynamic Light Scattering
2.4. DLS-Based Microrheology
3. Results
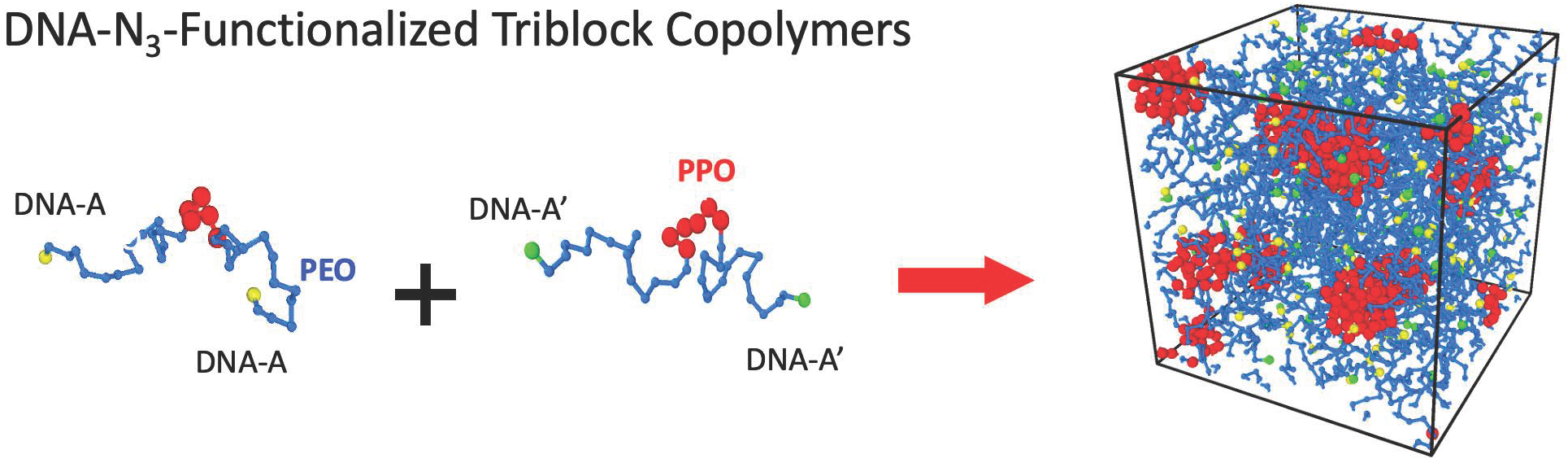
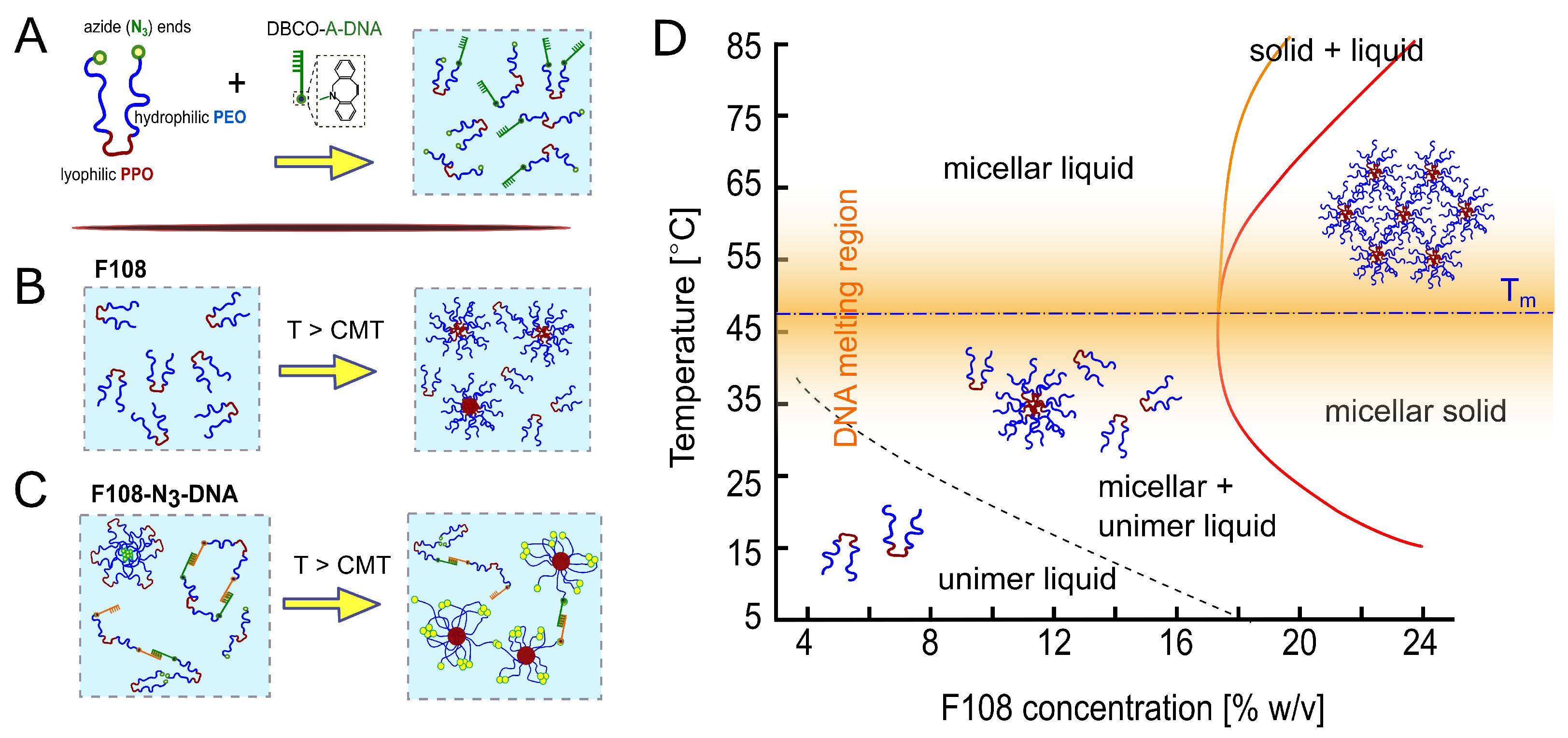
3.1. Design and Phase Diagram of the System
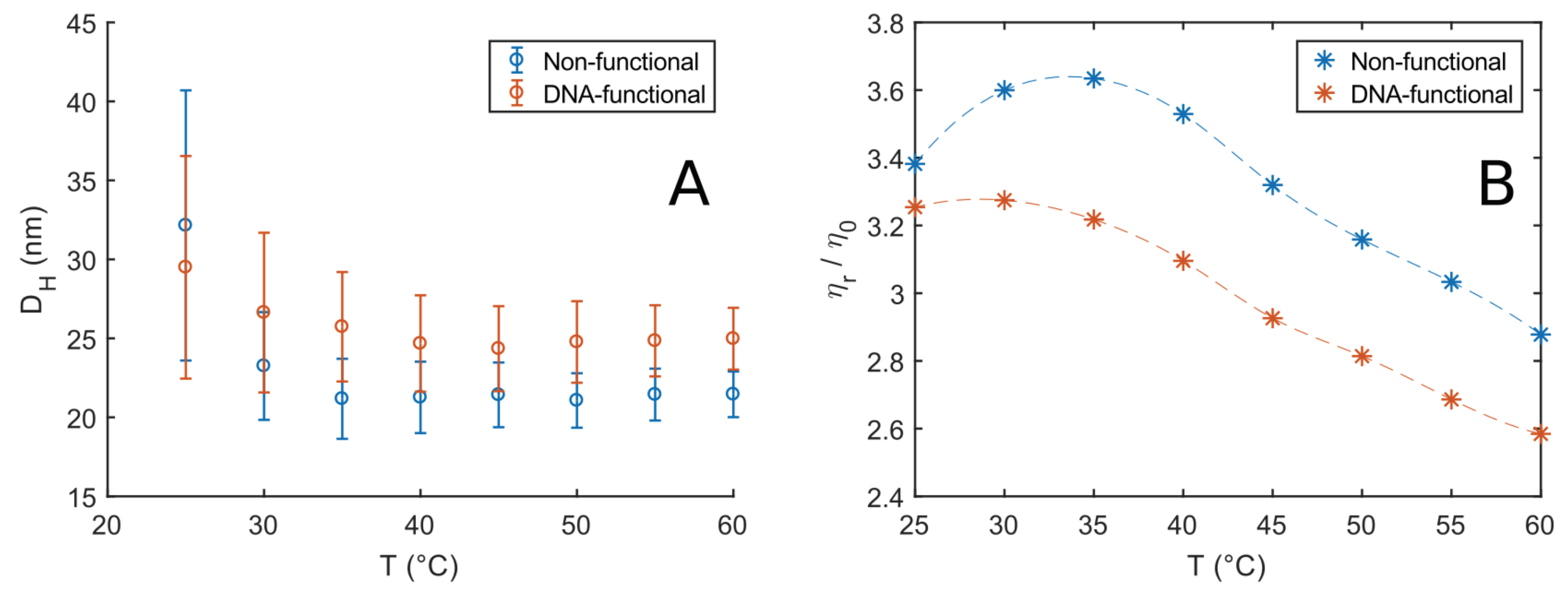
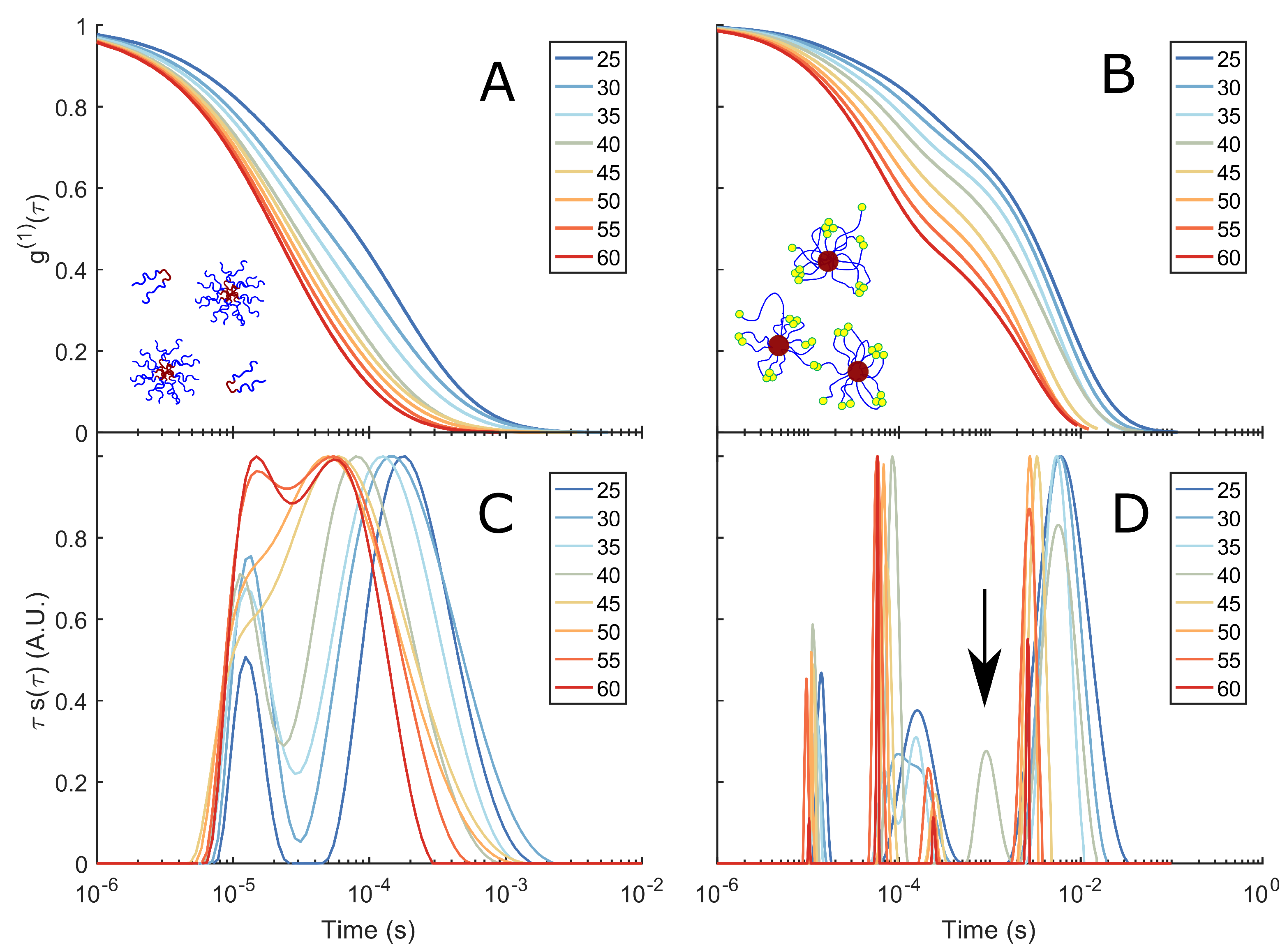
3.2. DLS and DLS-Microrheology Study of Semi-Dilute Solutions
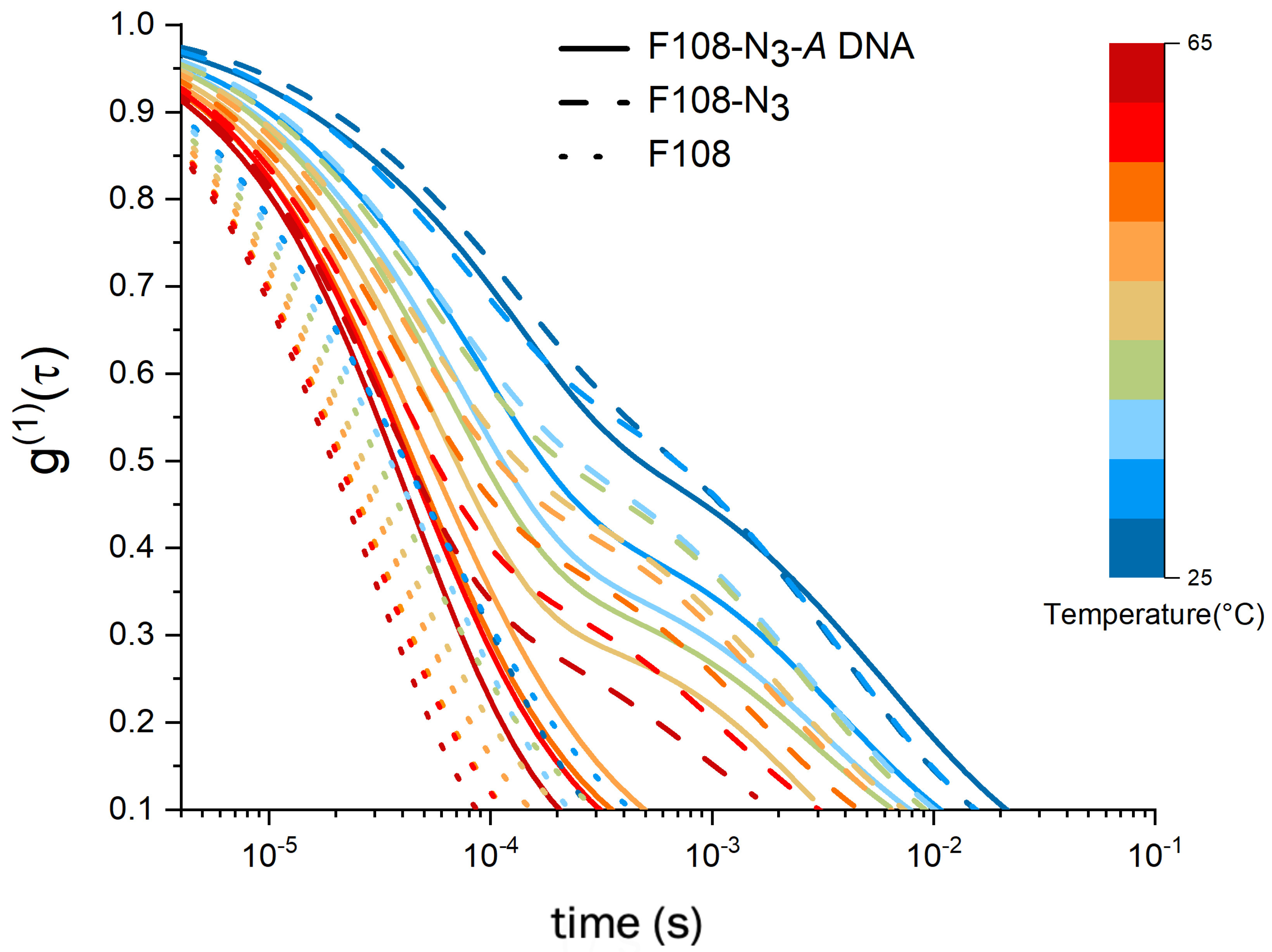
3.3. DLS and Microrheology Study of Semi-Concentrated Solutions
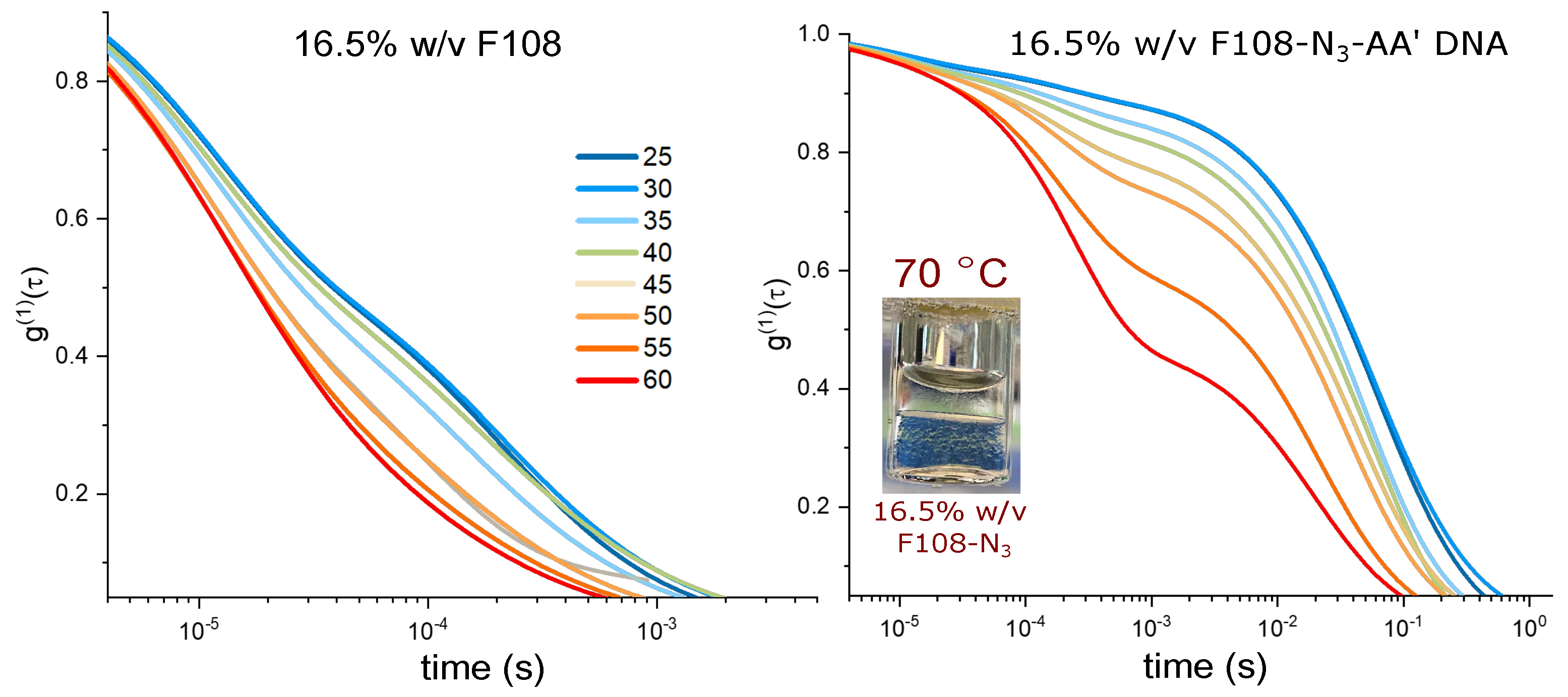
3.4. Microrheology of Concentrated Solutions
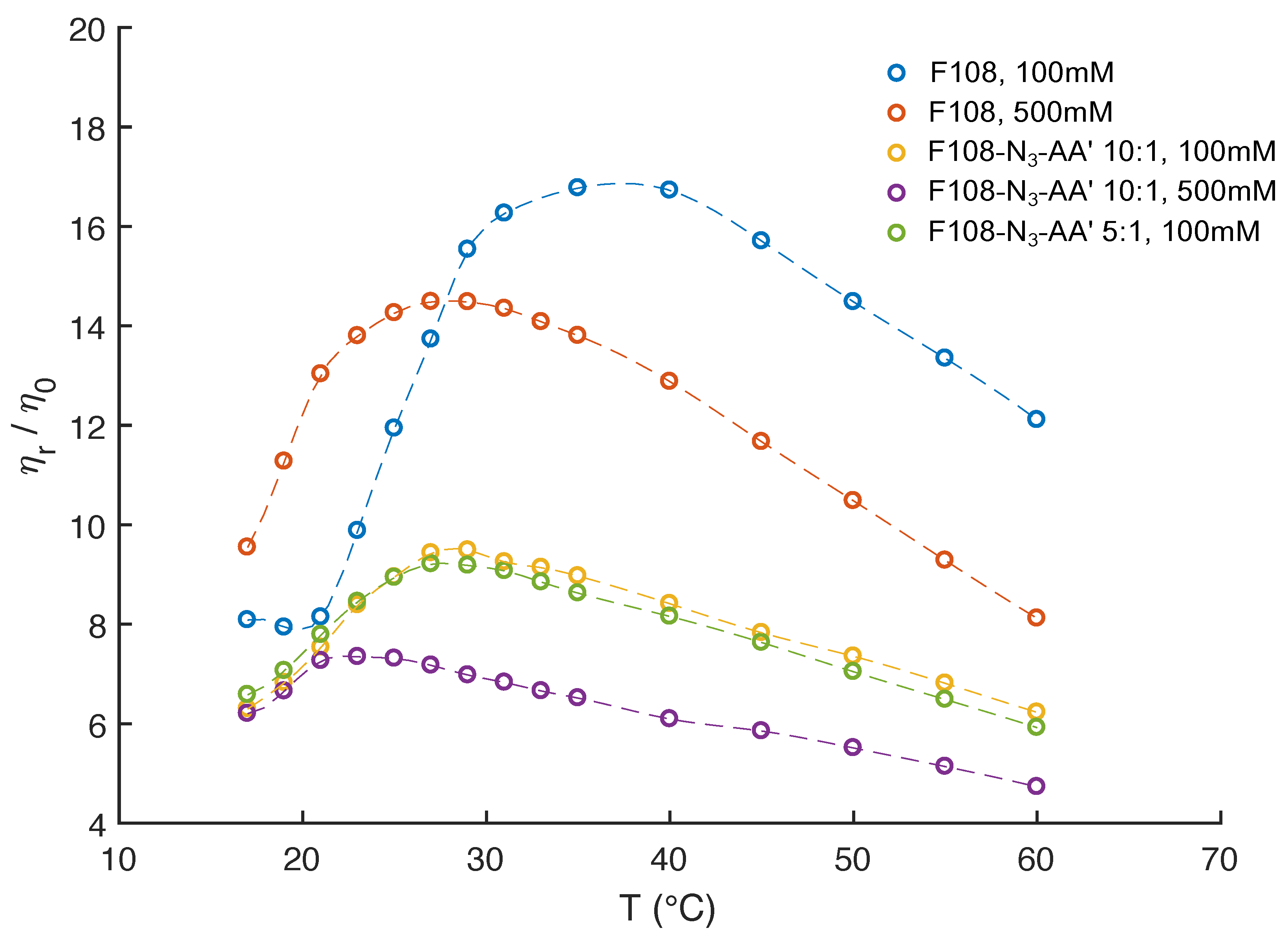
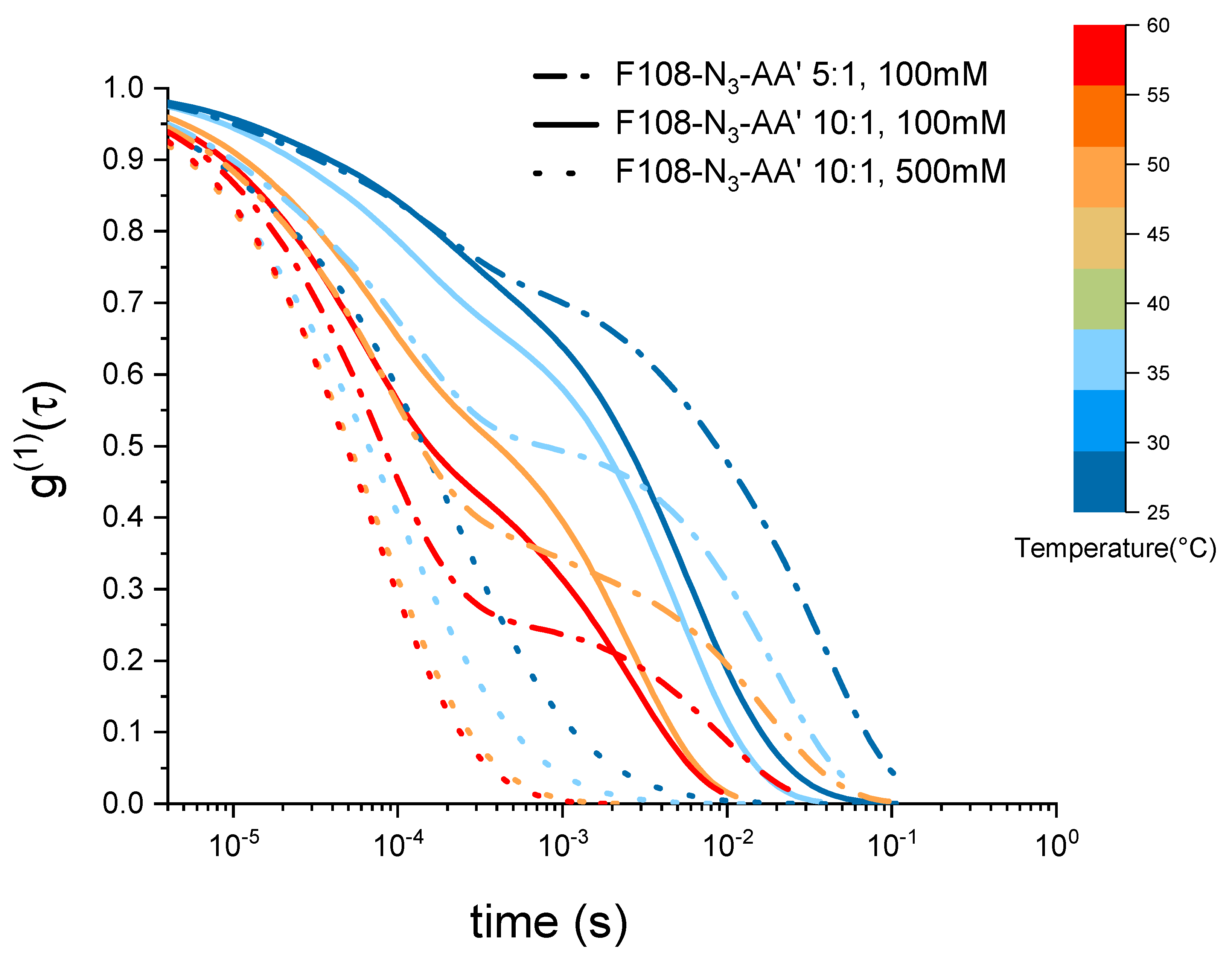
3.5. The Effect of Added Salt and Increased DNA Ratio
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Batrakova, E.V.; Kabanov, A.V. Pluronic block copolymers: Evolution of drug delivery concept from inert nanocarriers to biological response modifiers. J. Control. Release 2008, 130, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.S.; Gao, Z.; Bae, Y.H. Recent progress in tumor pH targeting nanotechnology. J. Control. Release 2008, 132, 164–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; del Pilar Rodriguez-Torres, M.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Eisenberg, A. Multiple morphologies of “crew-cut” aggregates of polystyrene-b-poly (acrylic acid) block copolymers. Science 1995, 268, 1728–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gröschel, A.H.; Schacher, F.H.; Schmalz, H.; Borisov, O.V.; Zhulina, E.B.; Walther, A.; Müller, A.H. Precise hierarchical self-assembly of multicompartment micelles. Nat. Commun. 2012, 3, 710. [Google Scholar] [CrossRef] [Green Version]
- Lunn, D.J.; Finnegan, J.R.; Manners, I. Self-assembly of “patchy” nanoparticles: A versatile approach to functional hierarchical materials. Chem. Sci. 2015, 6, 3663–3673. [Google Scholar] [CrossRef] [Green Version]
- Ravaine, S.; Duguet, E. Synthesis and assembly of patchy particles: Recent progress and future prospects. Curr. Opin. Colloid Interface Sci. 2017, 30, 45–53. [Google Scholar] [CrossRef]
- Yan, L.T. Self-Assembling Systems: Theory and Simulation; John Wiley & Sons: Chichester, UK, 2016. [Google Scholar]
- Teixeira, P.; Tavares, J. Phase behaviour of pure and mixed patchy colloids—Theory and simulation. Curr. Opin. Colloid Interface Sci. 2017, 30, 16–24. [Google Scholar] [CrossRef]
- Geerts, N.; Eiser, E. DNA-functionalized colloids: Physical properties and applications. Soft Matter 2010, 6, 4647–4660. [Google Scholar] [CrossRef]
- Di Michele, L.; Varrato, F.; Kotar, J.; Nathan, S.H.; Foffi, G.; Eiser, E. Multistep kinetic self-assembly of DNA-coated colloids. Nat. Commun. 2013, 4, 2007. [Google Scholar] [CrossRef]
- Feng, L.; Dreyfus, R.; Sha, R.; Seeman, N.C.; Chaikin, P.M. DNA patchy particles. Adv. Mater. 2013, 25, 2779–2783. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.R.; Macfarlane, R.J.; Lee, B.; Zhang, J.; Young, K.L.; Senesi, A.J.; Mirkin, C.A. DNA-nanoparticle superlattices formed from anisotropic building blocks. Nat. Mater. 2010, 9, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, R.J.; Lee, B.; Jones, M.R.; Harris, N.; Schatz, G.C.; Mirkin, C.A. Nanoparticle superlattice engineering with DNA. Science 2011, 334, 204–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirkin, C.A.; Letsinger, R.L.; Mucic, R.C.; Storhoff, J.J. A DNA-based method for rationally assembling nanoparticles into macroscopic materials. Nature 1996, 382, 607–609. [Google Scholar] [CrossRef]
- Alivisatos, A.P.; Johnsson, K.P.; Peng, X.; Wilson, T.E.; Loweth, C.J.; Bruchez, M.P.; Schultz, P.G. Organization of ‘nanocrystal molecules’ using DNA. Nature 1996, 382, 609–611. [Google Scholar] [CrossRef] [Green Version]
- Valignat, M.P.; Theodoly, O.; Crocker, J.C.; Russel, W.B.; Chaikin, P.M. Reversible self-assembly and directed assembly of DNA-linked micrometer-sized colloids. Proc. Natl. Acad. Sci. USA 2005, 102, 4225–4229. [Google Scholar] [CrossRef] [Green Version]
- Nykypanchuk, D.; Maye, M.M.; Van Der Lelie, D.; Gang, O. DNA-guided crystallization of colloidal nanoparticles. Nature 2008, 451, 549–552. [Google Scholar] [CrossRef]
- Hadorn, M.; Boenzli, E.; Sørensen, K.T.; Fellermann, H.; Hotz, P.E.; Hanczyc, M.M. Specific and reversible DNA-directed self-assembly of oil-in-water emulsion droplets. Proc. Natl. Acad. Sci. USA 2012, 109, 20320–20325. [Google Scholar] [CrossRef] [Green Version]
- Parolini, L.; Mognetti, B.M.; Kotar, J.; Eiser, E.; Cicuta, P.; Di Michele, L. Volume and porosity thermal regulation in lipid mesophases by coupling mobile ligands to soft membranes. Nat. Commun. 2015, 6, 5948. [Google Scholar] [CrossRef] [Green Version]
- Joshi, D.; Bargteil, D.; Caciagli, A.; Burelbach, J.; Xing, Z.; Nunes, A.S.; Pinto, D.E.; Araújo, N.A.; Brujic, J.; Eiser, E. Kinetic control of the coverage of oil droplets by DNA-functionalized colloids. Sci. Adv. 2016, 2, e1600881. [Google Scholar] [CrossRef]
- Zhang, Y.; McMullen, A.; Pontani, L.L.; He, X.; Sha, R.; Seeman, N.C.; Brujic, J.; Chaikin, P.M. Sequential self-assembly of DNA functionalized droplets. Nat. Commun. 2017, 8, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caciagli, A.; Zupkauskas, M.; Levin, A.; Knowles, T.P.; Mugemana, C.; Bruns, N.; O’Neill, T.; Frith, W.J.; Eiser, E. DNA-Coated Functional Oil Droplets. Langmuir 2018, 34, 10073–10080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Yoon, J.H.; Jo, I.S.; Oh, J.S.; Pine, D.J.; Shim, T.S.; Yi, G.R. DNA-functionalized 100 nm polymer nanoparticles from block copolymer micelles. Langmuir 2018, 34, 11042–11048. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yamauchi, Y. Block copolymer mediated synthesis of dendritic platinum nanoparticles. J. Am. Chem. Soc. 2009, 131, 9152–9153. [Google Scholar] [CrossRef] [PubMed]
- Tadros, T. Viscoelastic properties of sterically stabilised emulsions and their stability. Adv. Colloid Interface Sci. 2015, 222, 692–708. [Google Scholar] [CrossRef]
- Agnely, F.; Djedour, A.; Bochot, A.; Grossiord, J.L. Properties of various thermoassociating polymers: Pharmaceutical and cosmetic applications. J. Drug Deliv. Sci. Technol. 2006, 16, 3–10. [Google Scholar] [CrossRef]
- Chang, Y.; Chu, W.L.; Chen, W.Y.; Zheng, J.; Liu, L.; Ruaan, R.C.; Higuchi, A. A systematic SPR study of human plasma protein adsorption behavior on the controlled surface packing of self-assembled poly (ethylene oxide) triblock copolymer surfaces. J. Biomed. Mater. Res. Part A 2010, 93, 400–408. [Google Scholar] [CrossRef]
- Almeida, H.; Amaral, M.H.; Lobão, P.; Lobo, J.M.S. Pluronic® F-127 and Pluronic Lecithin Organogel (PLO): Main features and their applications in topical and transdermal administration of drugs. J. Pharm. Pharm. Sci. 2012, 15, 592–605. [Google Scholar] [CrossRef] [Green Version]
- Bodratti, A.M.; Alexandridis, P. Formulation of poloxamers for drug delivery. J. Funct. Biomater. 2018, 9, 11. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Castanon, J.; Bianchi, S.; Saglimbeni, F.; Di Leonardo, R.; Sciortino, F. Microrheology of DNA hydrogel gelling and melting on cooling. Soft Matter 2018, 14, 6431–6438. [Google Scholar] [CrossRef]
- Xing, Y.; Cheng, E.; Yang, Y.; Chen, P.; Zhang, T.; Sun, Y.; Yang, Z.; Liu, D. Self-assembled DNA hydrogels with designable thermal and enzymatic responsiveness. Adv. Mater. 2011, 23, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Xing, Z.; Caciagli, A.; Cao, T.; Stoev, I.; Zupkauskas, M.; O’Neill, T.; Wenzel, T.; Lamboll, R.; Liu, D.; Eiser, E. Microrheology of DNA hydrogels. Proc. Natl. Acad. Sci. USA 2018, 115, 8137–8142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.S.; Wang, Y.; Pine, D.J.; Yi, G.R. High-density PEO-b-DNA brushes on polymer particles for colloidal superstructures. Chem. Mater. 2015, 27, 8337–8344. [Google Scholar] [CrossRef]
- SantaLucia, J., Jr. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA 1998, 95, 1460–1465. [Google Scholar] [CrossRef] [Green Version]
- Zupkauskas, M.; Lan, Y.; Joshi, D.; Ruff, Z.; Eiser, E. Optically transparent dense colloidal gels. Chem. Sci. 2017, 8, 5559–5566. [Google Scholar] [CrossRef] [Green Version]
- Stoev, I.D.; Cao, T.; Caciagli, A.; Yu, J.; Ness, C.; Liu, R.; Ghosh, R.; O’Neill, T.; Liu, D.; Eiser, E. On the role of flexibility in linker-mediated DNA hydrogels. Soft Matter 2020, 16, 990–1001. [Google Scholar] [CrossRef] [Green Version]
- Caciagli, A. Design and Characterisation of DNA-Functionalised Soft Colloids. Ph.D. Thesis, University of Cambridge, Cavendish Laboratory, Cambridge, UK, 2022. [Google Scholar]
- Wanka, G.; Hoffman, H.; Ulbricht, W. Phase Diagrams and Aggregation Behavior of Poly(oxyethylene)-Poly(oxypropylene)-Poly(oxyethylene)-Block-Copolymers in Aqueous Solution. Macromolecules 1994, 27, 4145–4159. [Google Scholar] [CrossRef]
- Alexandridis, P.; Spontak, R.J. Solvent-regulated ordering in block copolymers. Curr. Opin. Colloid Interface Sci. 1999, 4, 130–139. [Google Scholar] [CrossRef]
- Salamone, J.C. Polymeric Materials Encyclopedia, Twelve Volume Set; CRC Press: Boca Raton, FL, USA, 1996. [Google Scholar]
- Diat, O.; Porte, G.; Berret, J.F. Orientation and twins separation in a micellar cubic crystal under oscillating shear. Phys. Rev. B 1992, 54, 14869–14872. [Google Scholar] [CrossRef]
- Wu, Y.L.; Sprik, R.; Poon, W.C.K.; Eiser, E. Effect of salt on the phase behaviour of F68 triblock PEO/PPO/PEO copolymer. J. Phys. Condens. Matter 2006, 18, 4461–4470. [Google Scholar] [CrossRef]
- Kumar, M.S.; Dash, S. Effect of salts on micellization and clouding behavior of Pluronic F108 in aqueous solution using Trypan blue dye. Surfaces Interfaces 2018, 12, 1–7. [Google Scholar] [CrossRef]
- Eiser, E.; Molino, F.; Porte, G.; Diat, O. Nonhomogeneous textures and banded flow in a soft cubic phase under shear. Phys. Rev. E 2000, 61, 6759–6764. [Google Scholar] [CrossRef]
- Liu, R. Designing Biocompatible Functional Hydrogels: Experiments and Simulations. Ph.D. Thesis, University of Cambridge, Cavendish Laboratory, Cambridge, UK, 2022. [Google Scholar]
- Mohan, P.H.; Bandyopadhyay, R. Phase behavior and dynamics of a micelle-forming triblock copolymer system. Phys. Rev. E 2008, 77, 041803. [Google Scholar] [CrossRef] [Green Version]
- Hamley, I.W.; Pople, J.A.; Fairclough, J.P.A.; Ryan, A.J.; Booth, C.; Yang, Y.W. Shear-Induced Orientational Transitions in the Body-Centered Cubic Phase of a Diblock Copolymer Gel. Macromolecules 1998, 31, 3906–3911. [Google Scholar] [CrossRef]
- Yu, J. Multi-Level Coarse-Grained Modelling on DNA Functionalised Building Blocks. Ph.D. Thesis, University of Cambridge, Cavendish Laboratory, Cambridge, UK, 2022. [Google Scholar]
- Patel, K.; Bahadur, P.; Guo, C.; Ma, J.; Liu, H.; Yamashita, Y.; Khanal, A.; Nakashima, K. Salt induced micellization of very hydrophilic PEO–PPO–PEO block copolymers in aqueous solutions. Eur. Polym. J. 2007, 43, 1699–1708. [Google Scholar] [CrossRef]
- NIST. NIST Standard Reference Database Number 69; NIST: Gaithersburg, MD, USA, 2016.
- Zhou, Z.; Chu, B. Light-scattering study on the association behavior of triblock polymers of ethylene oxide and propylene oxide in aqueous solution. J. Colloid Interface Sci. 1988, 126, 171–180. [Google Scholar] [CrossRef]
- Brown, W.; Schillen, K.; Hvid, S. Triblock copolymers in aqueous solution studied by static and dynamic light scattering and oscillatory shear measurements: Influence of relative block sizes. J. Phys. Chem. 1992, 96, 6038–6044. [Google Scholar] [CrossRef]
- Nolan, S.L.; Phillips, R.J.; Cotts, P.M.; Dungan, S.R. Light scattering study on the effect of polymer composition on the structural properties of PEO–PPO–PEO micelles. J. Colloid Interface Sci. 1997, 191, 291–302. [Google Scholar] [CrossRef]
- Mortensen, K. Phase Behaviour of Poly(ethylene oxide)-Poly(propylene oxide)-Poly(ethylene oxide) Triblock-Copolymer Dissolved in Water. Europhys. Lett. 1992, 19, 599–604. [Google Scholar] [CrossRef]
- Tinland, B.; Pluen, A.; Sturm, J.; Weill, G. Persistence length of single-stranded DNA. Macromolecules 1997, 30, 5763–5765. [Google Scholar] [CrossRef]
- Alexandridis, P.; Nivaggioli, T.; Hatton, T.A. Temperature Effects on Structural Properties of Pluronic P104 and F108 PEO-PPO-PEO Block Copolymer Solutions. Langmuir 1995, 11, 1468–1476. [Google Scholar] [CrossRef]
- Exerowa, D.; Sedev, R.; Ivanova, R.; Kolarov, T.; Thadros, T.F. Transition from electrostatic to steric stabilization in foam films from ABA triblock copolymers of poly(ethylene oxide) and poly(propylene oxide). Coll. Surf. A Physicochem. Eng. Asp. 1997, 123–124, 277–282. [Google Scholar] [CrossRef]
- Eiser, E.; Molino, F.; Porte, G.; Pithon, X. Flow in micellar cubic crystals. Rheol. Acta 2000, 39, 201–208. [Google Scholar] [CrossRef]
- Kjellander, R.; Florin, E. Water structure and changes in thermal stability of the system poly (ethylene oxide)–water. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1981, 77, 2053–2077. [Google Scholar] [CrossRef]
- Bae, Y.; Lambert, S.; Soane, D.; Prausnitz, J.M. Cloud-point curves of polymer solutions from thermooptical measurements. Macromolecules 1991, 24, 4403–4407. [Google Scholar] [CrossRef] [Green Version]
- Dormidontova, E.E. Role of competitive PEO- water and water- water hydrogen bonding in aqueous solution PEO behavior. Macromolecules 2002, 35, 987–1001. [Google Scholar] [CrossRef]
- Zupkauskas, M. Colloidal Self-Assembly: Particle Synthesis, Functionalization and Applications. Ph.D. Thesis, University of Cambridge, Cavendish Laboratory, Cambridge, UK, 2018. [Google Scholar]
- Nicoud, L.; Lattuada, M.; Yates, A.; Morbidelli, M. Impact of aggregate formation on the viscosity of protein solutions. Soft Matter 2015, 11, 5513–5522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijmans, C.M.; Eiser, E.; Frenkel, D. Simulation study of intra- and intermicellar ordering in triblock-copolymer systems. J. Chem. Phys. 2004, 120, 5839–5848. [Google Scholar] [CrossRef] [Green Version]
- Molino, F.; Berret, J.F.; Porte, G.; Diat, O.; Lindner, P. Identification of flow mechanisms for a soft crystal. Eur. Phys. J. B-Condens. Matter Complex Syst. 1998, 3, 59–72. [Google Scholar] [CrossRef]
- Eiser, E.; Klein, J.; Witten, T.A.; Fetters, L.J. Shear of telechelic brushes. Phys. Rev. Lett. 1999, 82, 5076. [Google Scholar] [CrossRef]
- Halperin, A.; Alexander, S. Polymeric micelles: Their relaxation kinetics. Macromolecules 1989, 22, 2403–2412. [Google Scholar] [CrossRef]
- Wang, Y.; Balaji, R.; Quirk, R.P.; Mattice, W.L. Detection of the rate of exchange of chains between micelles formed by diblock copolymers in aqueous solution. Polym. Bull. 1992, 28, 333–338. [Google Scholar] [CrossRef]
- Michel, E.; Appell, J.; Molino, F.; Kieffer, J.; Porte, G. Unstable flow and nonmonotonic flow curves of transient networks. J. Rheol. 2001, 45, 1465–1477. [Google Scholar] [CrossRef] [Green Version]
- Filali, M.; Ouazzani, M.J.; Michel, E.; Aznar, R.; Porte, G.; Appell, J. Robust phase behavior of model transient networks. J. Phys. Chem. B 2001, 105, 10528–10535. [Google Scholar] [CrossRef] [Green Version]
- Zilman, A.; Tlusty, T.; Safran, S. Entropic networks in colloidal, polymeric and amphiphilic systems. J. Phys. Condens. Matter 2002, 15, S57. [Google Scholar] [CrossRef]
- Curk, T.; Brackley, C.A.; Farrell, J.D.; Xing, Z.; Joshi, D.; Direito, S.; Bren, U.; Angioletti-Uberti, S.; Dobnikar, J.; Eiser, E.; et al. Computational design of probes to detect bacterial genomes by multivalent binding. Proc. Natl. Acad. Sci. USA 2020, 117, 8719–8726. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, R.; Caciagli, A.; Yu, J.; Tang, X.; Ghosh, R.; Eiser, E. Dynamic Light Scattering Based Microrheology of End-Functionalised Triblock Copolymer Solutions. Polymers 2023, 15, 481. https://doi.org/10.3390/polym15030481
Liu R, Caciagli A, Yu J, Tang X, Ghosh R, Eiser E. Dynamic Light Scattering Based Microrheology of End-Functionalised Triblock Copolymer Solutions. Polymers. 2023; 15(3):481. https://doi.org/10.3390/polym15030481
Chicago/Turabian StyleLiu, Ren, Alessio Caciagli, Jiaming Yu, Xiaoying Tang, Rini Ghosh, and Erika Eiser. 2023. "Dynamic Light Scattering Based Microrheology of End-Functionalised Triblock Copolymer Solutions" Polymers 15, no. 3: 481. https://doi.org/10.3390/polym15030481
APA StyleLiu, R., Caciagli, A., Yu, J., Tang, X., Ghosh, R., & Eiser, E. (2023). Dynamic Light Scattering Based Microrheology of End-Functionalised Triblock Copolymer Solutions. Polymers, 15(3), 481. https://doi.org/10.3390/polym15030481