
The Polymers of Diethynylarenes—Is Selective Polymerization at One Acetylene Bond Possible? A Review


Abstract
:1. Introduction
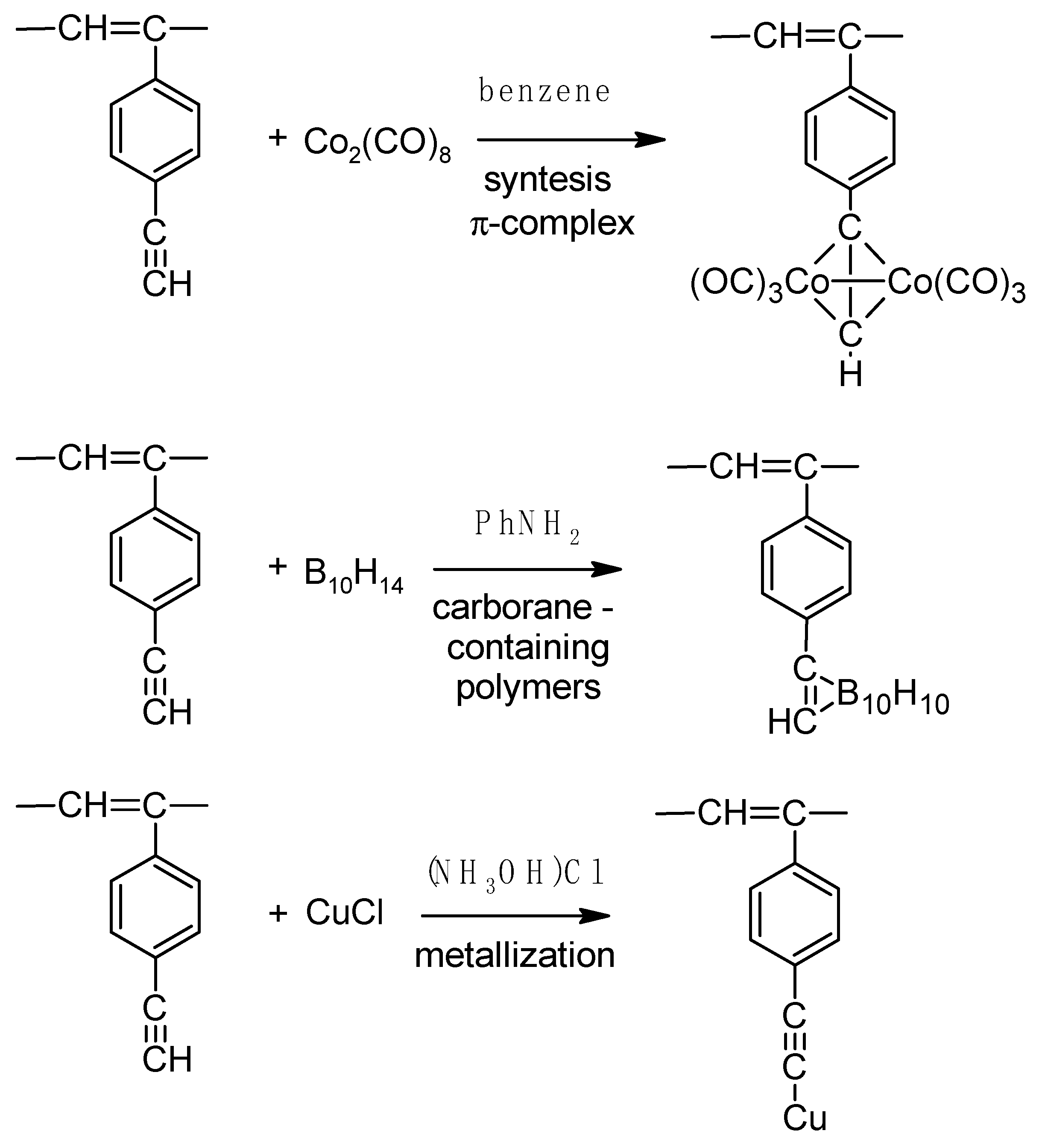
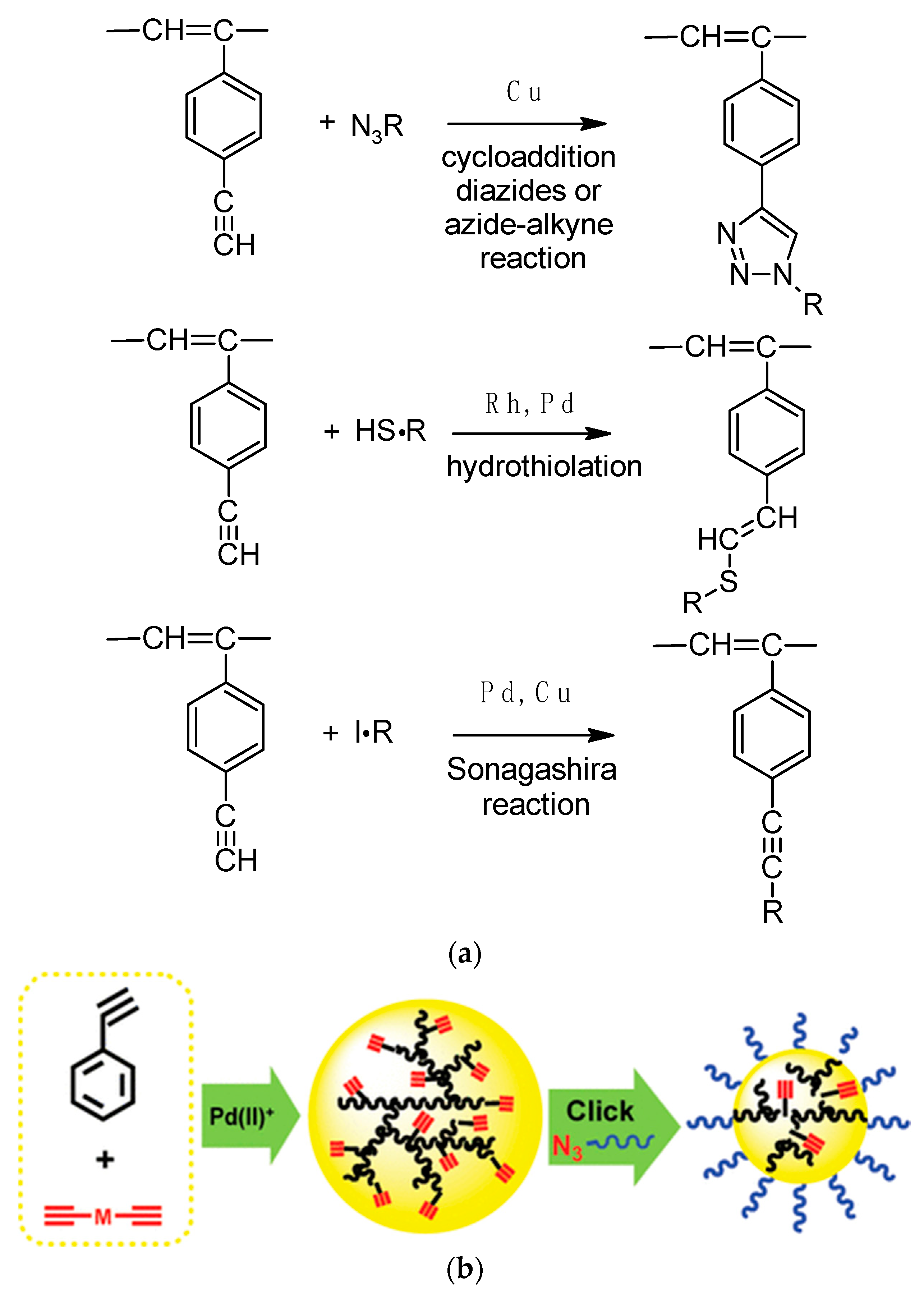
2. Synthesis, Structural Features, and Properties of p-Diethynylbenzene Polymers
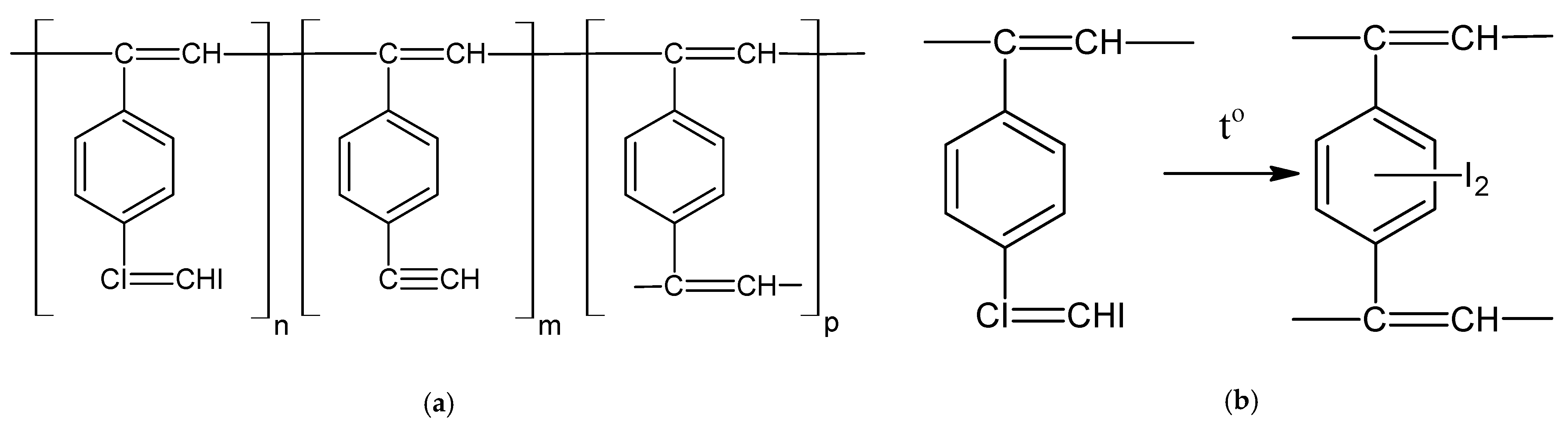
2.1. Solid-Phase Polymerization of p-DEB Initiated by Physical Methods of Exposure
2.2. Gas-Phase Polymerization of p-DEB
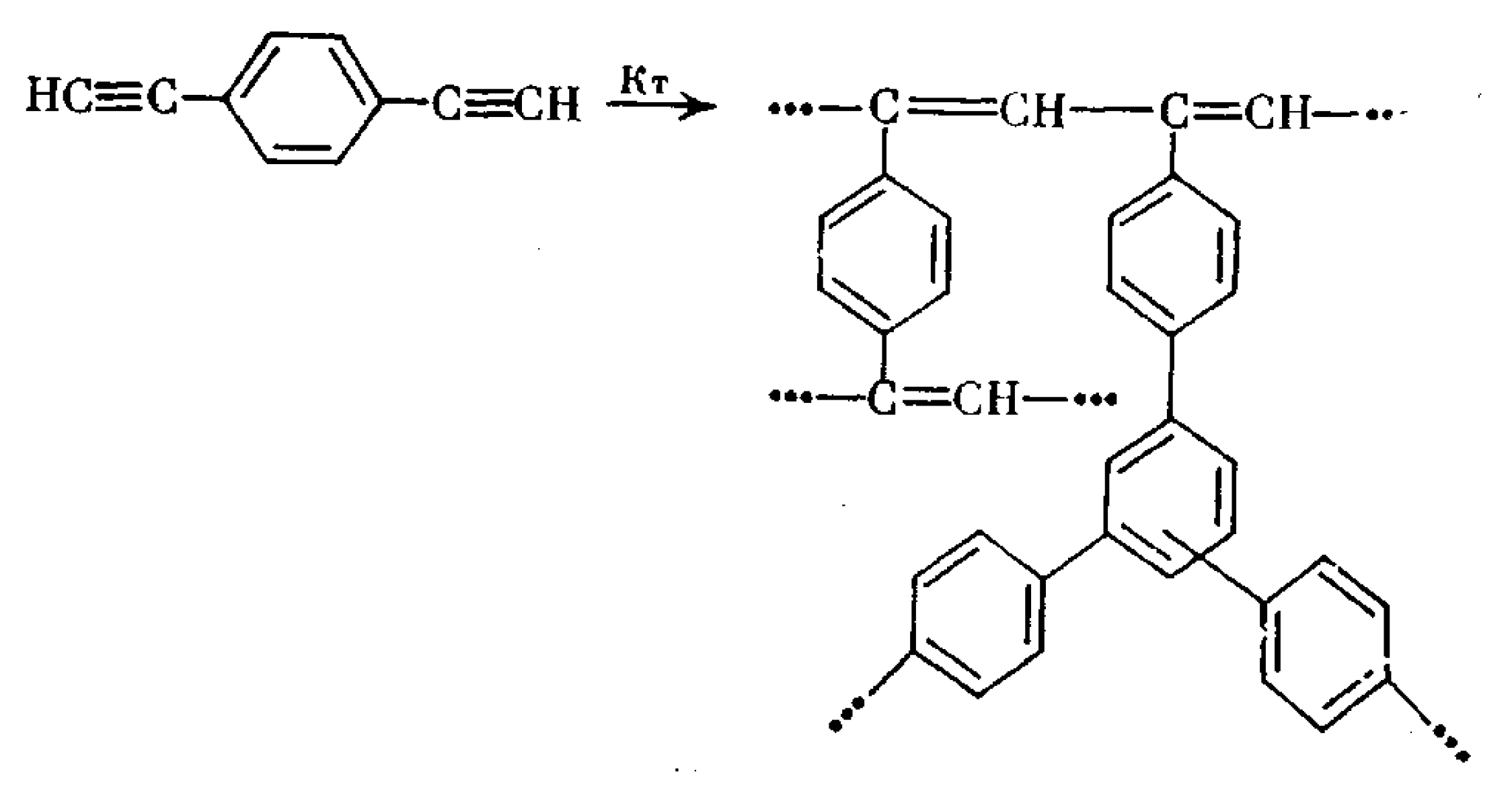
2.3. Liquid-Phase Polymerization of p-DEB without the Use of Catalysts
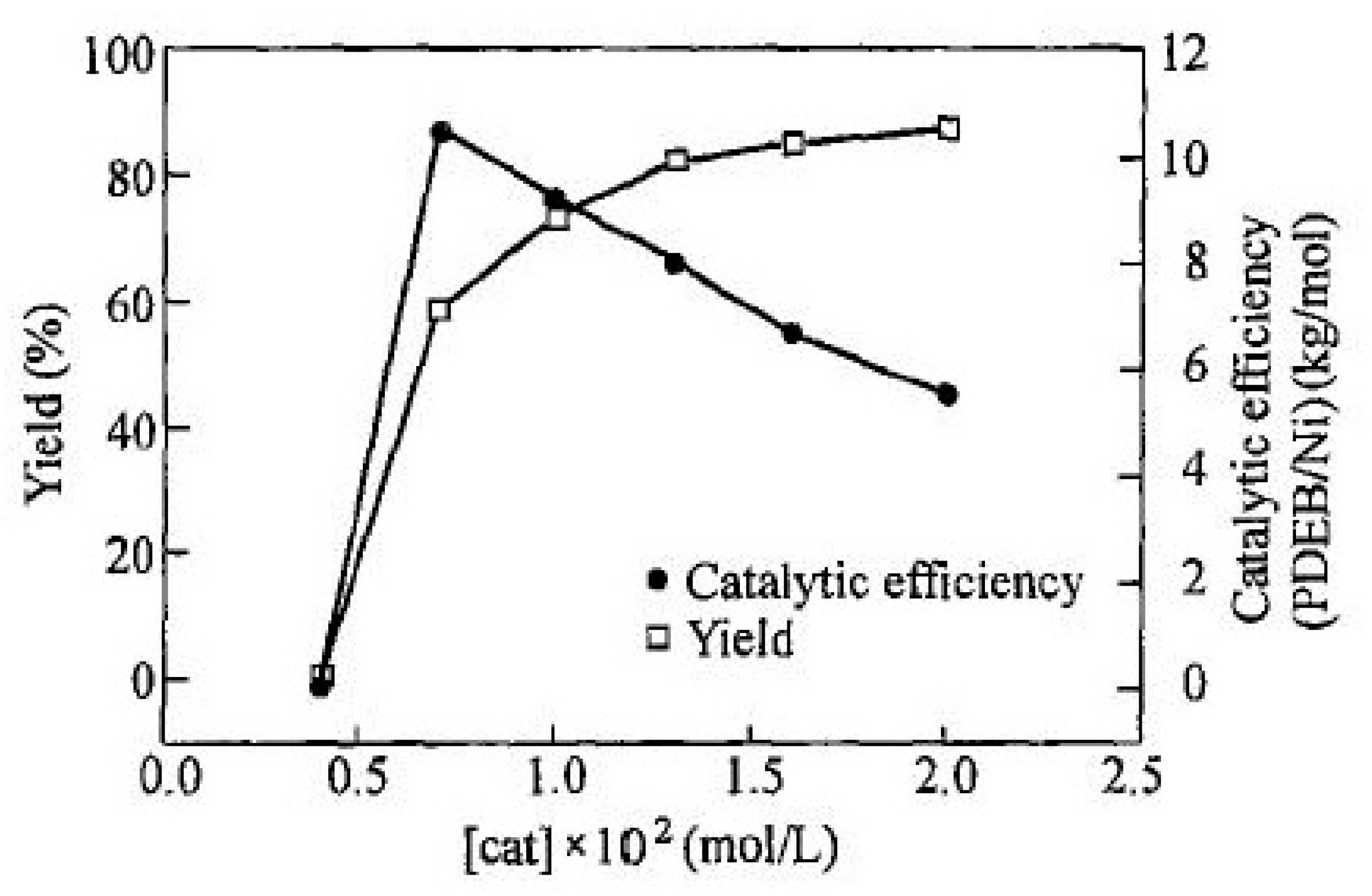
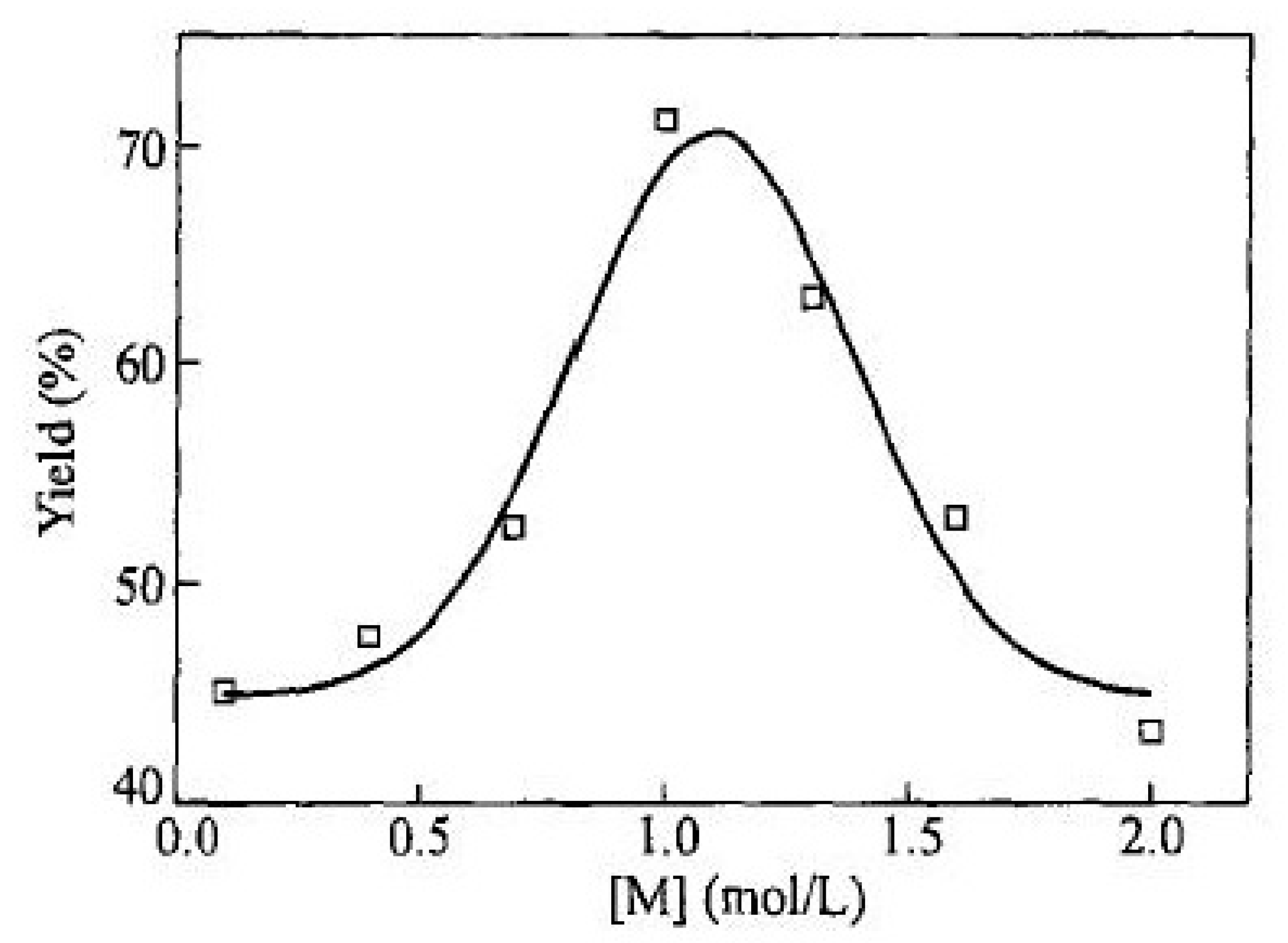
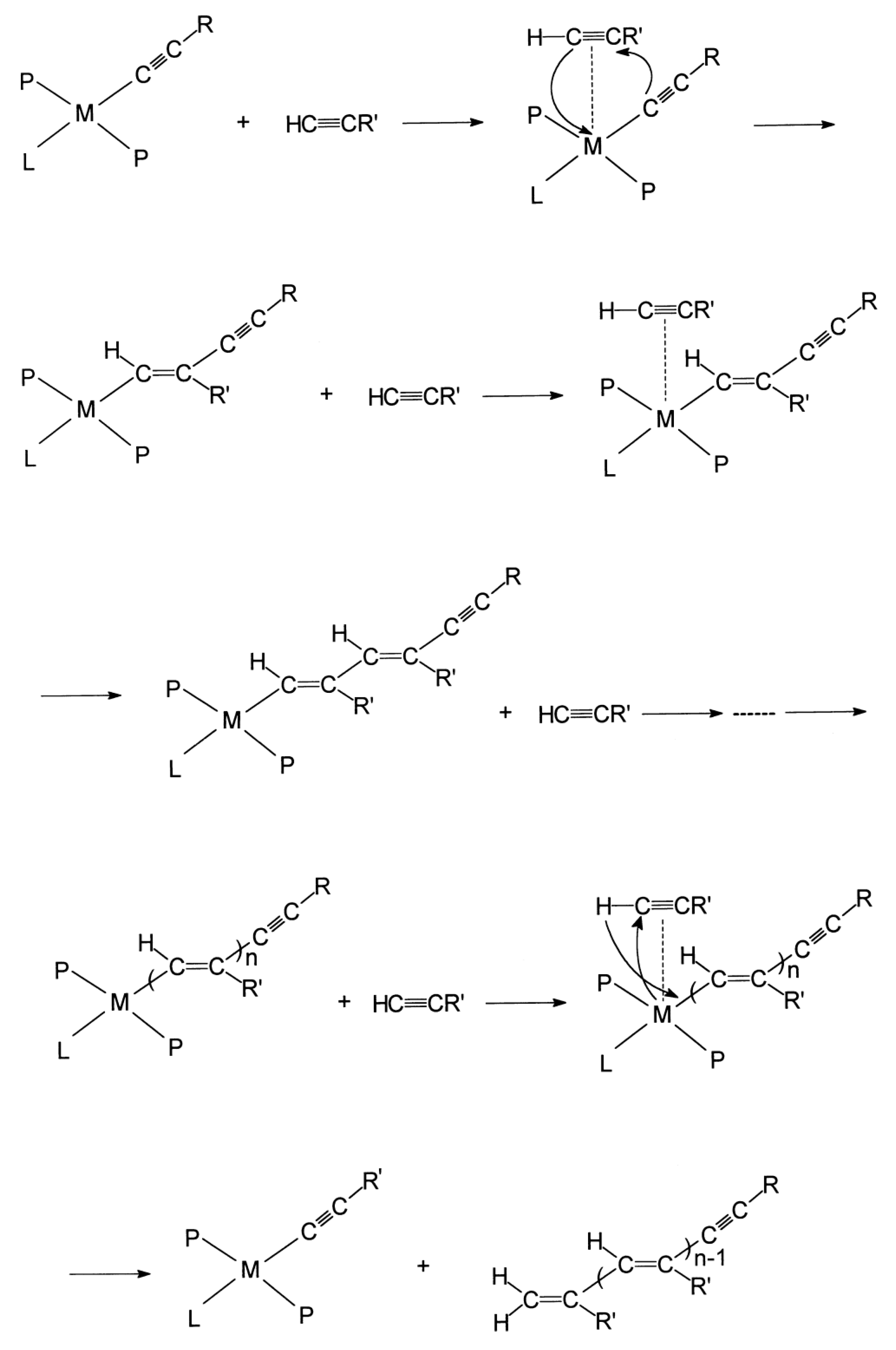
2.4. Liquid-Phase Polymerization of p-DEB with the Use of Catalysts
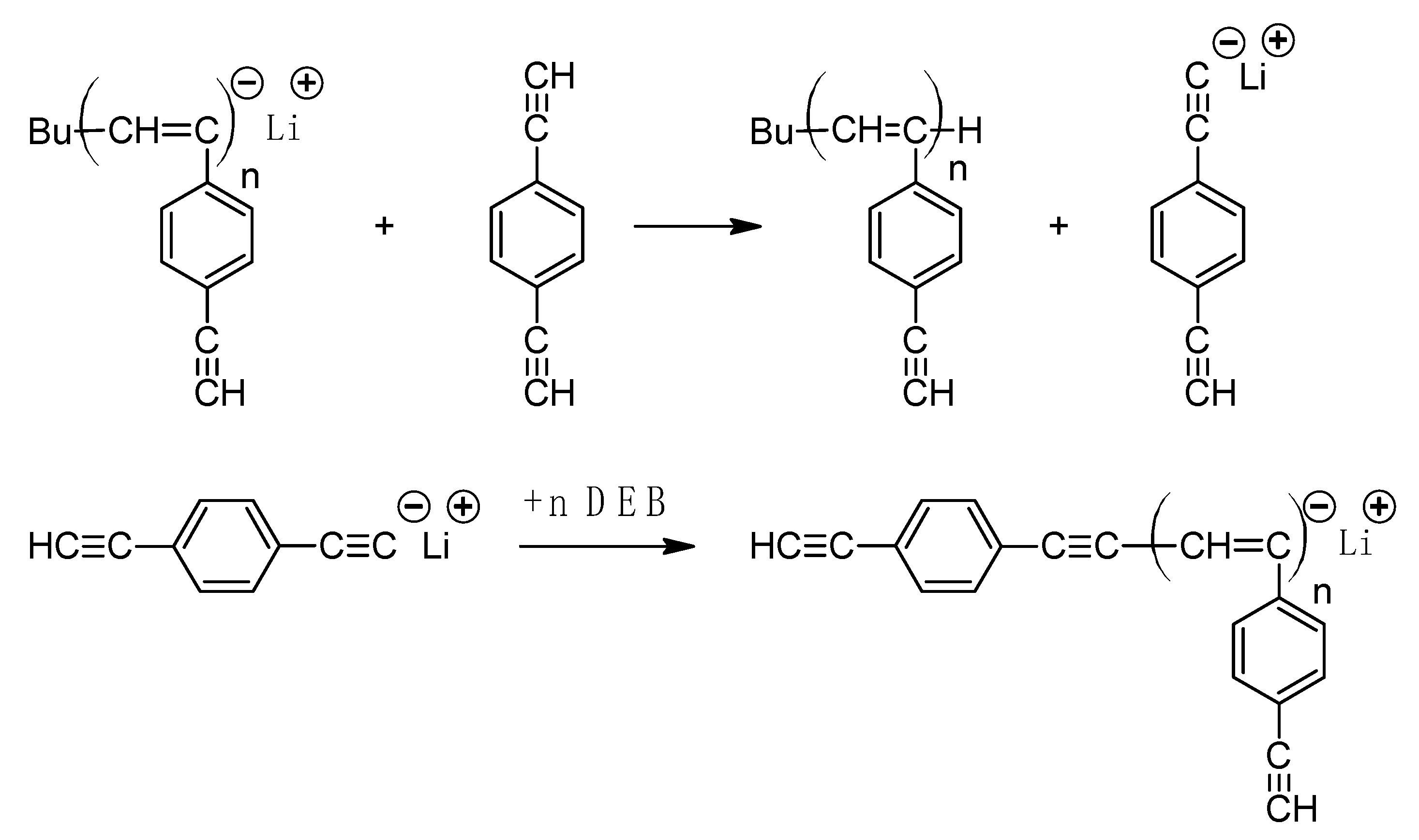
2.5. Synthesis of Linear Unbranched Poly-DEB
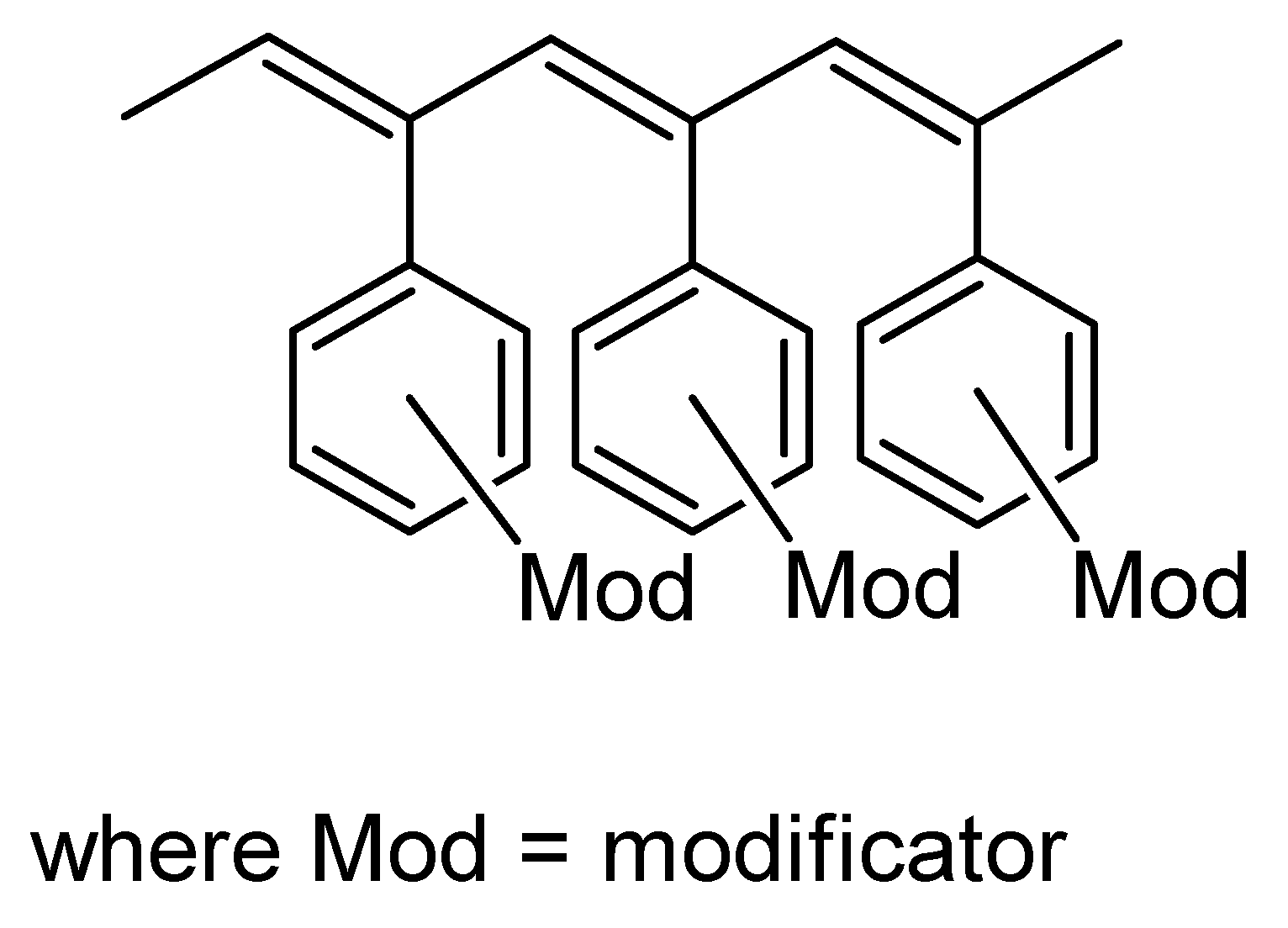
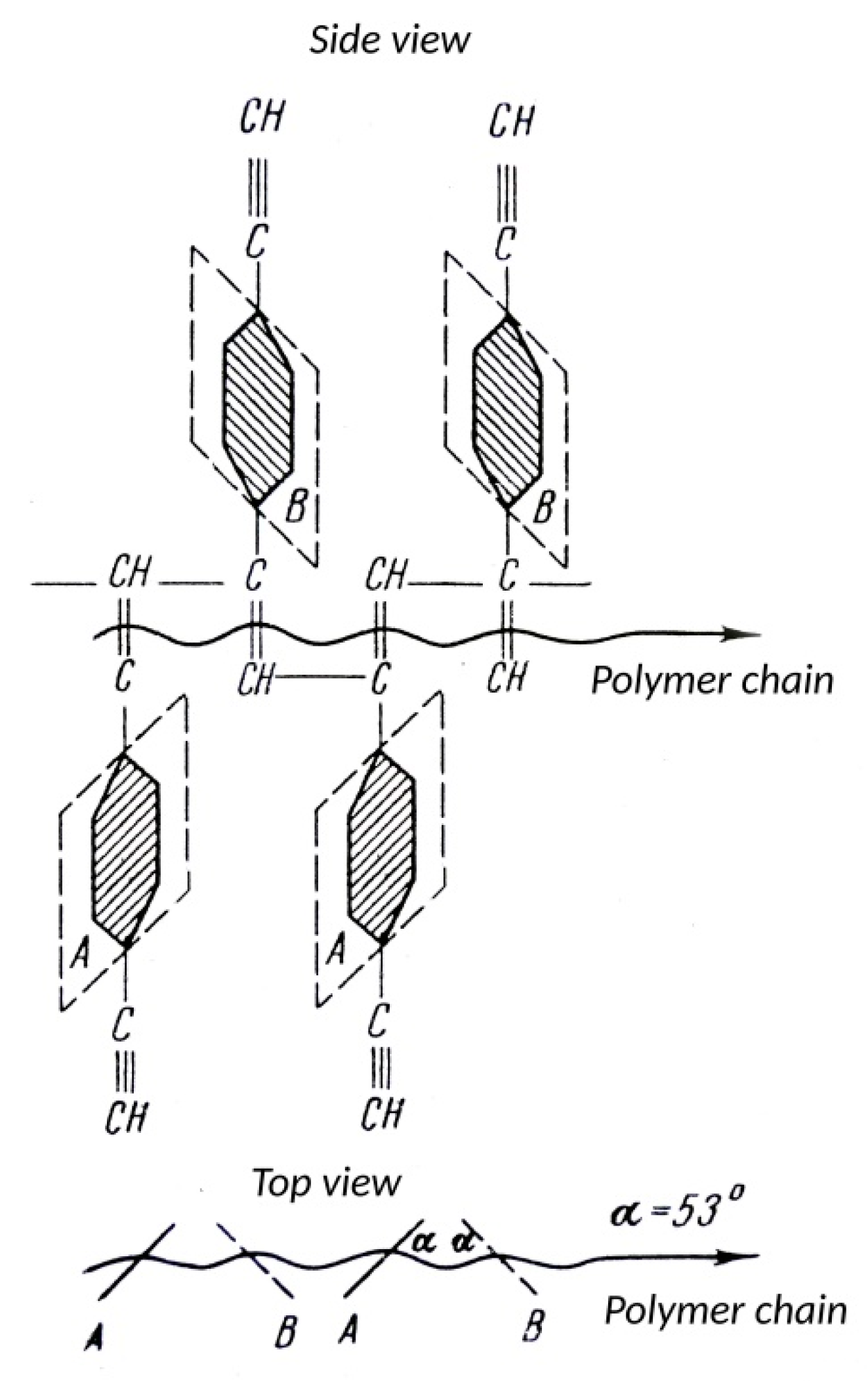
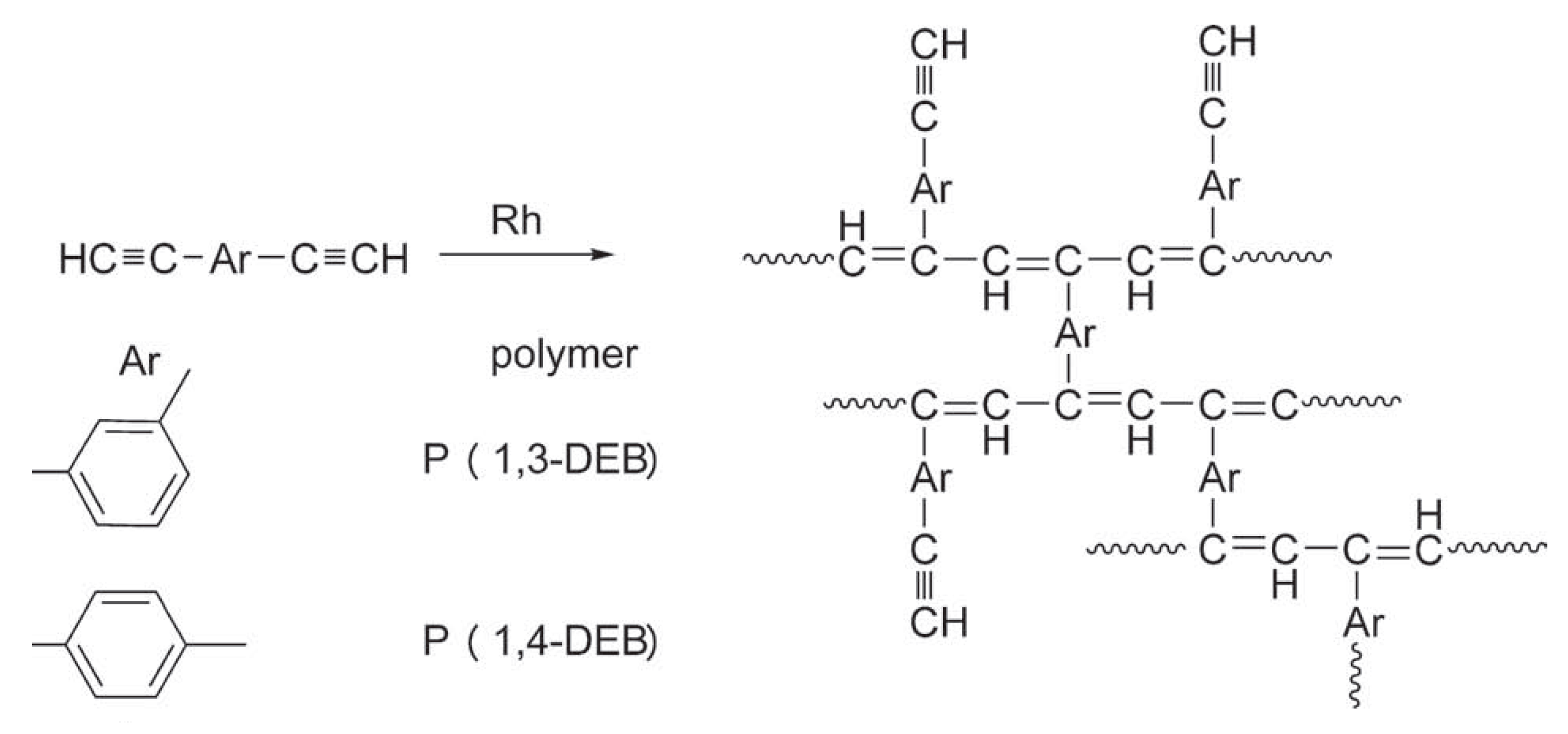
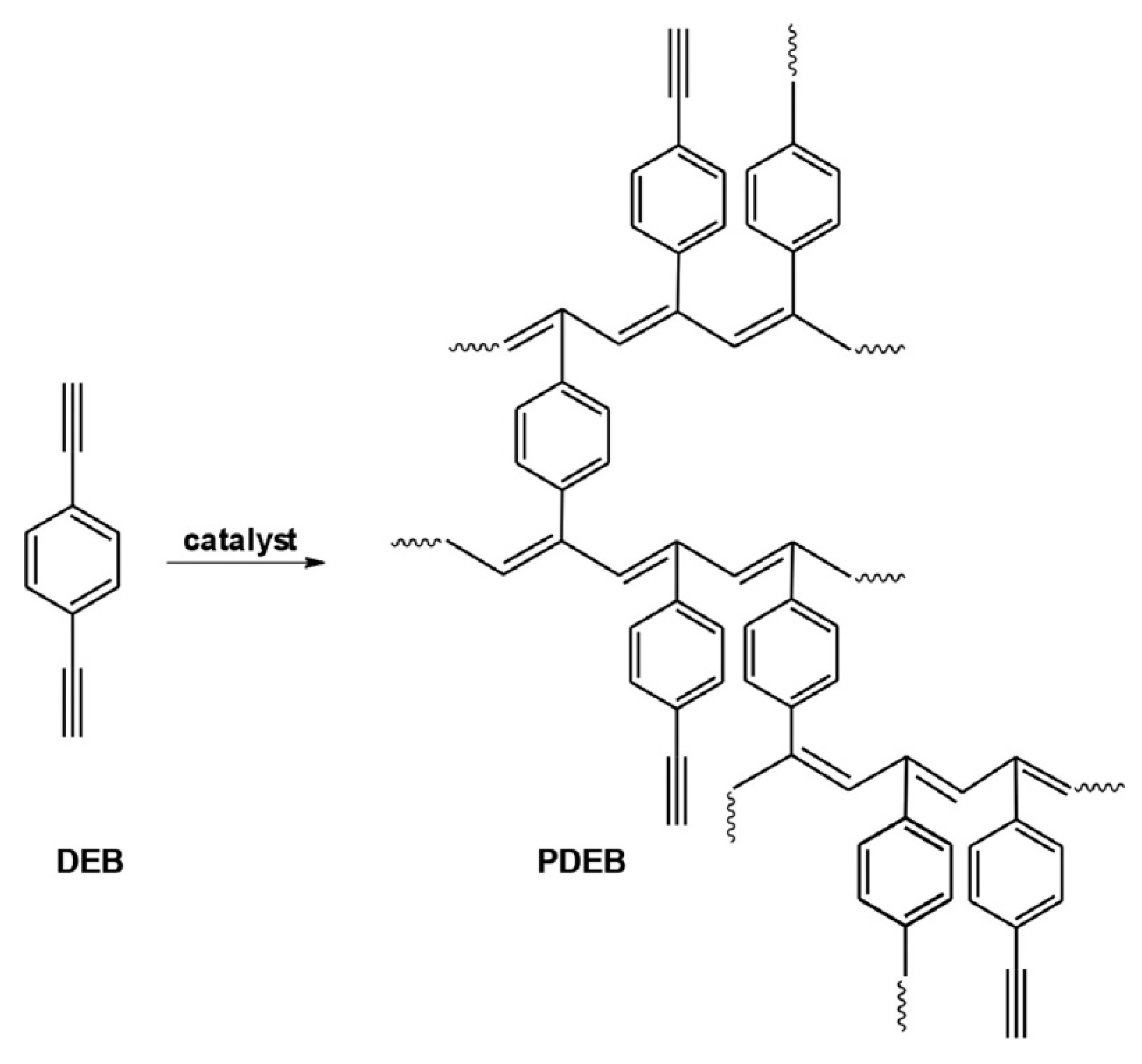
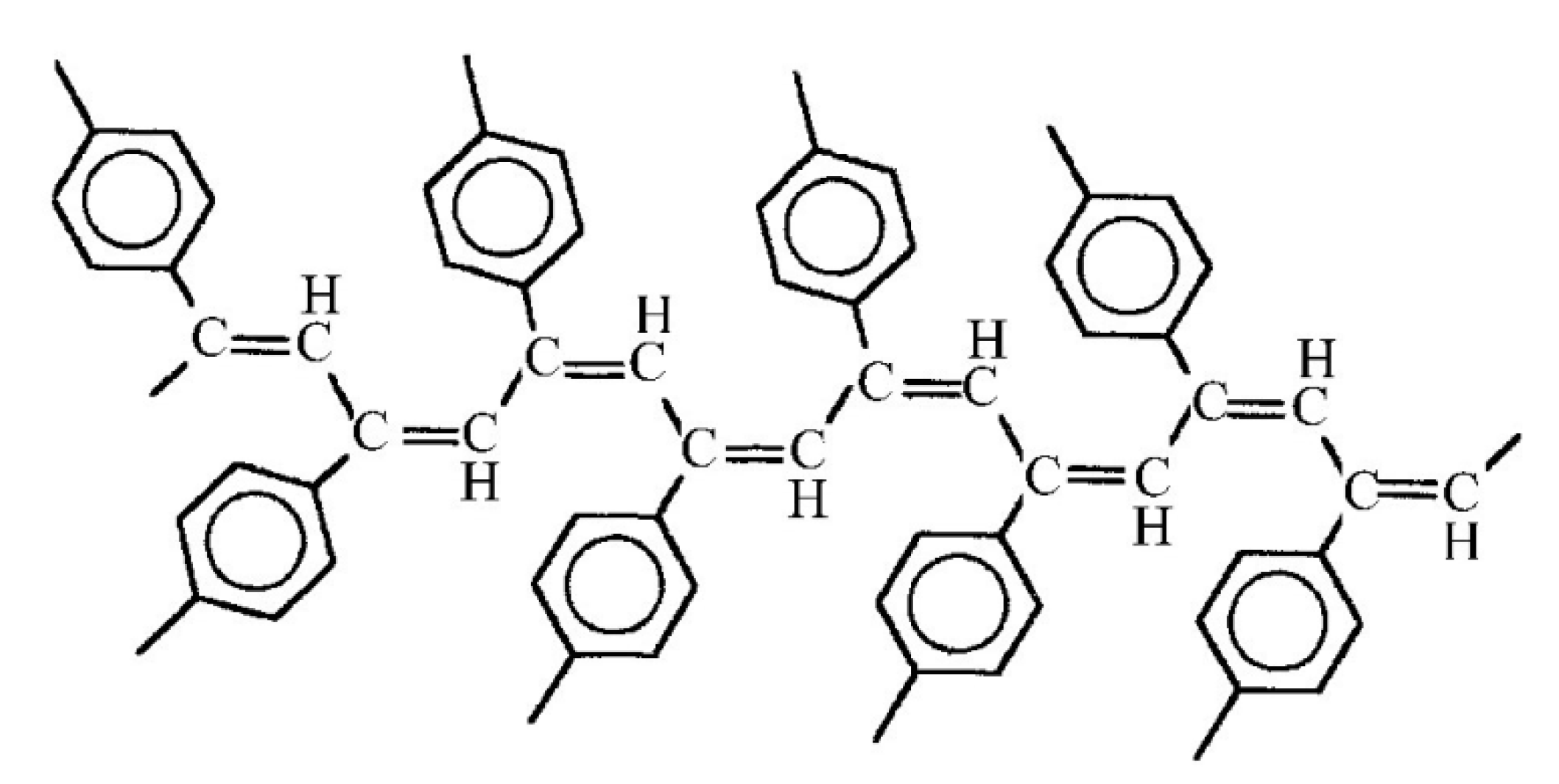
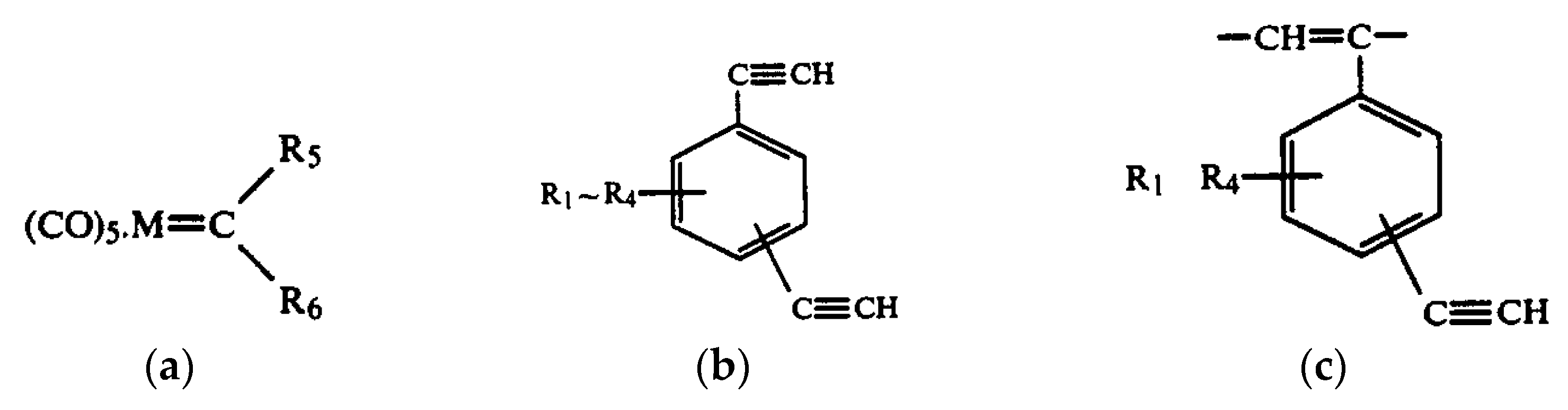
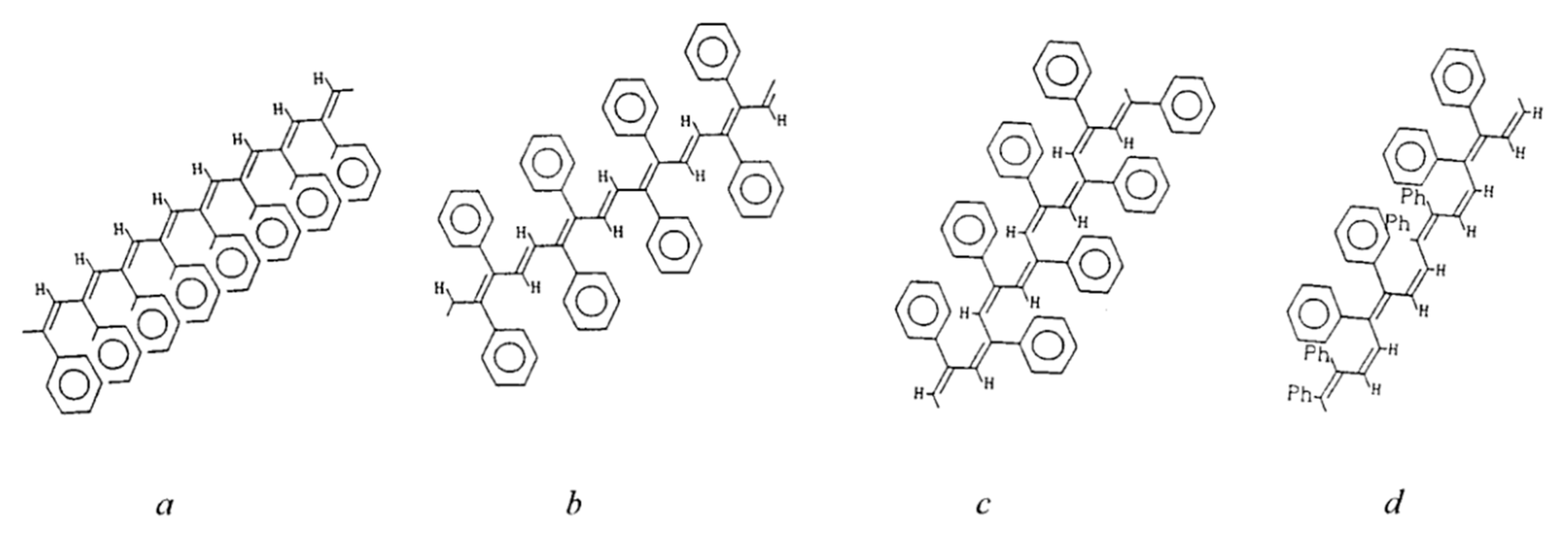
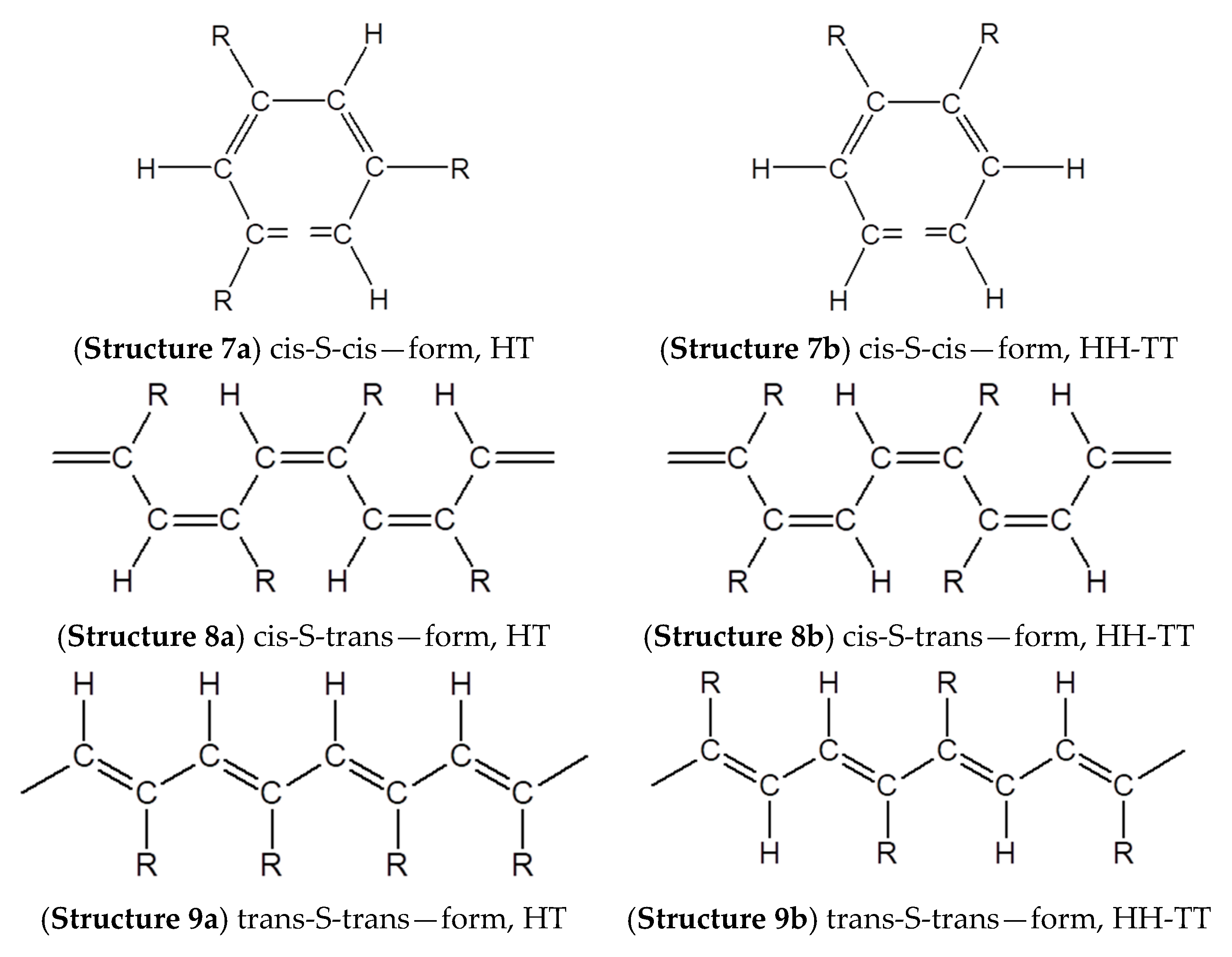
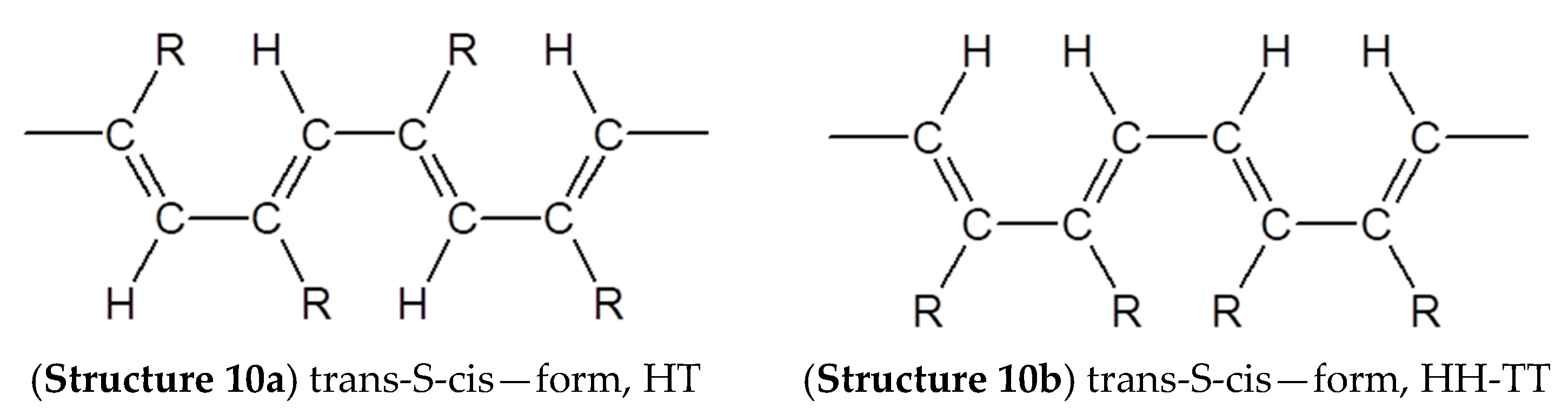
2.6. Intramolecular Structure of Substituted Polyacetylenes
2.7. Features of the Intramolecular Structure of Poly-p-DEB
- formation of cis-S-transoid in structure 8 is not possible;
- the trans-S-cisoid in structure 10b cannot be realized when connecting the head-head-tail-tail links;
- other types of in structures 7, 9, and 10a may be formed.
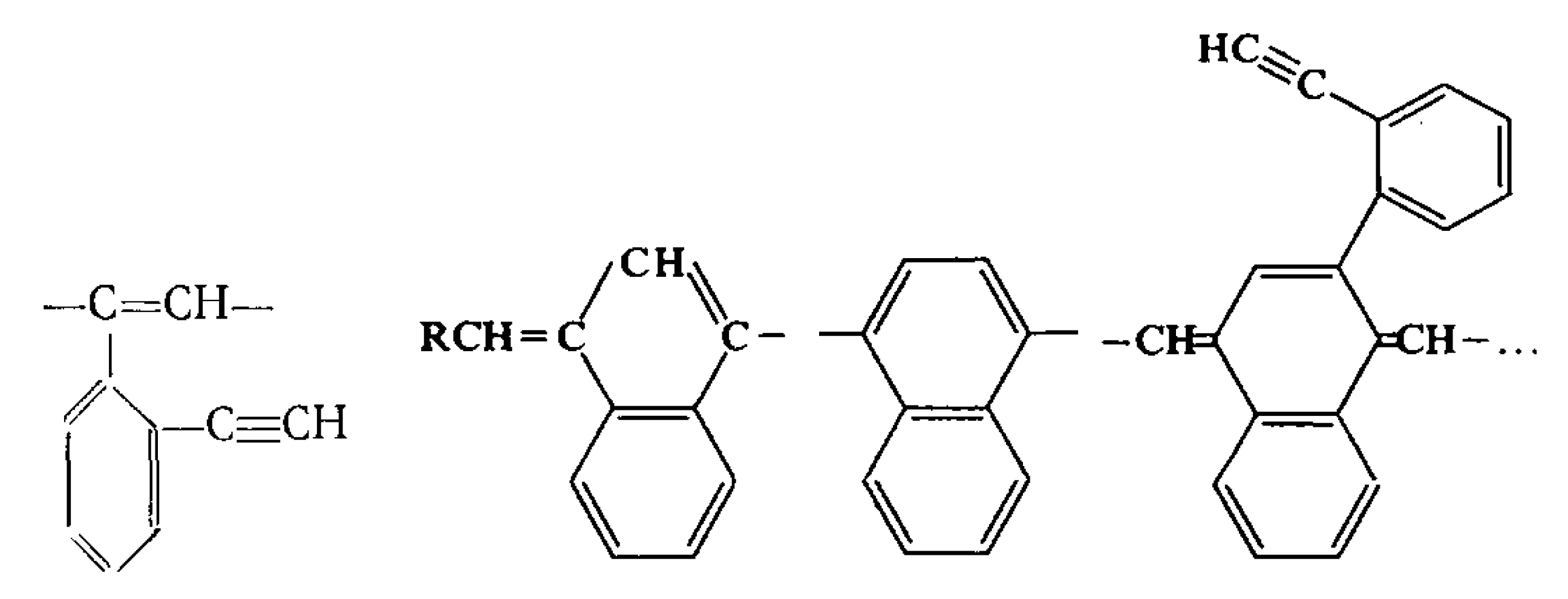
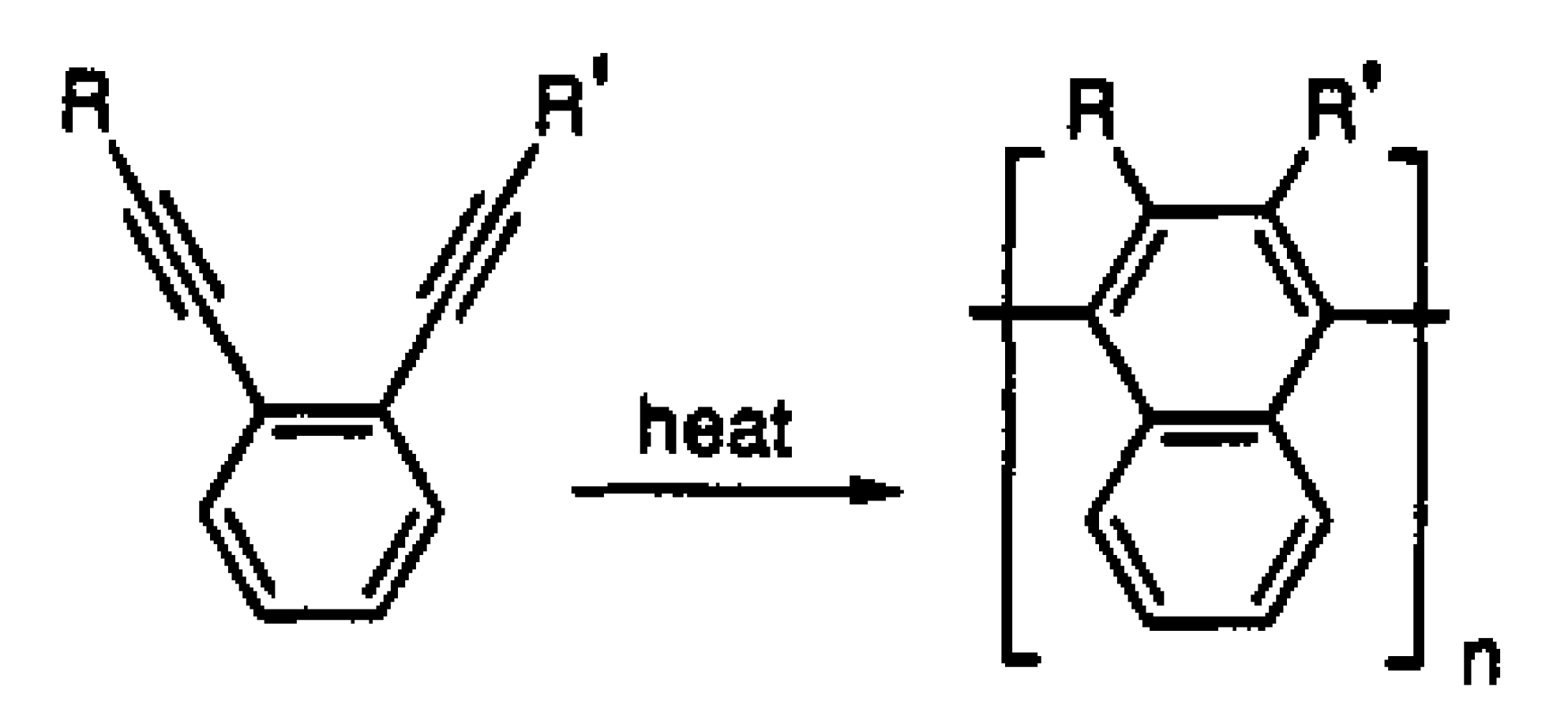
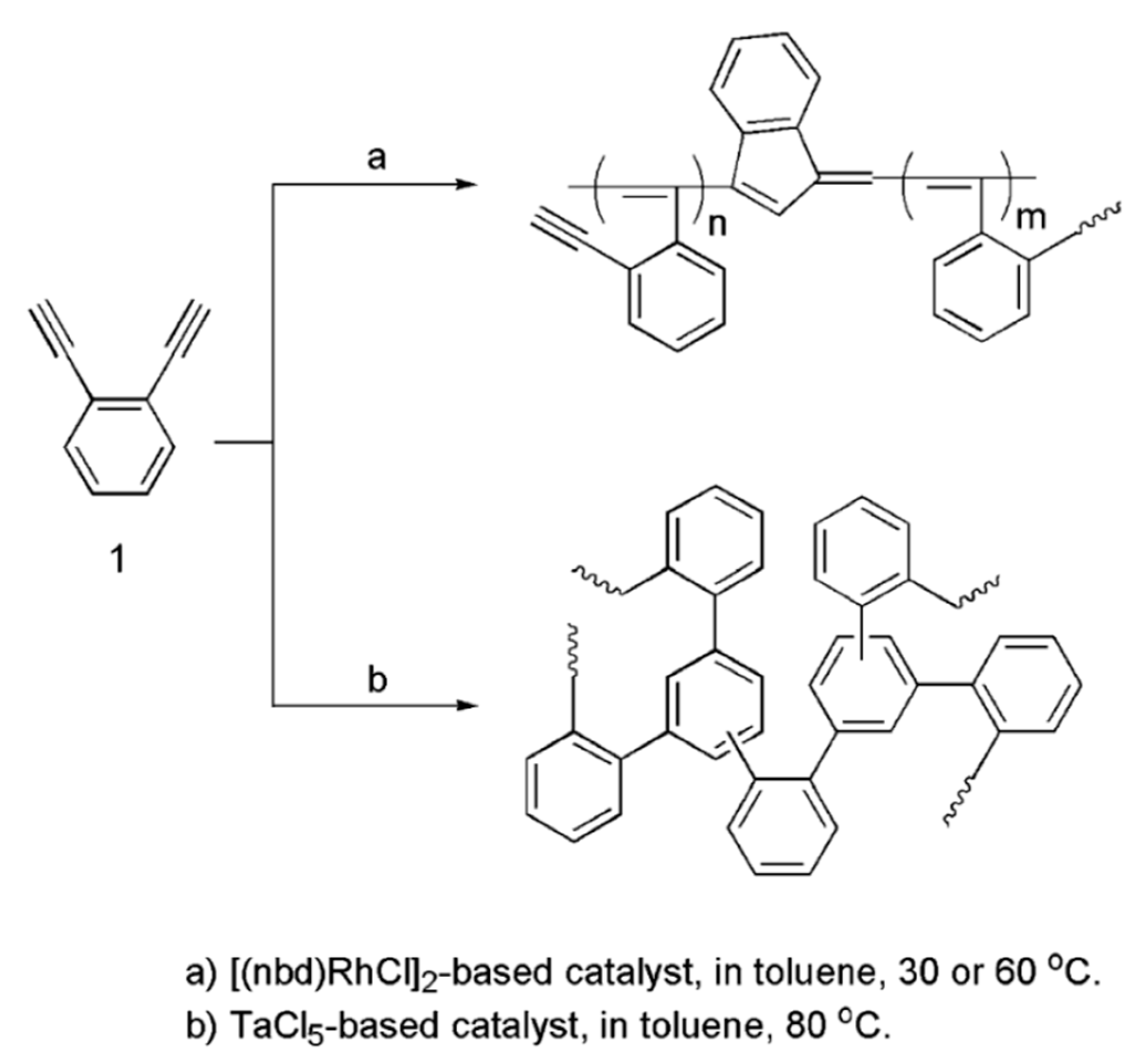
3. Synthesis, Structural Features, and Properties of o-Diethynylbenzene Polymers
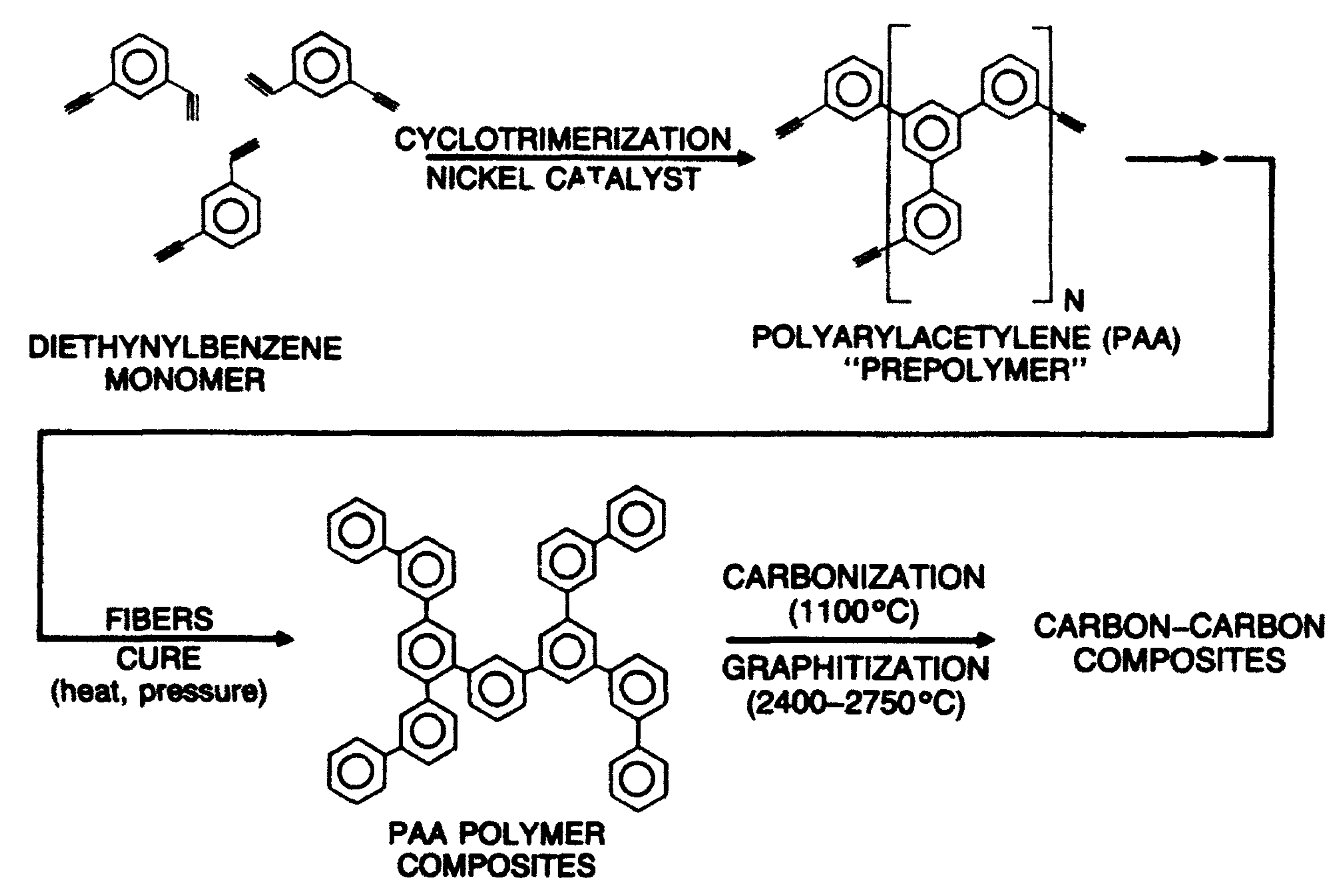
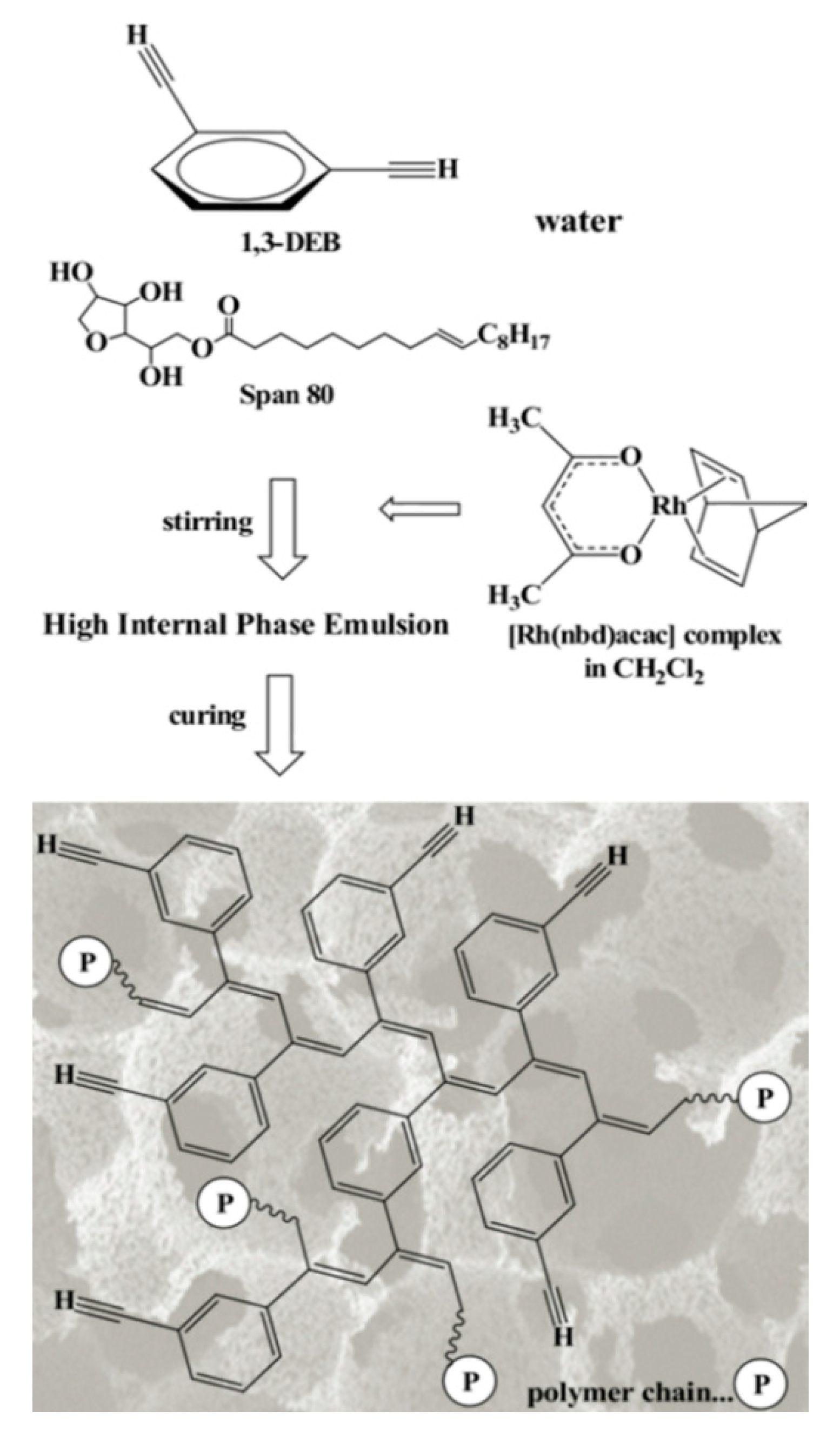
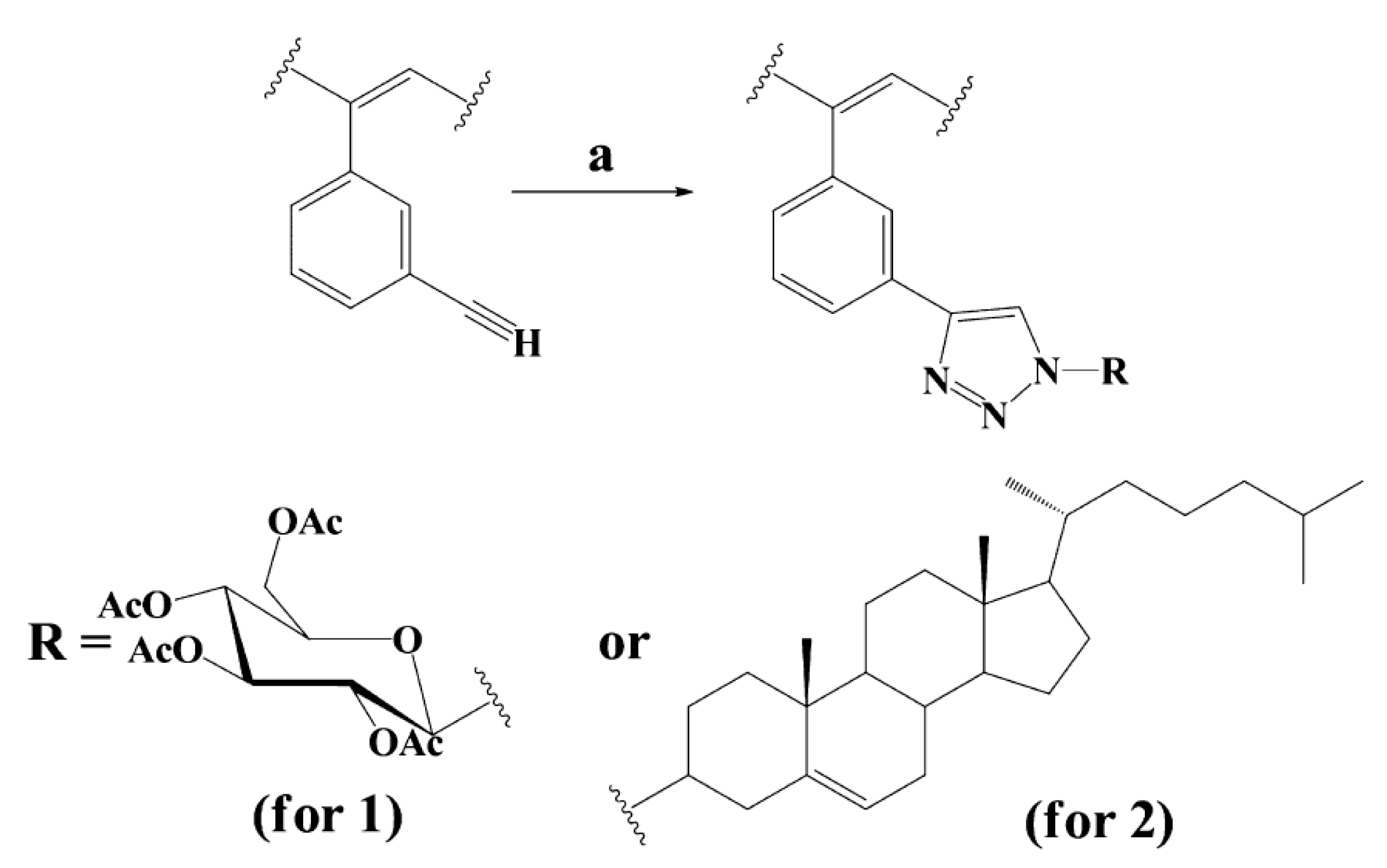
4. Synthesis, Structural Features and Properties of m-Diethynylbenzene Polymers
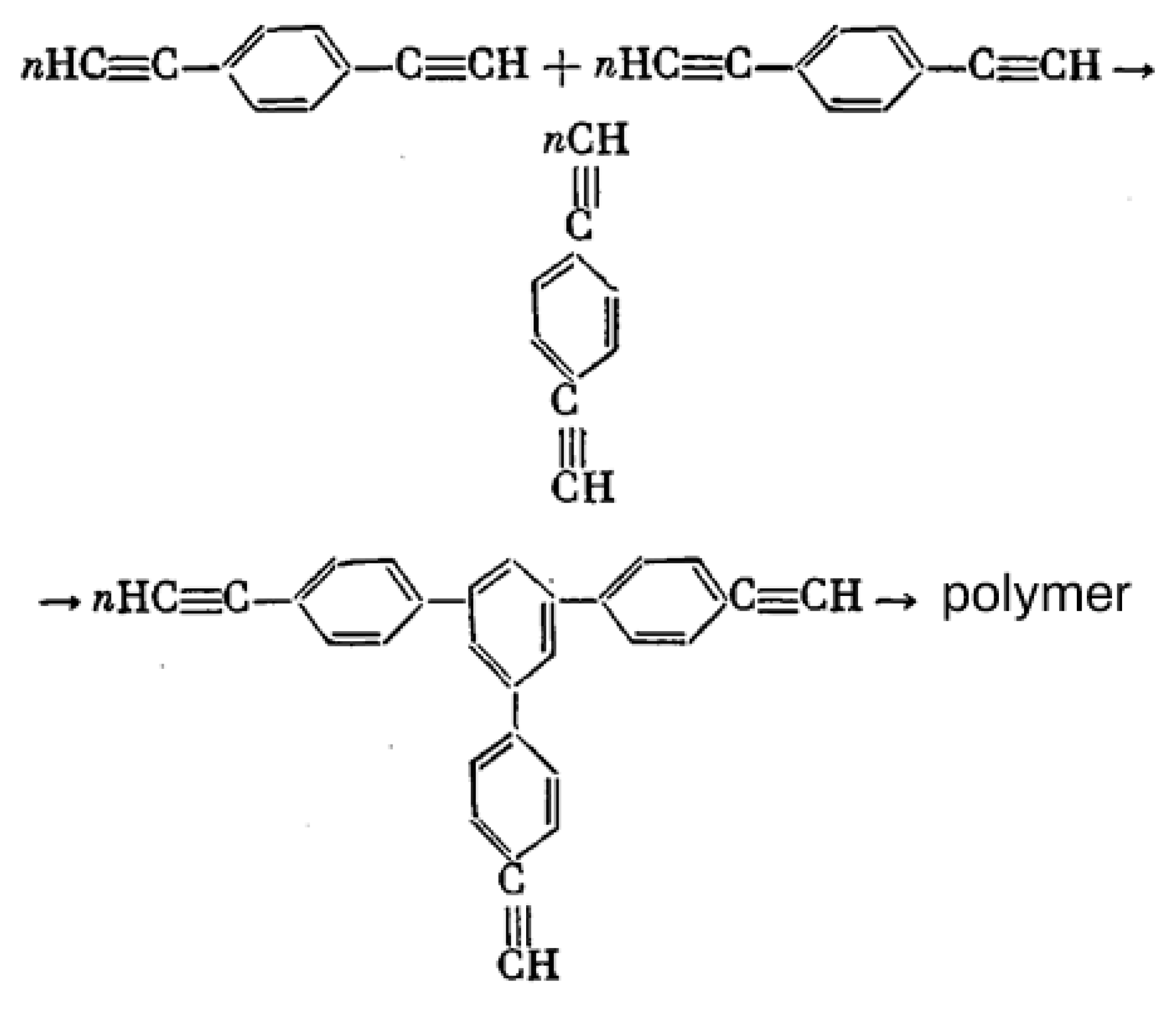
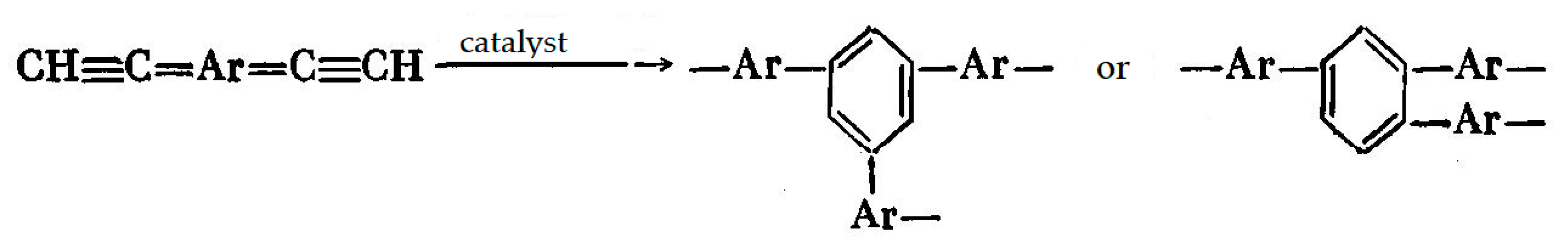
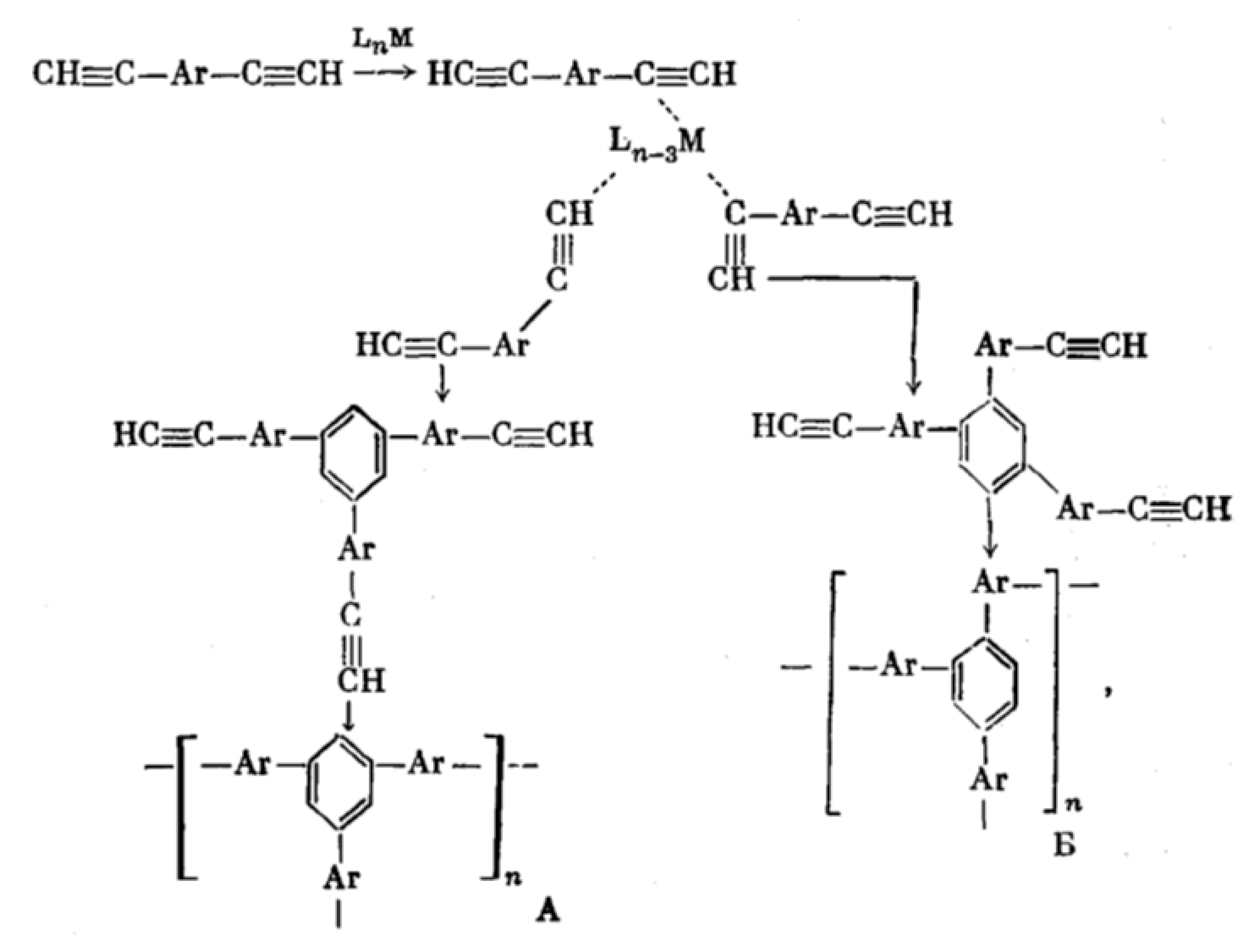
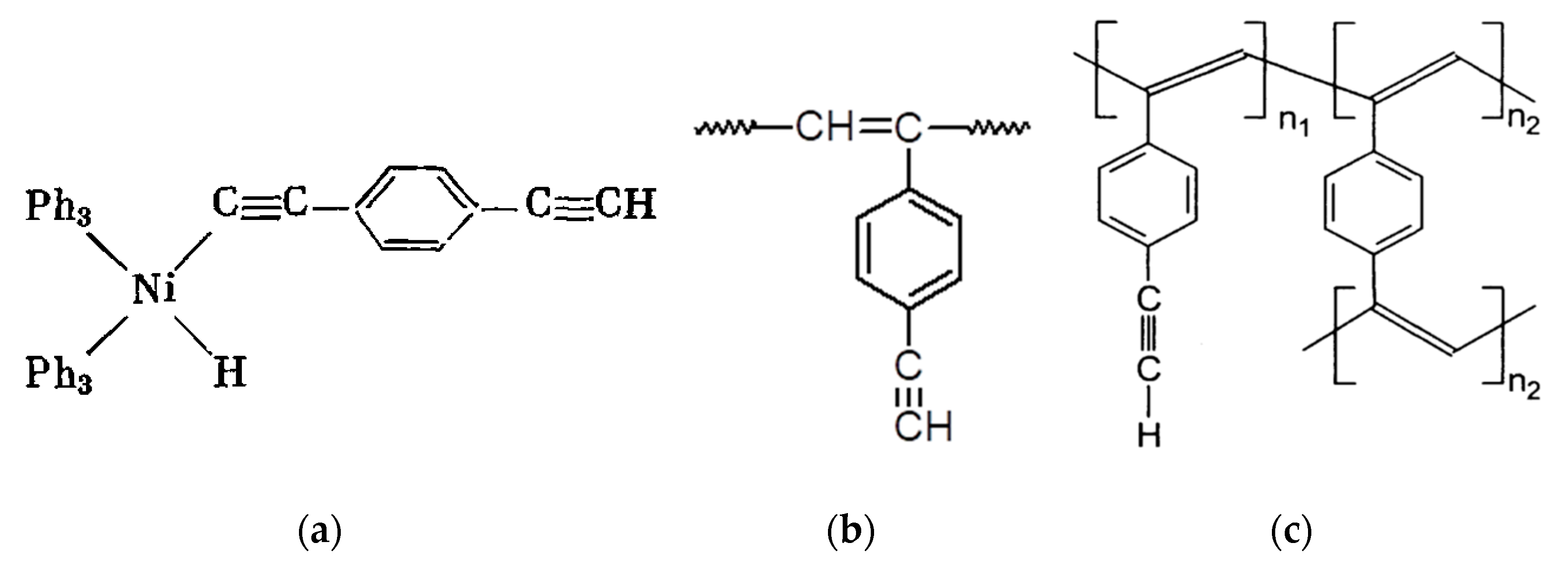
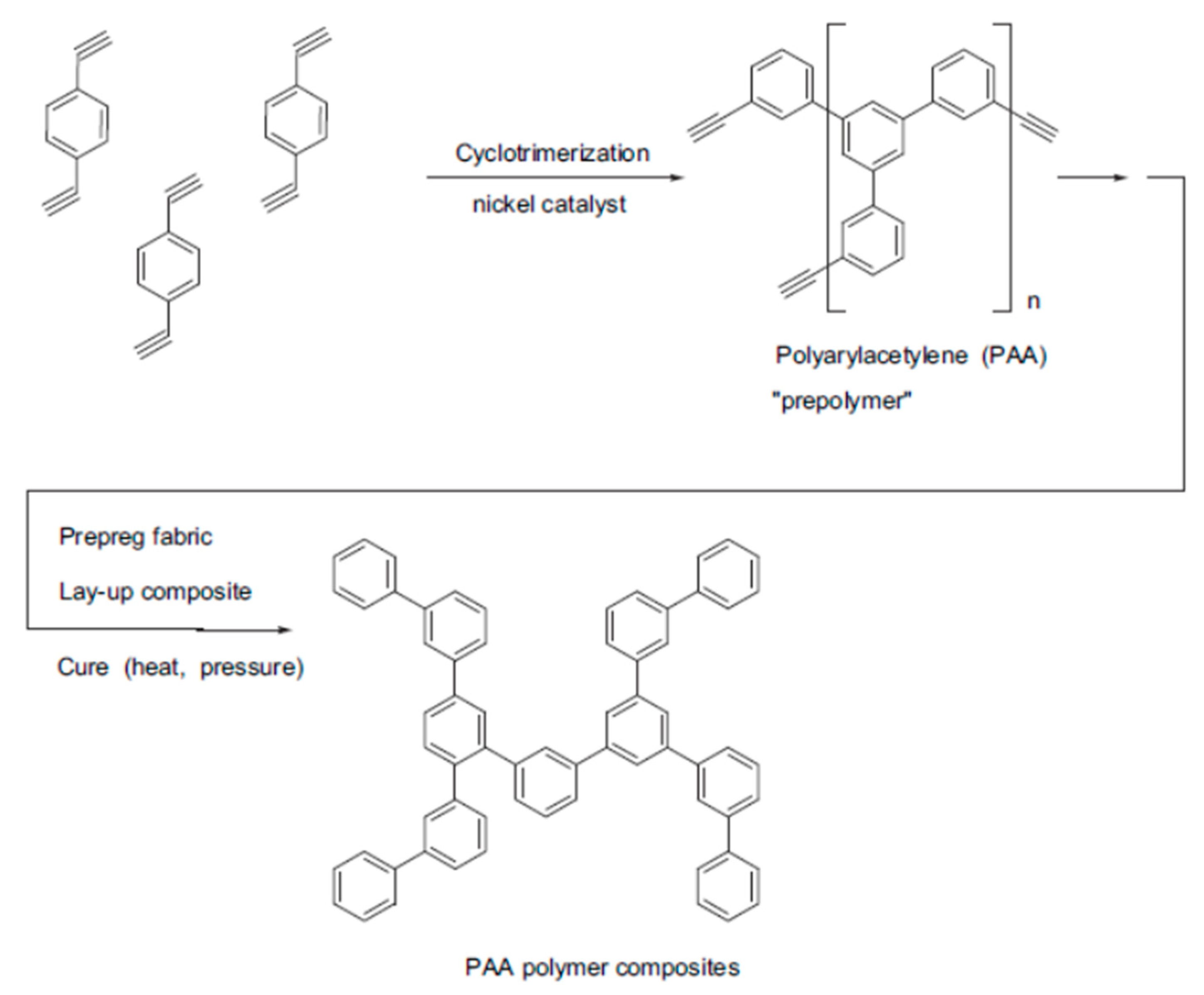
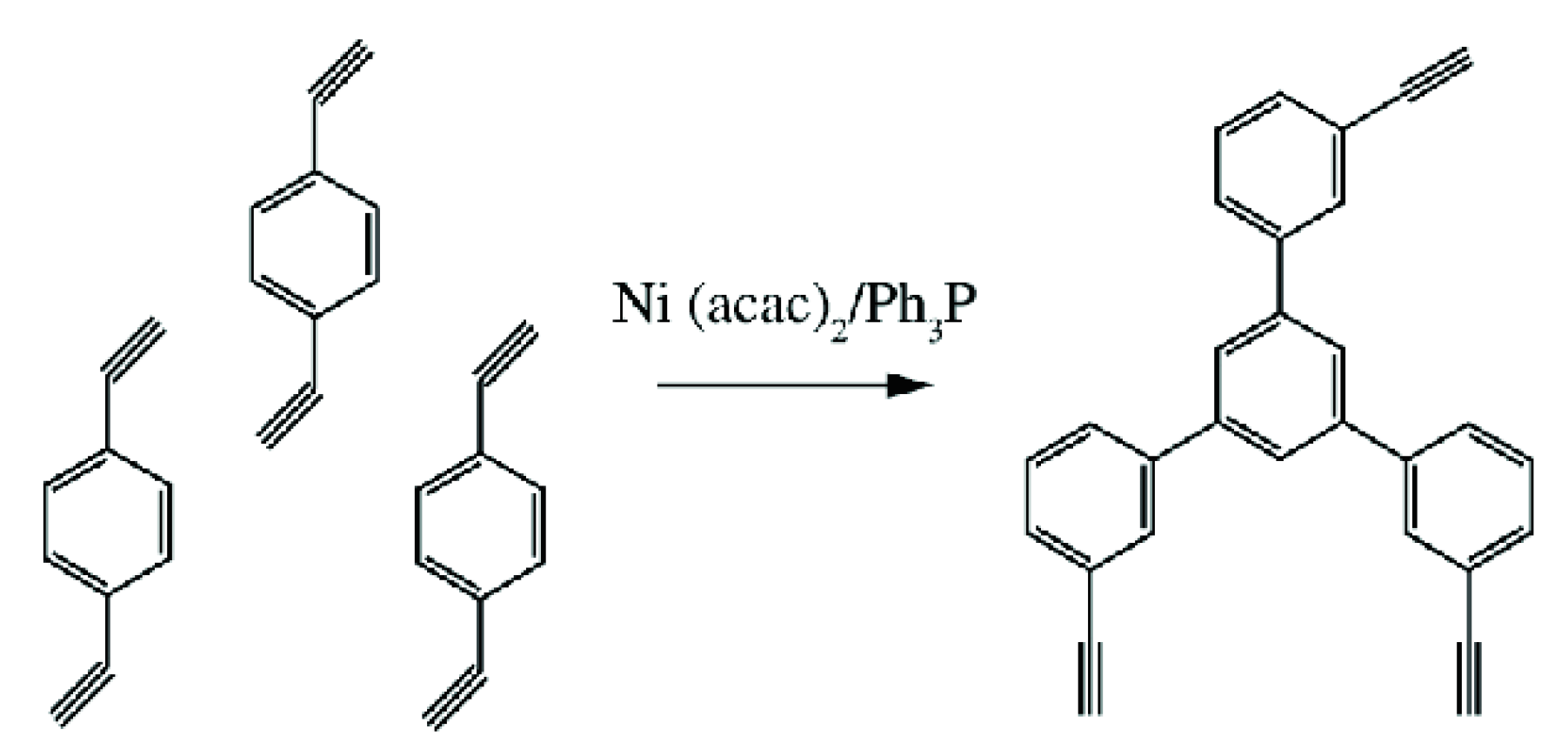
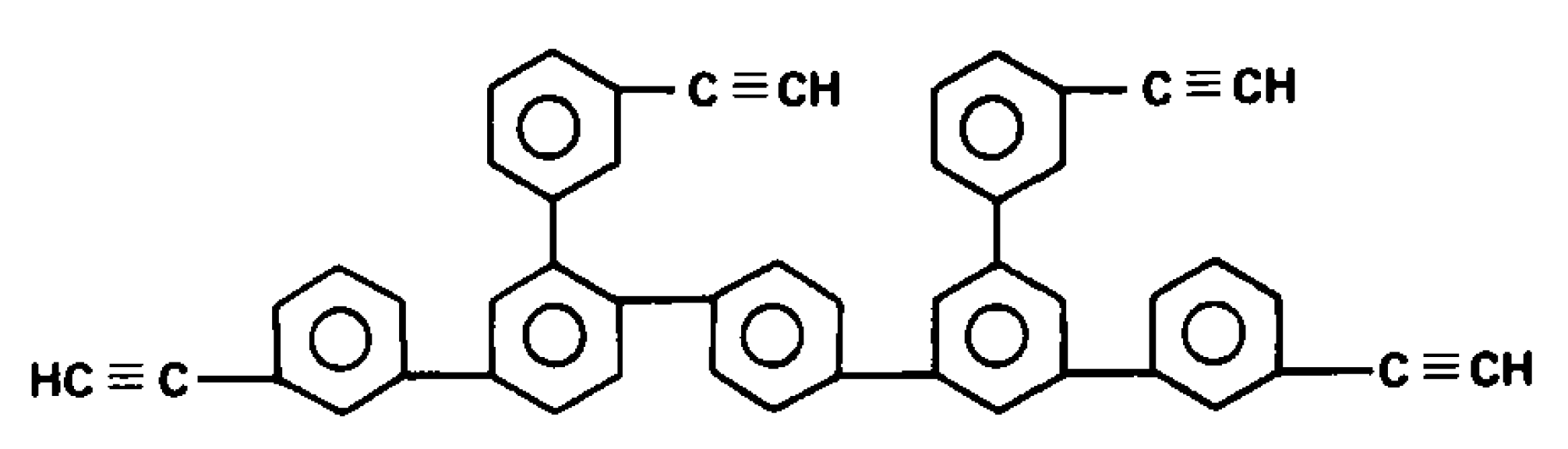
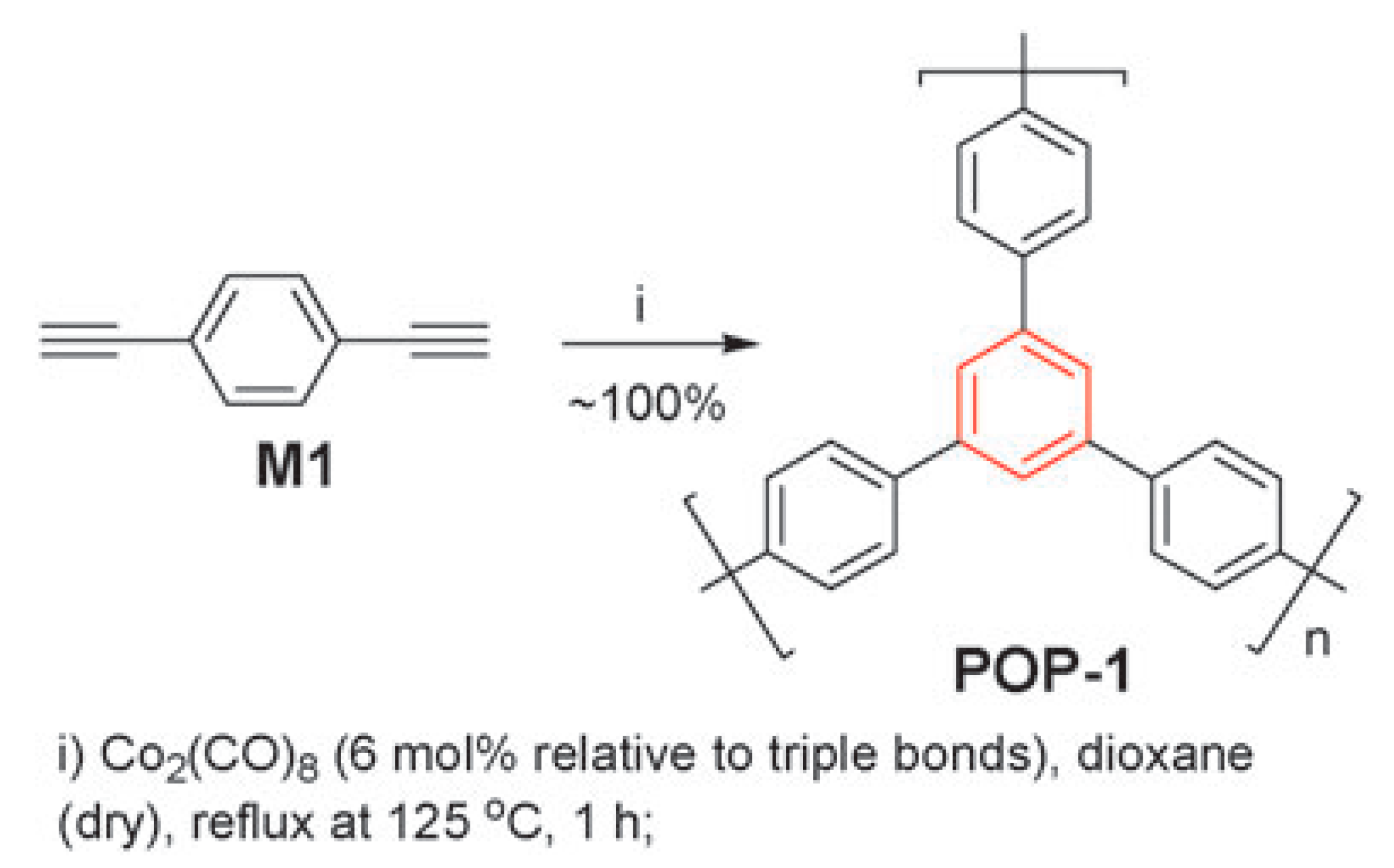
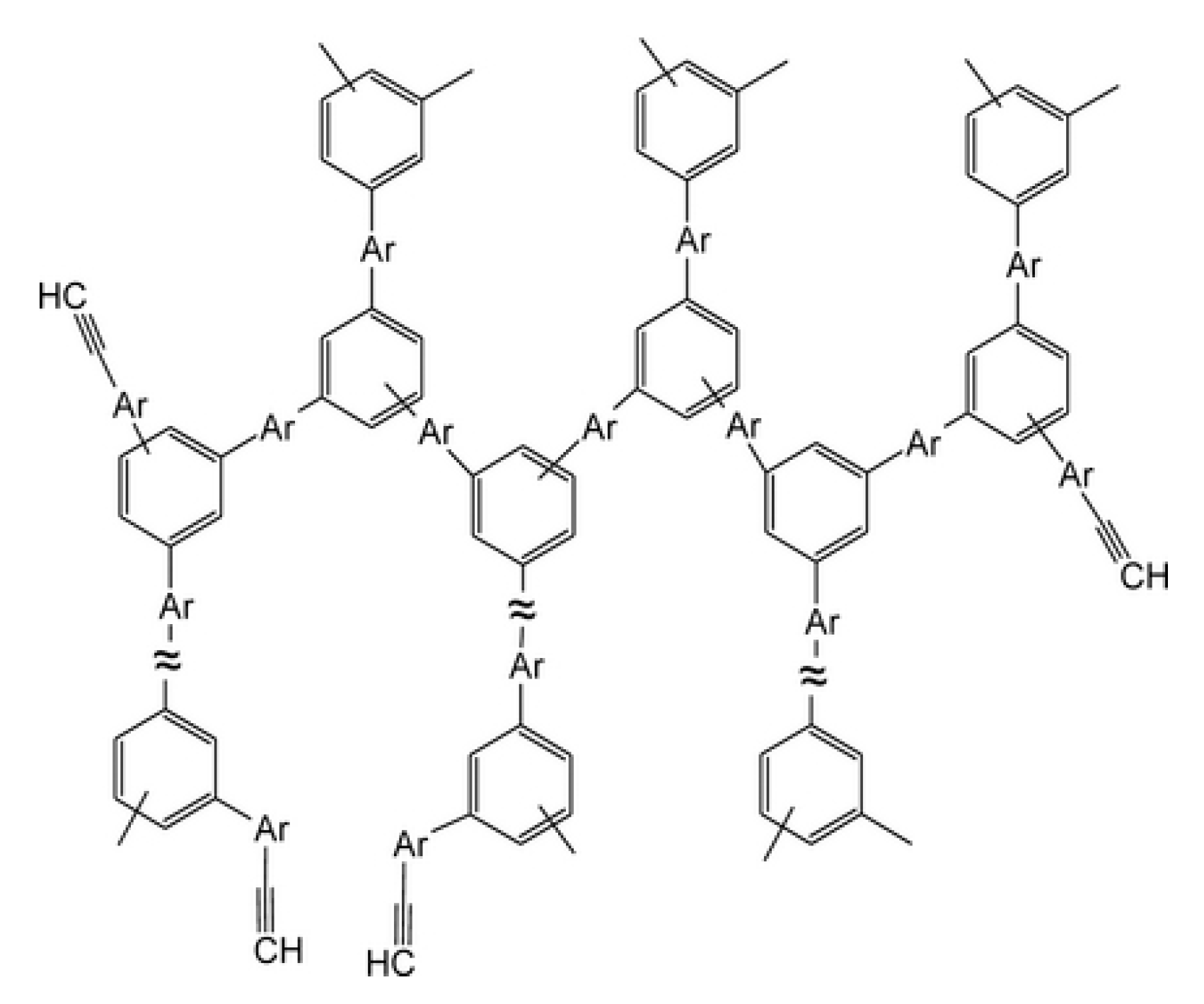
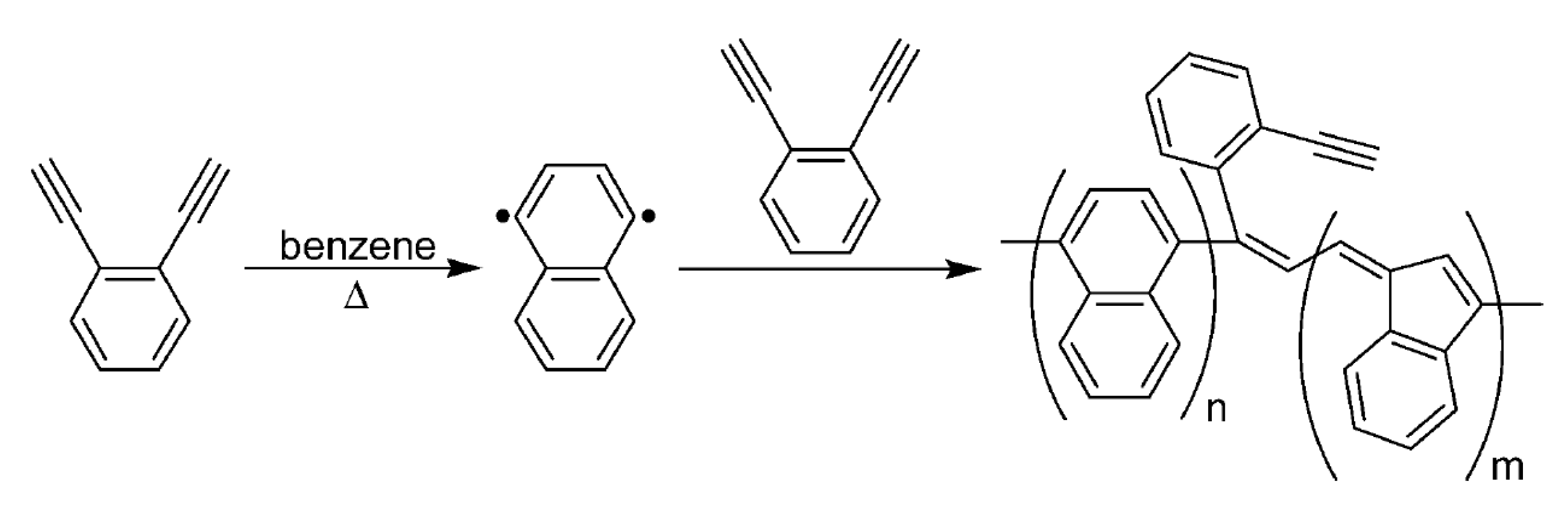
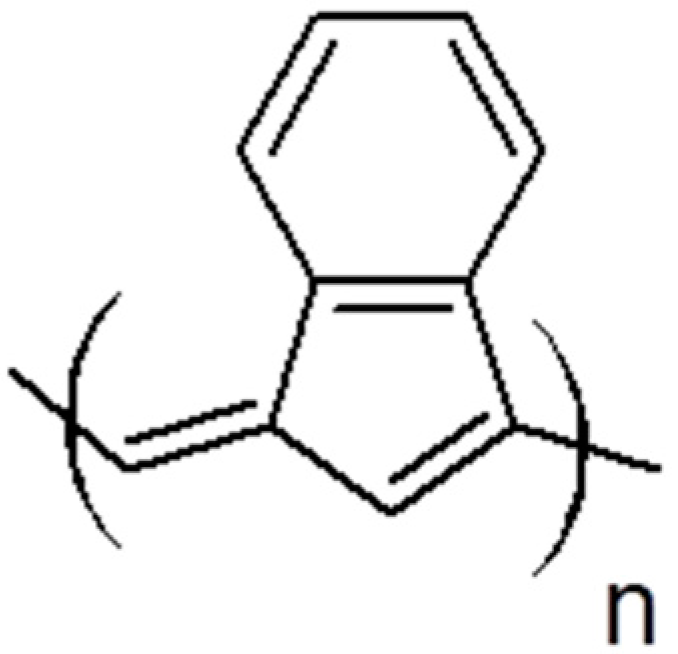
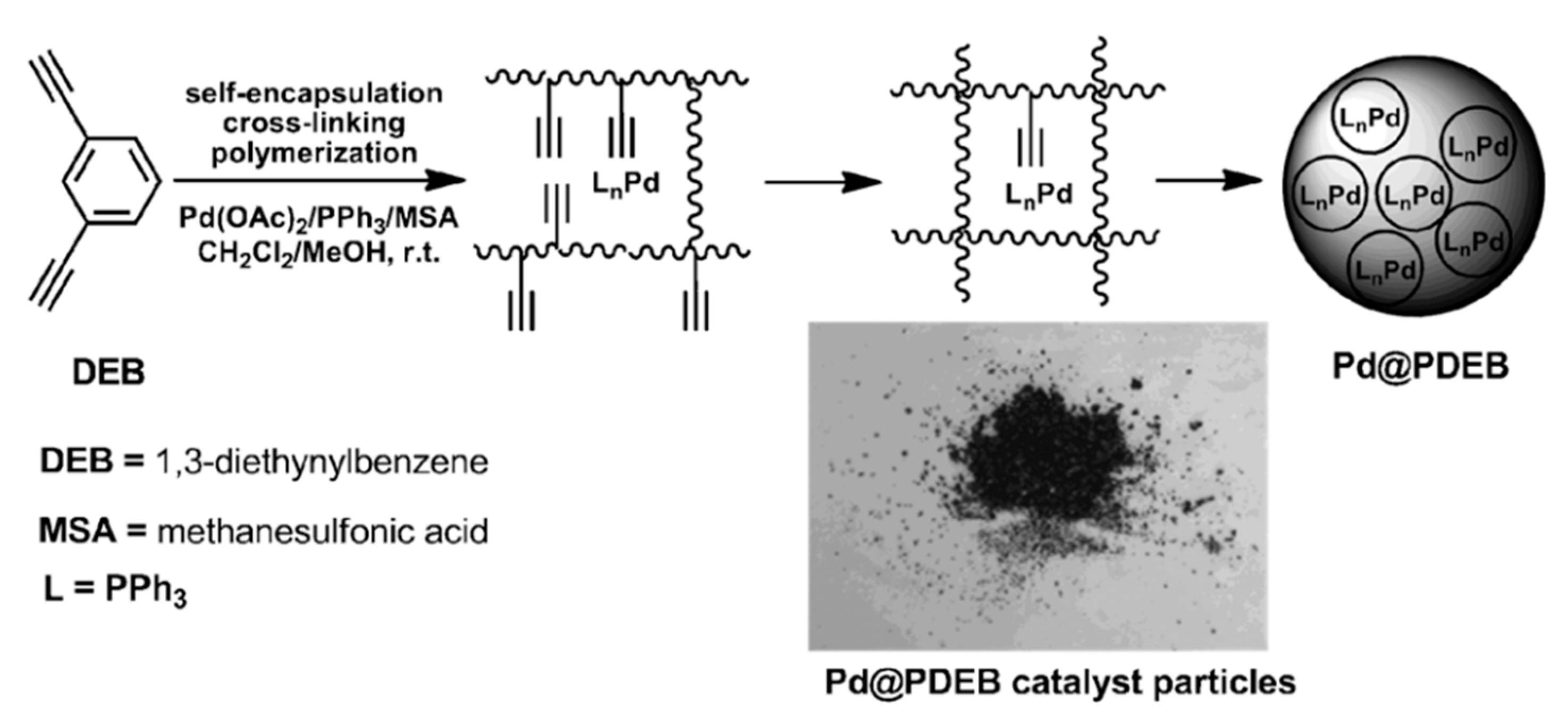
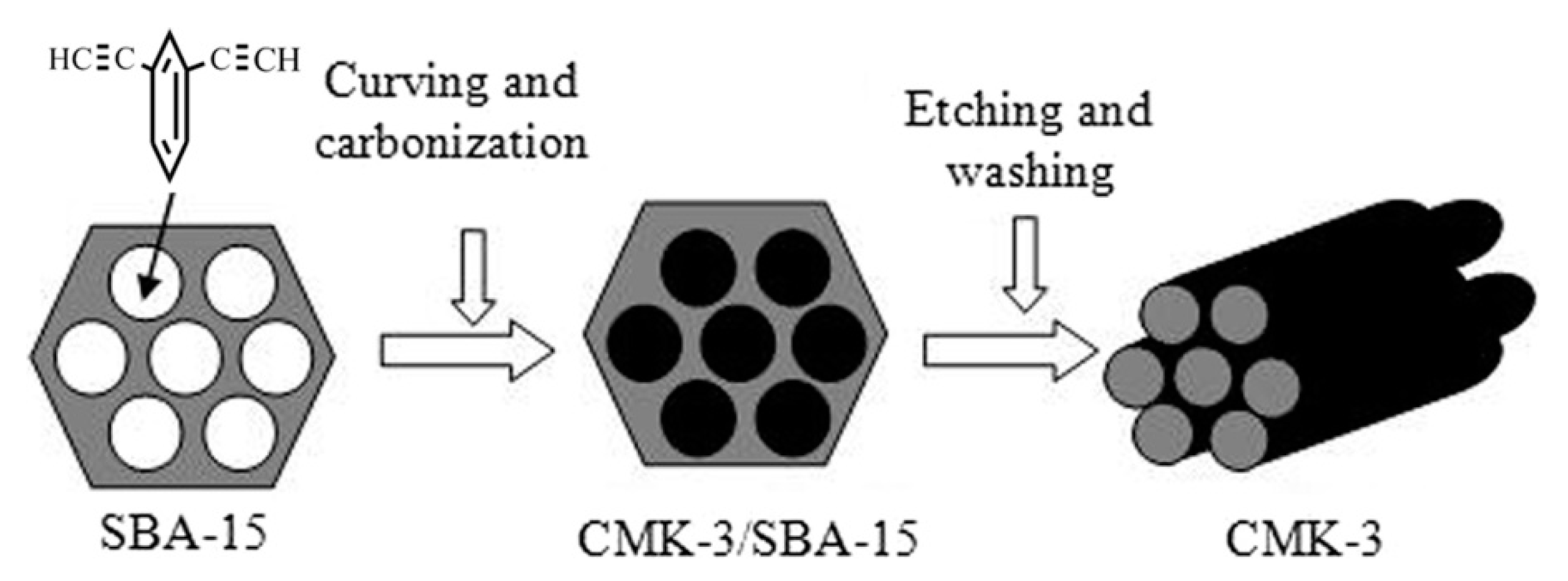
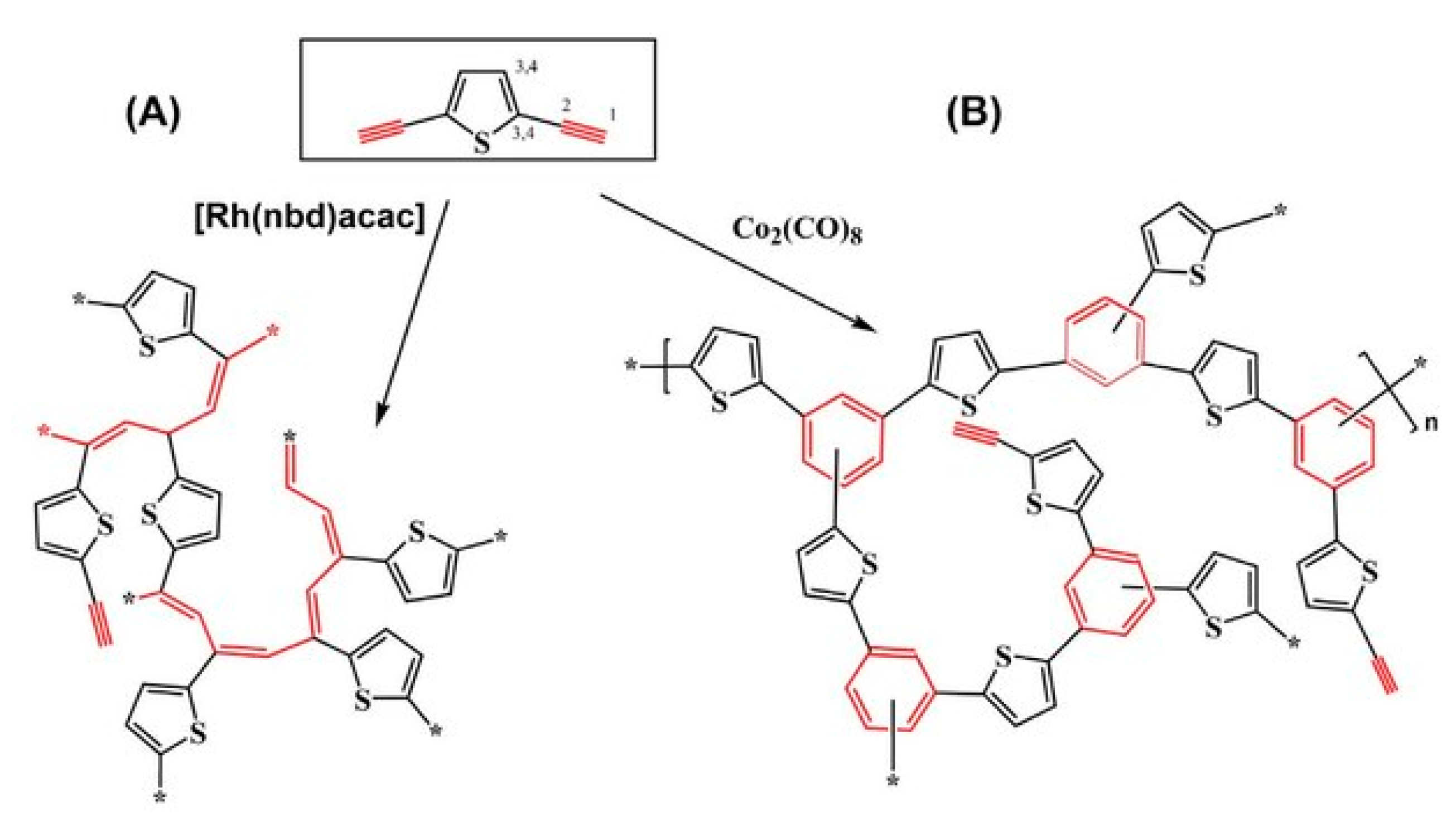
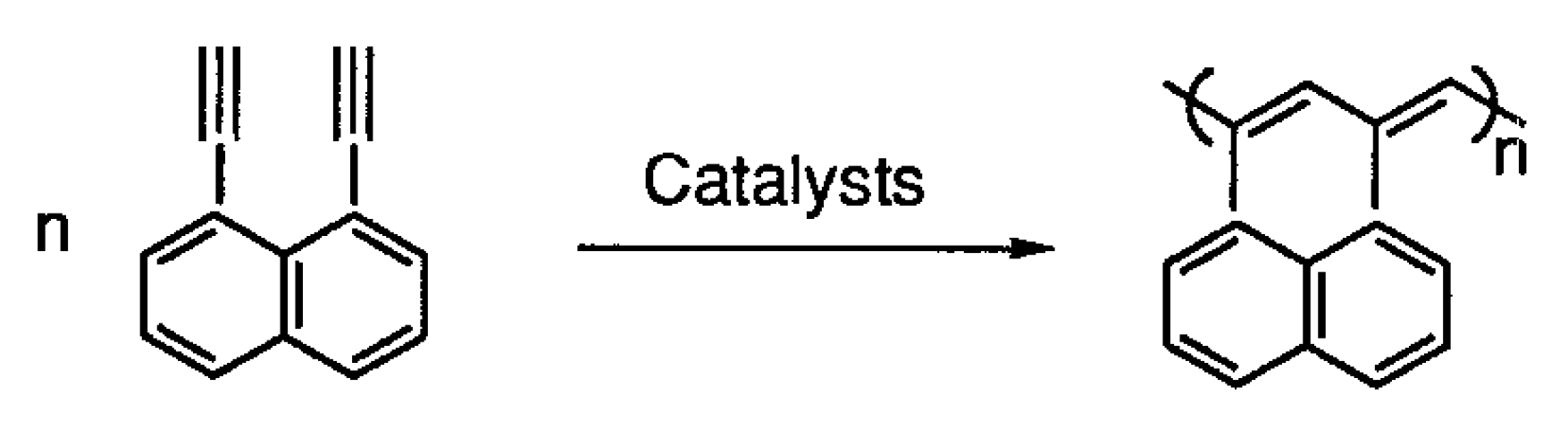
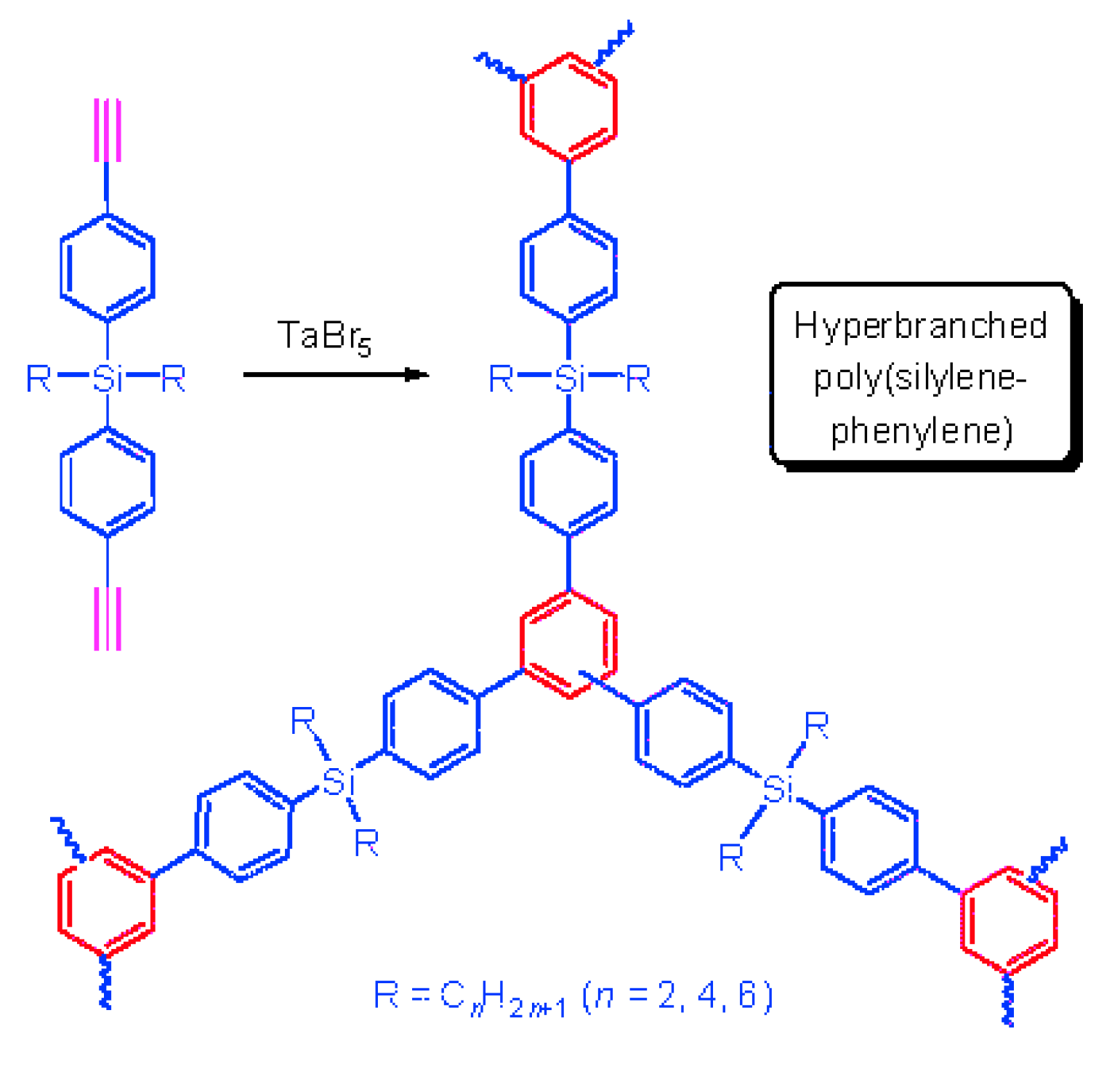
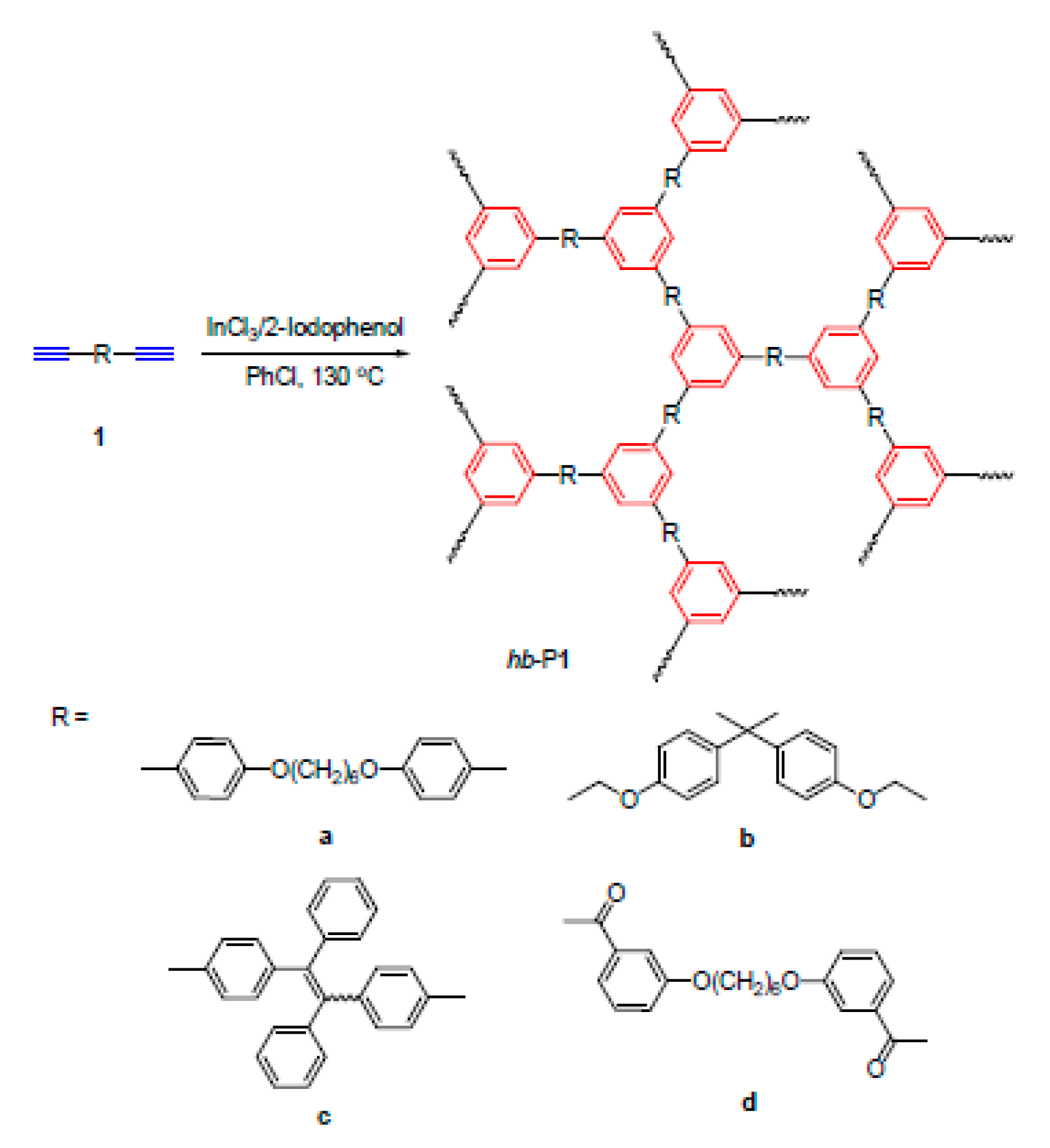
5. Polymers of Polynuclear Diethynylarenes
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chauser, M.G.; Rodionov, Y.M.; Misin, V.M.; Cherkashin, M.I. Polymerisation of Acetylenes. The Structure and Electrophysical Properties of Polyvinylenes. Russ. Chem. Rev. 1976, 45, 348–374. [Google Scholar] [CrossRef]
- Misin, V.M.; Cherkashin, M.I. The Solid-phase Polymerisation of Monomers with Conjugated Acetylenic Groups. Russ. Chem. Rev. 1985, 54, 562–593. [Google Scholar] [CrossRef]
- Nomura, R.; Masuda, T. Acetylenic Polymers, Substituted. In Encyclopedia of Polymer Science and Technology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2001; p. pst002. ISBN 978-0-471-44026-0. [Google Scholar]
- Häußler, M.; Qin, A.; Tang, B.Z. Acetylenes with multiple triple bonds: A group of versatile An-type building blocks for the construction of functional hyperbranched polymers. Polymer 2007, 48, 6181–6204. [Google Scholar] [CrossRef] [Green Version]
- Voit, B.I.; Lederer, A. Hyperbranched and Highly Branched Polymer Architectures—Synthetic Strategies and Major Characterization Aspects. Chem. Rev. 2009, 109, 5924–5973. [Google Scholar] [CrossRef]
- Liu, Y.; Qin, A.; Tang, B.Z. Polymerizations based on triple-bond building blocks. Prog. Polym. Sci. 2018, 78, 92–138. [Google Scholar] [CrossRef]
- Shiotsuki, M.; Sanda, F.; Masuda, T. Polymerization of substituted acetylenes and features of the formed polymers. Polym. Chem. 2011, 2, 1044–1058. [Google Scholar] [CrossRef]
- Chen, H.; Kong, J. Hyperbranched polymers from A2 + B3 strategy: Recent advances in description and control of fine topology. Polym. Chem. 2016, 7, 3643–3663. [Google Scholar] [CrossRef]
- Sedláček, J.; Balcar, H. Substituted Polyacetylenes Prepared with Rh Catalysts: From Linear to Network-Type Conjugated Polymers. Polym. Rev. 2017, 57, 31–51. [Google Scholar] [CrossRef]
- Qiu, Z.; Han, T.; Lam, J.W.Y.; Tang, B.Z. Recent New Methodologies for Acetylenic Polymers with Advanced Functionalities. Top. Curr. Chem. (Z) 2017, 375, 70. [Google Scholar] [CrossRef]
- Masuda, T.; Higashimura, T. Polyacetylenes with substituents: Their synthesis and properties. In Catalytical and Radical Polymerization; Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 1986; Volume 81, pp. 121–165. ISBN 978-3-540-16754-9. [Google Scholar]
- Bilow, N.; Landis, A.L.; Austin, W.B.; Woolley, D.D. Arylacetylenes as high char forming matrix resins. SAMPE J. 1982, 18, 19–24. [Google Scholar]
- Dong, Z.; Ye, Z. Reusable, Highly Active Heterogeneous Palladium Catalyst by Convenient Self-Encapsulation Cross-Linking Polymerization for Multiple Carbon—Carbon Cross-Coupling Reactions at ppm to ppb Palladium Loadings. Adv. Synth. Catal. 2014, 356, 3401–3414. [Google Scholar] [CrossRef]
- Hanková, V.; Slováková, E.; Zedník, J.; Vohlídal, J.; Sivkova, R.; Balcar, H.; Zukal, A.; Brus, J.; Sedláček, J. Polyacetylene-Type Networks Prepared by Coordination Polymerization of Diethynylarenes: New Type of Microporous Organic Polymers. Macromol. Rapid Commun. 2012, 33, 158–163. [Google Scholar] [CrossRef]
- Slováková, E.; Zukal, A.; Brus, J.; Balcar, H.; Brabec, L.; Bondarev, D.; Sedláček, J. Transition-Metal-Catalyzed Chain-Growth Polymerization of 1,4-Diethynylbenzene into Microporous Crosslinked Poly(phenylacetylene)s: The Effect of Reaction Conditions. Macromol. Chem. Phys. 2014, 215, 1855–1869. [Google Scholar] [CrossRef]
- Slováková, E.; Ješelnik, M.; Žagar, E.; Zedník, J.; Sedláček, J.; Kovačič, S. Chain-Growth Insertion Polymerization of 1,3-Diethynylbenzene High Internal Phase Emulsions into Reactive π-Conjugated Foams. Macromolecules 2014, 47, 4864–4869. [Google Scholar] [CrossRef]
- Zhang, W.; Shiotsuki, M.; Masuda, T. Polymerization of o-diethynylbenzene and its derivatives controlled by transition metal catalyst systems. Polymer 2006, 47, 2956–2961. [Google Scholar] [CrossRef]
- Bolasco, A.; Chimenti, F.; Frezza, A.; Furlani, A.; Infante, G.; Muraglia, E.; Ortaggi, G.; Polzonetti, G.; Russo, M.V.; Sleiter, G. Polymerization of p-diethynylbiphenyl via oxidative or catalytic reactions. Polymer 1992, 33, 3049–3054. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, J.; Du, S.; Zhang, L.; Li, G.; Zhang, Y.; Tang, B.Z.; Gao, H.-J. Direct Visualization of Surface-Assisted Two-Dimensional Diyne Polycyclotrimerization. J. Am. Chem. Soc. 2014, 136, 5567–5570. [Google Scholar] [CrossRef]
- Bondarev, D.; Sivkova, R.; Šuly, P.; Polášková, M.; Krejčí, O.; Křikavová, R.; Trávníček, Z.; Zukal, A.; Kubů, M.; Sedláček, J. Microporous conjugated polymers via homopolymerization of 2,5-diethynylthiophene. Eur. Polym. J. 2017, 92, 213–219. [Google Scholar] [CrossRef]
- Häußler, M.; Lam, J.W.Y.; Zheng, R.; Peng, H.; Luo, J.; Chen, J.; Law, C.C.W.; Tang, B.Z. Hyperbranched polyarylenes. Comptes Rendus Chim. 2003, 6, 833–842. [Google Scholar] [CrossRef]
- Cessna, L.C.; Jabloner, H. A New Class of Easily Moldable Highly Stable Thermosetting Resins. J. Elastomers Plast. 1974, 6, 103–113. [Google Scholar] [CrossRef]
- Hergenrother, P.M. Acetylene-Containing Precursor Polymers. J. Macromol. Sci. Polym. Rev. 1980, 19, 1–34. [Google Scholar] [CrossRef]
- Katzman, H.A.; Mallon, J.J.; Barry, W.T. Polyarylacetylene-matrix composites for solid rocket motor components. J. Adv. Mater. 1995, 3, 21–27. [Google Scholar]
- Zhang, X.; Huang, Y.; Wang, T.; Liu, L. Influence of fibre surface oxidation–reduction followed by silsesquioxane coating treatment on interfacial mechanical properties of carbon fibre/polyarylacetylene composites. Compos. Part A 2007, 38, 936–944. [Google Scholar] [CrossRef]
- Rondeau-Gagné, S.; Morin, J.-F. Preparation of carbon nanomaterials from molecular precursors. Chem. Soc. Rev. 2014, 43, 85–98. [Google Scholar] [CrossRef]
- Li, Q.; Yu, R.; Zhu, C.; Jiao, Z. 60Co γ-rays irradiation modified p-diethynylbenzene as prepolymers to prepare polyarylacetylene with excellent heat resistance. Polym. Degrad. Stab. 2015, 114, 81–88. [Google Scholar] [CrossRef]
- Cai, W.; Li, M.; Wang, S.; Gu, Y.; Li, Q.; Zhang, Z. Strong, flexible and thermal-resistant CNT/polyarylacetylene nanocomposite films. RSC Adv. 2016, 6, 4077–4084. [Google Scholar] [CrossRef]
- Glagolev, N.N.; Misin, V.M.; Cherkashin, M.I. Synthesis of Organometallic Acetylene Compounds Based on an Oligomer with a Free Ethynyl Group; USSR: Aghveran, Armenia, 1984; p. 211. [Google Scholar]
- Sergeyev, V.A.; Kirilenko, Y.K.; Plyashkevich, L.A.; Kalinin, V.N.; Perepechkina, Y.P.; Shitikov, V.K.; Kudryavtsev, G.I.; Parfenov, B.P.; Zakharkin, L.I. Carborane-containing poly(arylacetylenes). Polym. Sci. USSR 1986, 28, 2638–2641. [Google Scholar] [CrossRef]
- Misin, V.M.; Glagolev, N.N.; Misin, M.V. Structure and properties of polymeric ethyne-hexacarbonyldicobalt π complexes. Russ. J. Appl. Chem. 2007, 80, 1910–1913. [Google Scholar] [CrossRef]
- Misin, V.M.; Stoyanov, O.V.; Ordyan, A.E. Steric obstacles in the synthesis of metal-containing polyphenyldiacetylene. Vestn. Kazan. Tekhnol. Univ. 2016, 19, 50–59. [Google Scholar]
- Misin, V.M.; Maltseva, I.E.; Maltsev, A.A.; Naumkin, A.V.; Kazakov, M.E. Anionic Polymerization of Para-Diethynylbenzene: Synthesis of a Strictly Linear Polymer. Polymers 2022, 14, 900. [Google Scholar] [CrossRef]
- Bakbak, S.; Leech, P.J.; Carson, B.E.; Saxena, S.; King, W.P.; Bunz, U.H.F. 1,3-Dipolar Cycloaddition for the Generation of Nanostructured Semiconductors by Heated Probe Tips. Macromolecules 2006, 39, 6793–6795. [Google Scholar] [CrossRef]
- Brombosz, S.M.; Appleton, A.L.; Zappas II, A.J.; Bunz, U.H.F. Water-soluble benzo- and naphtho-thiadiazole-based bistriazoles and their metal-binding properties. Chem. Commun. 2010, 46, 1419. [Google Scholar] [CrossRef]
- Liu, J.; Lam, J.W.Y.; Jim, C.K.W.; Ng, J.C.Y.; Shi, J.; Su, H.; Yeung, K.F.; Hong, Y.; Faisal, M.; Yu, Y.; et al. Thiol−Yne Click Polymerization: Regio- and Stereoselective Synthesis of Sulfur-Rich Acetylenic Polymers with Controllable Chain Conformations and Tunable Optical Properties. Macromolecules 2011, 44, 68–79. [Google Scholar] [CrossRef]
- Bao, L.; Sun, H.; Zhu, Z.; Liang, W.; Mu, P.; Zang, J.; Li, A. Synthesis and properties of tubular-shape conjugated microporous polymers with high purity. Mater. Lett. 2016, 178, 5–9. [Google Scholar] [CrossRef]
- Bangcuyo, C.G.; Ellsworth, J.M.; Evans, U.; Myrick, M.L.; Bunz, U.H.F. Quinoxaline-Based Poly(aryleneethynylene)s. Macromolecules 2003, 36, 546–548. [Google Scholar] [CrossRef]
- Dong, Z.; Ye, Z. Synthesis of Hyperbranched Poly(phenylacetylene)s Containing Pendant Alkyne Groups by One-Pot Pd-Catalyzed Copolymerization of Phenylacetylene with Diynes. Macromolecules 2012, 45, 5020–5031. [Google Scholar] [CrossRef]
- Kapustyan, V.M.; Zharov, A.A.; Enikolopyan, N.S. Polymerization of monomers in the solid phase under conditions of high pressures and shear stresses. Dokl. Akad. Nauk SSSR 1968, 179, 627–628. [Google Scholar]
- Broude, V.L.; Gol’danskii, V.I.; Gordon, D.A. Photopolymerization of p-diethynylbenzene single crystals. Khim. Vys. Energ. 1968, 2, 165–171. [Google Scholar]
- Gordon, D.A.; Migunova, I.I.; Mikhailov, A.I. Solid-phase reaction of monomer molecule addition to a radical at 77.deg.K in the presence of ionizing radiation. Dokl. Akad. Nauk SSSR 1973, 213, 368–371. [Google Scholar]
- Gordon, D.A.; Mikhailov, A.I. Localization of the unpaired electron in the macroradical of the growing polymer chain in low temperature, solid phase polymerization of p-diethynylbenzene. Polym. Sci. USSR 1975, 17, 1910–1919. [Google Scholar] [CrossRef]
- Gordon, D.A.; Mikhailov, A.I. On the localization of the unpaired electron in a macroradical of a propagating polymer chain during low-temperature, solid-phase polymerization of p-diethynylbenzene. J. Photochem. Photobiol. A 1995, 86, 253–257. [Google Scholar] [CrossRef]
- Gordon, D.A.; Mikhailov, A.I. Low Temperature Polymerization in p-Diethynylbenzene Crystal: Localization of Unpaired Electron in Poly-Conjugated System. J. Low Temp. Phys. 2005, 139, 675–681. [Google Scholar] [CrossRef]
- Gordon, D.A.; Mikhaylov, A.I. On kinetic features of photo- or γ-induced polymerization in p-diethynylbenzene crystals in the temperature range 4.2–300K. Low Temp. Phys. 2009, 35, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.A.; Mikhailov, A.I. The growth and interaction of conjugated polymer chains, the unpaired electron localization. J. Polym. Sci. B Polym. Phys. 1994, 32, 2405–2411. [Google Scholar] [CrossRef]
- Gordon, D.A.; Volodina, V.A.; Mikhailov, A.I. Characteristics of the radical polymerization of acetylene monomers. Russ. J. Phys. Chem. 2014, 88, 2129–2136. [Google Scholar] [CrossRef]
- Gordon, D.A.; Volodina, V.A.; Mikhailov, A.I. Unpaired-electron localization and delocalization in macromolecules with a system of conjugated C=C bonds. High Energy Chem. 2014, 48, 5–10. [Google Scholar] [CrossRef]
- Hagihara, M.; Yamamoto, Y.; Takahashi, S.; Hayashi, K. Radiation-induced polymerization of crystalline diethynylbenzene derivatives. Int. J. Radiat. Appl. Instrum. Part C. Radiat. Phys. Chem. 1986, 28, 165–167. [Google Scholar] [CrossRef]
- Kozlovskii, A.A.; Gordon, D.A.; Volodina, V.A.; Bol’shakov, A.I.; Mikhailov, A.I. Initiation of free-radical polymerization of acetylene monomers with molecular chlorine at low temperatures. Russ. J. Phys. Chem. 2010, 84, 1439–1444. [Google Scholar] [CrossRef]
- Gordon, D.A.; Volodina, V.A.; Mikhailov, A.I. Reactivity of free radical intermediates that form spontaneously during molecular chlorine action on acetylene and vinyl monomers at low temperatures. Low Temp. Phys. 2013, 39, 565–570. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.A.; Mikhailov, A.I. Features of the processes occurring during low-temperature reactions between molecular chlorine and monomers and during the heating of mixtures. Russ. J. Phys. Chem. B 2016, 10, 699–706. [Google Scholar] [CrossRef]
- Eichhorn, J.; Heckl, W.M.; Lackinger, M. On-surface polymerization of 1,4-diethynylbenzene on Cu(111). Chem. Commun. 2013, 49, 2900. [Google Scholar] [CrossRef]
- Volkova, N.N.; Zholudev, A.F.; Kislov, M.B.; Suslova, E.V.; Yanovskiy, L.S. Microstructure of 1,4-diethynylbenzene frontal polymerization products. IOP Conf. Ser. Mater. Sci. Eng. 2019, 693, 012027. [Google Scholar] [CrossRef] [Green Version]
- Cherkashin, M.I.; Kisilitsa, P.P.; Berlin, A.A. On the production of highly conducting polymers based on para-diethynylbenzene. Russ. Chem. Bull. 1967, 16, 2335–2337. [Google Scholar] [CrossRef]
- Berlin, A.A.; Vonsiatsky, V.A.; Liogonkiy, B.I. About quasi-radical block copolymerization. Dokl. AN SSSR 1962, 144, 1316–1319. [Google Scholar]
- Berlin, A.A.; Skachkova, V.K.; Grigorovskaya, V.A.; Kuzayev, A.I.; Nikitayev, A.T.; Nekipelov, V.M. On the interaction of oligoarylenes with p-diethynylbenzene. Polym. Sci. USSR 1979, 21, 2256–2268. [Google Scholar] [CrossRef]
- Yanovskii, L.S.; Lempert, D.B.; Raznoschikov, V.V.; Averkov, I.S.; Zyuzin, I.N.; Zholudev, A.F.; Kislov, M.B. Prospects for the use of diethynylbenzene as a fuel dispersant for rocket ramjet engines. Russ. Chem. Bull. 2019, 68, 1848–1855. [Google Scholar] [CrossRef]
- Kazakov, A.I.; Lempert, D.B.; Nabatova, A.V.; Larikova, T.S.; Molokanov, A.A.; Zyuzin, I.N.; Volkova, N.N.; Kislov, M.B.; Zholudev, A.F.; Yanovskii, L.S. Heat Release Kinetics in Thermally Initiated 1,4-Diethynylbenzene Polymerization. Russ. J. Appl. Chem. 2021, 94, 977–985. [Google Scholar] [CrossRef]
- Berlin, A.A.; Cherkashin, M.I.; Kisilitsa, P.P. Method for Producing Poly-p-diethynylbenzene. USSR Patent SU-191800-A, 22 September 1965. [Google Scholar]
- Sastri, S.B.; Keller, T.M.; Jones, K.M.; Armistead, J.P. Studies on cure chemistry of new acetylenic resins. Macromolecules 1993, 26, 6171–6174. [Google Scholar] [CrossRef]
- Grigoryan, S.G.; Avetisyan, K.G.; Martirosyan, G.V.; Arzumanyan, A.M.; Matnishyan, A.A. Polymer complexes of arylacetylenes with iodine. Polym. Sci. USSR 1989, 31, 649–654. [Google Scholar] [CrossRef]
- Tseng, W.-C.; Chen, Y.; Chang, G.-W. Curing conditions of polyarylacetylene prepolymers to obtain thermally resistant materials. Polym. Degrad. Stab. 2009, 94, 2149–2156. [Google Scholar] [CrossRef]
- Wang, Y.-G.; Shi, T.-J.; Li, Z.; Tan, D.-X. Preparation and Characterization of Wood ceramics from Polyarylacetylene Resin/Fir Powder. Chin. J. Appl. Chem. 2010, 27, 418–423. [Google Scholar] [CrossRef]
- Korshak, V.V.; Volpin, M.E.; Sergeev, V.A.; Shitikov, V.K.; Kolomnikov, I.S. Polymers of Polyphenylene Type and Method of Producing the Same. U.S. Patent US-3705131-A, 27 November 1970. [Google Scholar]
- Korshak, V.V.; Sergeev, V.A.; Shitikov, V.K.; Volpin, M.E.; Kolomnikov, I.S. Polycyclotrimerization as a new method of aromatic polymer producing. Dokl. Akad. Nauk SSSR 1971, 201, 112–114. [Google Scholar]
- Ermakova, V.D.; Cherkashin, M.I.; Berlin, A.A. Copolymerization of p-diethynylbenzene and phenylacetylene on the catalytic system Al(i-C4H9)3-TiCl4. Russ. Chem. Bull. 1972, 21, 1139–1141. [Google Scholar] [CrossRef]
- Yermakova, V.D.; Sel’skaya, O.G.; Berlin, A.A.; Chauser, M.G.; Cherkashin, M.I. The structure and properties of p-diethynylbenzene-phenylacetylene copolymers. Polym. Sci. USSR 1974, 16, 752–761. [Google Scholar] [CrossRef]
- Korshak, V.V.; Sergeev, V.A.; Chernomordik, Y.A. Synthesis of polyphenylenes by polycyclotrimerization reaction in the presence of a catalytic system (iso-C4H9)3Al-TiCl4. Vysokomol. Soedin. Ser. B 1972, 14, 886–888. [Google Scholar]
- Sergeyev, V.A.; Shitikov, V.K.; Chernomordik, Y.A.; Korshak, V.V. Reactive oligomers based on the polycyclotrimerization reaction of acetylene compounds. Appl. Polym. Symp. 1975, 26, 237–248. [Google Scholar]
- Sergeev, V.A.; Chernomordik, Y.A.; Kurapov, A.S.; Alaev, S.B.; Korshak, V.V. Mechanism of the polycyclotrimerization of acetylenic compounds. Russ. Chem. Bull. 1980, 29, 1785–1788. [Google Scholar] [CrossRef]
- Donda, A.F.; Cervone, E.; Biancifiori, M.A. Cyclic trimerization of phenylacetylene with the catalytic system TiCl4/Al(C2H5)3. Recl. Trav. Chim. Pays-Bas 2010, 81, 585–590. [Google Scholar] [CrossRef]
- Korshak, V.V.; Sergeev, V.A.; Shitikov, V.K.; Volpin, M.E.; Kolomnikov, I.S. Polycyclotrimerization is a new method for obtaining polymers with aromatic cycles in the main chain. Vysokomol. Soedin. Ser. B 1971, 13, 973. [Google Scholar]
- Korshak, V.V.; Sergeyev, V.A.; Shitikov, V.K.; Danilov, V.G. Synthesis and properties of polyphenylenes obtained by polyclotrimerization of diethynylbenzene. Polym. Sci. USSR 1973, 15, 28–36. [Google Scholar] [CrossRef]
- Korshak, V.V.; Sergeyev, V.A.; Danilov, V.G.; Shitikov, V.K. Some thermal characteristics of branched and crosslinked polyphenylenes. Polym. Sci. USSR 1973, 15, 2465–2470. [Google Scholar] [CrossRef]
- Korshak, V.V. Polycyclotrimerization as a general route to polymers with rings in the chains. Pure Appl. Chem. 1974, 39, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Korshak, V.V. The principal characteristics of polycyclotrimerization. Polym. Sci. USSR 1974, 16, 1066–1087. [Google Scholar] [CrossRef]
- Sergeev, V.A.; Shitikov, V.K.; Pankratov, V.A. Synthesis of Polymers by Polycyclotrimerisation. Russ. Chem. Rev. 1979, 48, 79–93. [Google Scholar] [CrossRef]
- Cessna, L.C., Jr. Thermosetting Composition Containing Poly(arylacetylenes). U.S. Patent US-3882073-A, 18 December 1972. [Google Scholar]
- Cessna, L.C. Thermosetting Compositions Containing Poly(arylacetylenes). U.S. Patent US-4026859-A, 3 June 1974. [Google Scholar]
- Jabloner, H. Poly(arylacetylene) Molding Compositions. U.S. Patent US-4070333-A, 2 January 1974. [Google Scholar]
- Jabloner, H. Poly(arylacetylenes) and Thermoset Resins Therefrom. U.S. Patent US-4097460-A, 23 July 1971. [Google Scholar]
- Kirilenko, Y.K.; Plyashkevich, L.A.; Bogdanov, M.N.; Kudryavtsev, G.I.; Chauser, M.G.; Cherkashin, M.I. Method for the Production of Polyaryl Acetylene Fibers. USSR Patent SU-771207-A, 17 October 1978. [Google Scholar]
- Kirilenko, Y.K.; Moisa, V.A.; Plyashkevich, L.A.; Bogdanov, M.N.; Kudryavtsev, G.I.; Chauser, M.G.; Cherkashin, M.I. A Method of Obtaining p-Diethynylbenzene Polymers. USSR Patent SU-833994-A, 25 October 1979. [Google Scholar]
- Kirilenko, Y.K.; Plyashkevich, L.A.; Shitikov, V.K.; Kalinin, V.N.; Kudryavtsev, G.I.; Sergeyev, V.A.; Zakharkin, L.I.; Parfenov, B.P. The synthesis and study of the properties of carbonate-containing polyarylacetylenes. Polym. Sci. USSR 1984, 26, 2122–2127. [Google Scholar] [CrossRef]
- Cherkashin, M.I.; Chauser, M.G.; Dyumaev, K.M.; Kirilenko, Y.K.; Vlasenko, T.Y.; Plyashkevich, L.A.; Kudryavtsev, G.I.; Belitsin, M.N. Synthesis of a fibre-forming carbon-chain polymer based on para-diethynylbenzene. Vysokomol. Soedin. Ser. B 1985, 27, 9–11. [Google Scholar]
- Kirilenko, Y.K.; Batik’yan, B.A.; Plyashkevich, L.A.; Kudryavtsev, G.I.; Perepechkina, E.P. Preparation of polyarylacetylene fibres by the wet spinning method. Fibre Chem. 1985, 16, 429–430. [Google Scholar] [CrossRef]
- Oishi, S.S.; Botelho, E.C.; Luscombe, C.K.; Rezende, M.C. Synthesis and characterization of polyarylacetylene for use in the monolithic vitreous carbon processing. Polímeros 2014, 24, 541–546. [Google Scholar] [CrossRef] [Green Version]
- Sekerová, L.; Vyskočilová, E.; Červený, L.; Sedláček, J. A novel application of terminal alkynes as the homogeneous catalysts for acetalization and esterification. Tetrahedron 2019, 75, 2877–2882. [Google Scholar] [CrossRef]
- Sekerová, L.; Lhotka, M.; Vyskočilová, E.; Faukner, T.; Slováková, E.; Brus, J.; Červený, L.; Sedláček, J. Hyper-Cross-Linked Polyacetylene-Type Microporous Networks Decorated with Terminal Ethynyl Groups as Heterogeneous Acid Catalysts for Acetalization and Esterification Reactions. Chem. Eur. J. 2018, 24, 14742–14749. [Google Scholar] [CrossRef]
- Casalbore-Miceli, G. Charge transport mechanism in pressed pellets of polymer proton conductors. Solid State Ionics 1997, 100, 217–224. [Google Scholar] [CrossRef]
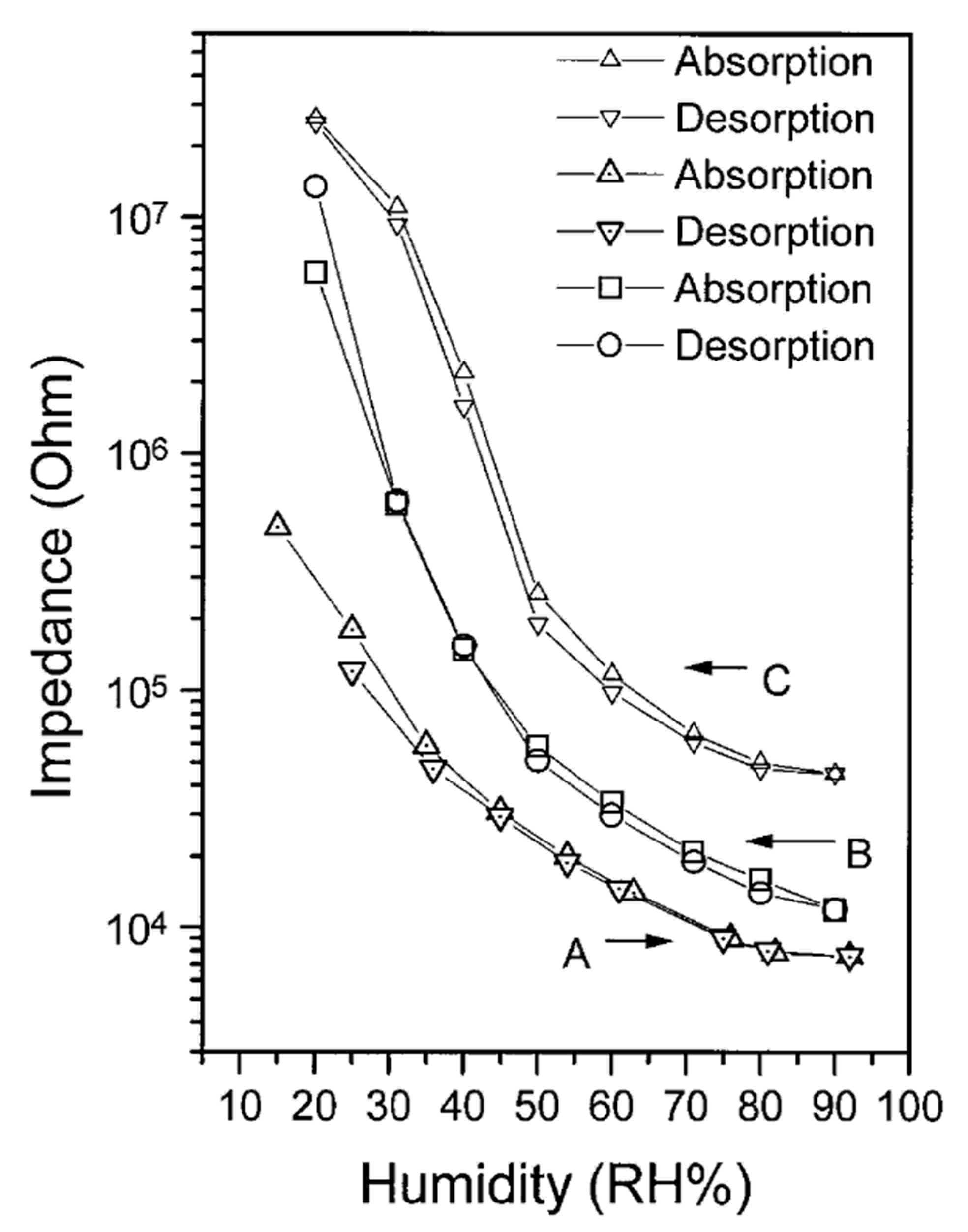
- Yang, M.; Zhan, X.; Li, Y.; Ling, M.; Sun, H.; Zhu, Y. A novel humidity-sensitive material based on PDEB with nickel complex catalyst. Chin. Chem. Lett. 1998, 9, 223–225. [Google Scholar]
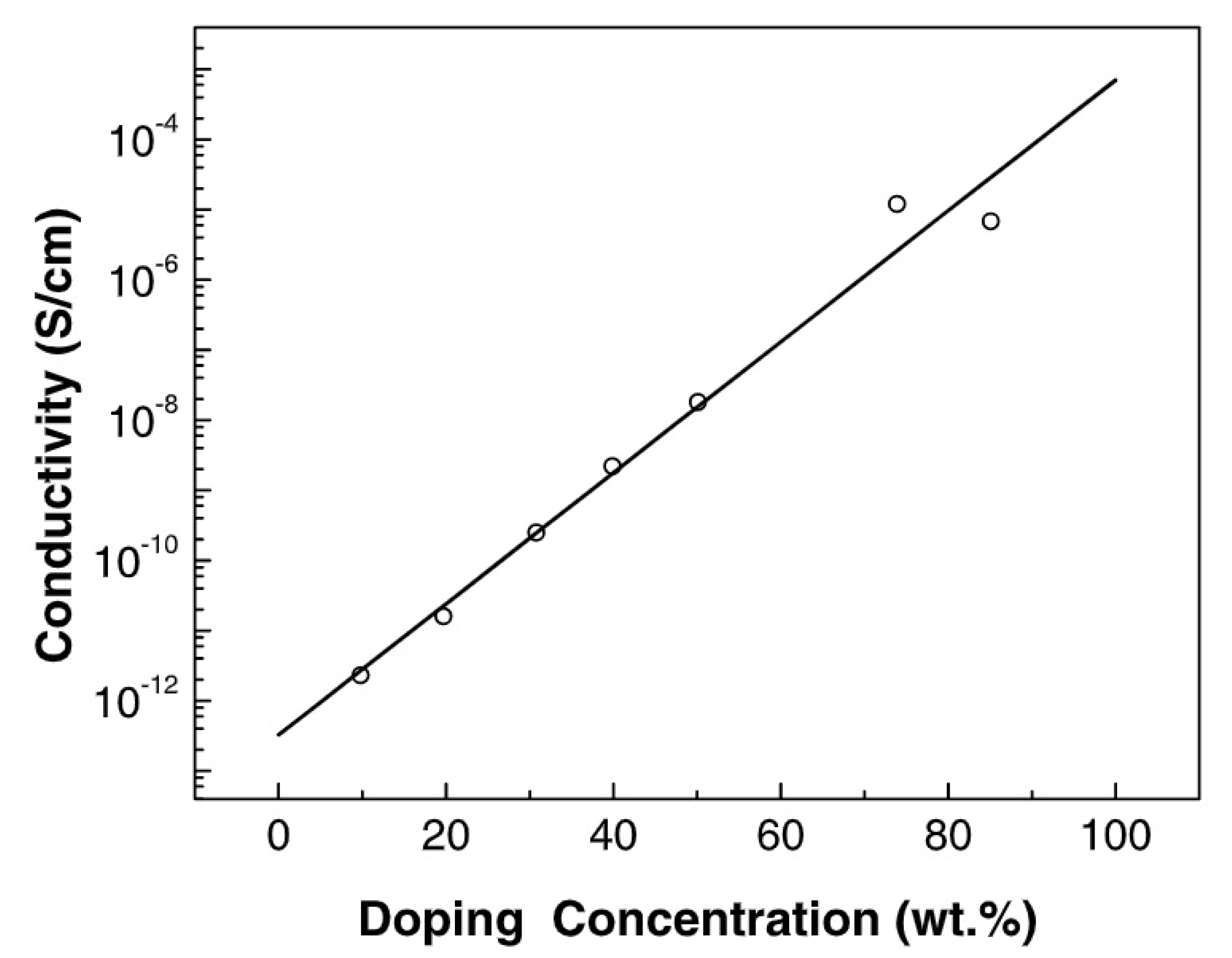
- Zhan, X.; Yang, M.; Wan, M. Electrical properties and spectroscopic studies of HClO4-doped poly(p-diethynylbenzene). Synth. Met. 1998, 94, 249–253. [Google Scholar] [CrossRef]
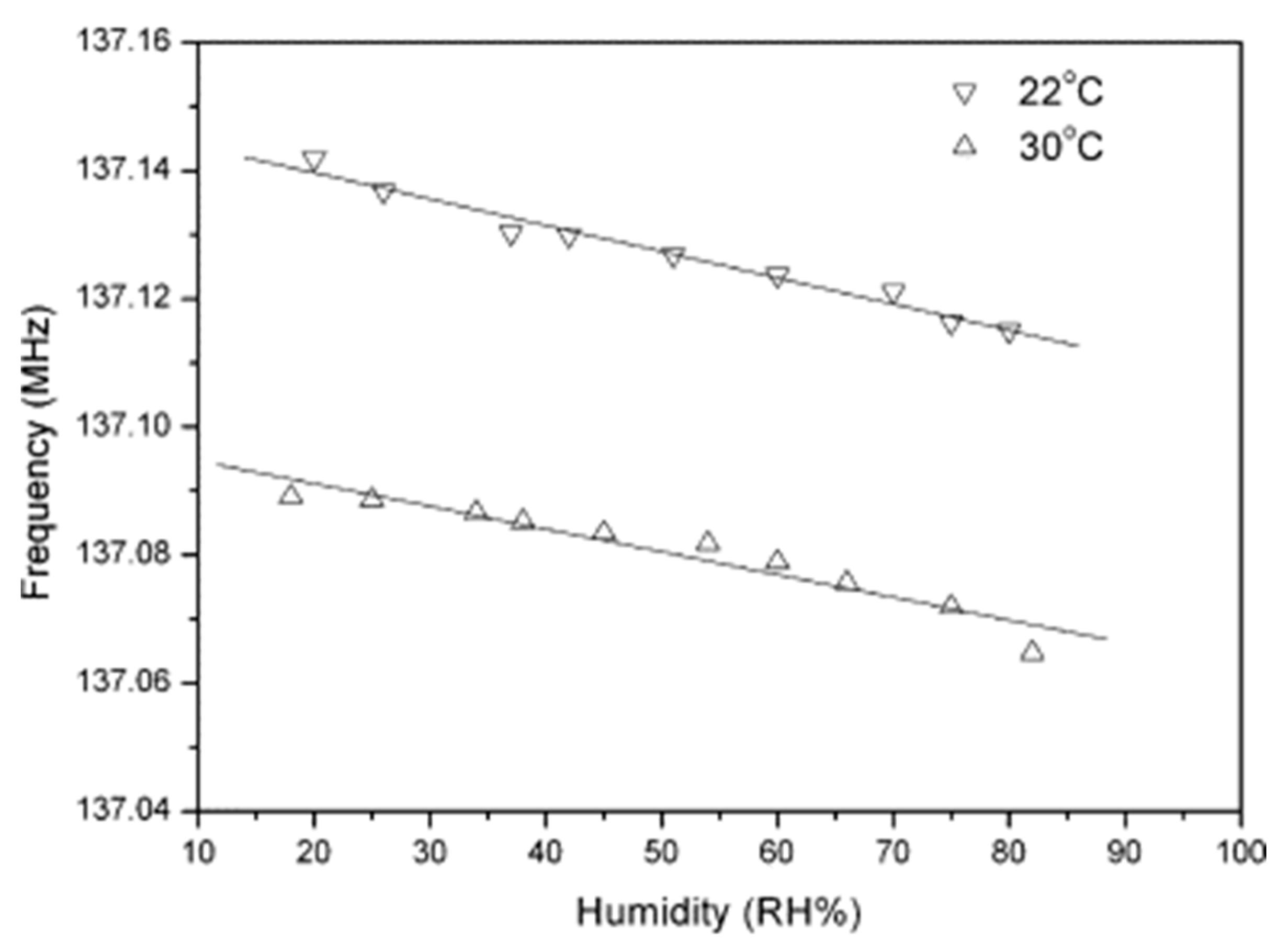
- Yang, M.; Li, Y.; Zhan, X.; Ling, M. A novel resistive-type humidity sensor based on poly(p-diethynylbenzene). J. Appl. Polym. Sci. 1999, 74, 2010–2015. [Google Scholar] [CrossRef]
- Yu, G.; Liu, Y.; Zhan, X.; Li, H.; Yang, M.; Zhu, D. Thermally stable light-emitting polymers of substituted polyacetylenes. Thin Solid Films 2000, 363, 126–129. [Google Scholar] [CrossRef]
- Zhan, X.; Yang, M.; Lei, Z.; Li, Y.; Liu, Y.; Yu, G.; Zhu, D. Photoluminescence, Electroluminescence, Nonlinear Optical, and Humidity Sensitive Properties of Poly(p-diethynylbenzene) Prepared with a Nickel Acetylide Catalyst. Adv. Mater. 2000, 12, 51–53. [Google Scholar] [CrossRef]
- Yang, M.; Zhan, X. Polymerization of p-diethynylbenzene initiated by Ni(C≡CC6H4C≡CH)2(PPH3)2. Chin. J. Polym. Sci. 2001, 19, 303–309. [Google Scholar]
- Zhan, X.; Yang, M.; Sun, H. Polymerization of Substituted Acetylenes Carrying Non-Polar and Polar Groups with Transition Metal Acetylide Catalysts. Macromol. Rapid Commun. 2001, 22, 530–534. [Google Scholar] [CrossRef]
- Zhan, X.; Yang, M. Transition metal acetylide catalysts for polymerization of alkynes: 1. Effect of ligands on catalytic activity of nickel complexes. J. Mol. Catal. A Chem. 2001, 169, 27–31. [Google Scholar] [CrossRef]
- Zhan, X.; Yang, M. Transition metal acetylide catalysts for polymerization of alkynes: 3. Polymerization mechanism. J. Mol. Catal. A Chem. 2001, 169, 57–62. [Google Scholar] [CrossRef]
- Zhan, X.; Xu, S.; Yang, M.; Shen, Y.; Wan, M. Vibration and X-ray photoelectron spectroscopies of FeCl3-doped poly(p-diethynylbenzene). Eur. Polym. J. 2002, 38, 2057–2061. [Google Scholar] [CrossRef]
- Li, Y.; Yang, M.; Ling, M.; Zhu, Y. Surface acoustic wave humidity sensors based on poly(p-diethynylbenzene) and sodium polysulfonesulfonate. Sens. Actuators B 2007, 122, 560–563. [Google Scholar] [CrossRef]
- Zhan, X.; Yang, M. Polymerization of p-diethynylbenzene and its derivatives with nickelocene acetylide catalysts containing different phosphine and alkynyl ligands. Macromol. Rapid Commun. 2000, 21, 1263–1266. [Google Scholar] [CrossRef]
- Zhan, X.; Yang, M. Polymerization of p-diethynylbenzene catalyzed by (π-C5H5)(PPh3)Ni(C≡CC6H4C≡CH). Eur. Polym. J. 2001, 37, 1649–1654. [Google Scholar] [CrossRef]
- Zhan, X.; Yang, M.; Shen, Y.; Wan, M. Vibration and photoelectron spectroscopies of iodine-doped poly(p-diethynylbenzene). Eur. Polym. J. 2002, 38, 2349–2353. [Google Scholar] [CrossRef]
- Ding, X.; Qi, H.; Zhuang, Y.; Wang, J.; Xu, B.; Jiao, Y. Curing Kinetics and Structure Characterization of Arylacetylene Polymer. J. East China Univ. Sci. Technol. 2001, 27, 161–164. [Google Scholar]
- Ding, X.; Wang, W.; Qi, H.; Huang, Y.; Zhuang, Y.; Wang, J.; Jiao, Y. Rheological Behavior of Polyarylacetylene Prepolymer. J. Funct. Polym. 2001, 14, 105–108. [Google Scholar]
- Zhan, X.; Yang, M.; Lei, Z. Transition metal acetylide catalysts for polymerization of p-diethynylbenzene 4: Effect of transition metals on catalytic activity of complexes. J. Mol. Catal. A Chem. 2002, 184, 139–145. [Google Scholar] [CrossRef]
- Zhan, X.; Yang, M.; Xu, G.; Liu, X.; Ye, P. Poly(p-diethynylbenzene) Derivatives for Nonlinear Optics. Macromol. Rapid Commun. 2001, 22, 358–362. [Google Scholar] [CrossRef]
- Zhan, X.; Yang, M.; Sun, H. Transition metal acetylide catalysts for polymerization of alkynes: 2. Effect of ligands on catalytic activity of palladium complexes. J. Mol. Catal. A Chem. 2001, 169, 63–66. [Google Scholar] [CrossRef]
- Yuan, S.; Dorney, B.; White, D.; Kirklin, S.; Zapol, P.; Yu, L.; Liu, D.-J. Microporous polyphenylenes with tunable pore size for hydrogen storage. Chem. Commun. 2010, 46, 4547. [Google Scholar] [CrossRef]
- Misin, V.M.; Maltseva, I.E.; Maltsev, A.A. Synthesis of homo- and copolymers of para-diethynylbenzene in the presence of the cobalt carbonyl-diphenylbutadiyne complex. ChemistrySelect 2022, 7, e202103612. [Google Scholar] [CrossRef]
- Glagolev, N.N.; Misin, V.M.; Khandozhko, V.N.; Cherkashin, M.I. Polymers Based on Phenyl-Substituted Diacetylenes; USSR: Aghveran, Armenia, 1984; pp. 30–31. [Google Scholar]
- Srinrivasan, R.; Farona, M.F. Polymerization of terminal diynes by niobium(V) catalysts. Polym. Bull. 1988, 20, 359–364. [Google Scholar] [CrossRef]
- Zukal, A.; Slováková, E.; Balcar, H.; Sedláček, J. Polycyclotrimers of 1,4-Diethynylbenzene, 2,6-Diethynylnaphthalene, and 2,6-Diethynylanthracene: Preparation and Gas Adsorption Properties. Macromol. Chem. Phys. 2013, 214, 2016–2026. [Google Scholar] [CrossRef]
- Masuda, T.; Mouri, T.; Higashimura, T. Cyclotrimerization of Phenylacetylene Catalyzed by Halides of Niobium and Tantalum. Bull. Chem. Soc. Jpn. 1980, 53, 1152–1155. [Google Scholar] [CrossRef] [Green Version]
- Štěpnička, P.; Císařová, I.; Sedláček, J.; Vohlídal, J.; Polášek, M. Synthesis of Triferrocenylbenzenes by Tantalum(V)-Catalyzed Cyclotrimerization of Ethynylferrocene. The Crystal Structure of 1,3,5-Triferrocenylbenzene. Collect. Czech. Chem. Commun. 1997, 62, 1577–1584. [Google Scholar] [CrossRef]
- Gasparyan, G.T.; Durgaryan, N.A.; Ovakimyan, M.Z.; Indjikyan, M.G. Polymerization of p-diethylbenzene under the action of compounds of tricoordinated phosphorus. Armyanskij Him. Zhurnal 1988, 41, 716–717. [Google Scholar]
- Szwarc, M.; Van Beylen, M. Ionic Polymerization and Living Polymers; Springer: Dordrecht, The Netherlands, 1993; ISBN 978-94-010-4649-7. [Google Scholar]
- Grovu-Ivanoiu, M.; Bulacovschi, V.; Simionescu, C.I. Electroinitiated polymerization of p-diethynylbenzene. Makromol. Chem. 1985, 186, 2247–2254. [Google Scholar] [CrossRef]
- Sergeev, V.A.; Chernomordik, Y.A.; Korshak, V.V. Synthesis and properties of polymers with polyvinylene-polyphenylene structure. Vysokomol. Soedin. Ser. B 1977, 19, 899–903. [Google Scholar]
- Ciampi, S.; Luais, E.; James, M.; Choudhury, M.H.; Darwish, N.A.; Gooding, J.J. The rapid formation of functional monolayers on silicon under mild conditions. Phys. Chem. Chem. Phys. 2014, 16, 8003–8011. [Google Scholar] [CrossRef] [Green Version]
- Misin, V.M.; Cherkashin, M.I. Synthesis of linear soluble polymer of para-diethynylbenzene. Vysokomol. Soedin. Ser. B 1981, 23, 130–131. [Google Scholar]
- Glagolev, N.N.; Misin, V.M.; Cherkashin, M.I. Method for Obtaining a Soluble Linear Polymer of p-Diethylbenzene. USSR Patent SU-910662-A1, 15 July 1980. [Google Scholar]
- Glagolev, N.N.; Misin, V.M.; Zaichenko, N.L.; Cherkashin, M.I. Structure and properties of the soluble p-diethynylbenzene polymer. Sov. J.Chem.Phys. 1990, 5, 1628–1640. [Google Scholar]
- Misin, V.M.; Glagolev, N.N.; Kuzaev, A.I.; Misina, V.P. Thin-Film Sublimates Based on Polyarylacetylenes; Ac. Sci. USSR: Gomel, Russia, 1990; pp. 83–84. [Google Scholar]
- Misin, V.M.; Kuzaev, A.I.; Misin, M.V. Preparation and characteristics of thin films of phenyl-containing polyacetylenes. Russ. J. Appl. Chem. 2006, 79, 1325–1328. [Google Scholar] [CrossRef]
- Razumovskiĭ, S.D.; Zaikov, G.E. Ozone and Its Reactions with Organic Compounds; Studies in Organic Chemistry; Elsevier Science Pub., Co.: Amsterdam, The Netherlands; New York, NY, USA, 1984; ISBN 978-0-444-42369-6. [Google Scholar]
- Misin, V.M.; Glagolev, N.N.; Misin, M.V. Chapter 24. Thermostability and Coke Formation Ability of Diphenyldiacetylene and P-Diethynylbenzene Polymers. In Monomers, Oligomers, Polymers, Composites and Nanocomposites Research: Synthesis, Properties and Applications (Polymer Yearbook, Volume 23); Pethrick, R.A., Petkov, P., Zaikov, G.E., Rakovsky, S.K., Eds.; Polymer Science and Technology; NOVA Science Publishers: New York, NY, USA, 2010; Volume 23, ISBN 978-1-60876-029-9. [Google Scholar]
- Misin, V.M.; Glagolev, N.N.; Misin, M.V. Modification of commercial oligomers with diphenyldiacetylene and p-diethynylbenzene polymers. Russ. J. Appl. Chem. 2008, 81, 2008–2013. [Google Scholar] [CrossRef]
- Misin, V.M.; Glagolev, N.N.; Misin, M.V. Polyphenyldiacetylenes thermostability and coke formation ability. Oxid. Commun. 2011, 34, 888–899. [Google Scholar]
- Kazakov, M.E.; Kirilenko, Y.K.; Trushnikov, A.M.; Misin, V.M.; Kalachev, A.I.; Kudryavtsev, G.I.; Valetsky, P.M.; Cherkashin, M.I. Structural transformations of a carborane-containing composition upon modification of oriented carbon. Dokl. AN SSSR 1986, 287, 1139–1141. [Google Scholar]
- Cherkashin, M.I.; Misin, V.M.; Kazakov, M.E.; Kirilenko, Y.K.; Stanko, V.I.; Valetsky, P.M. Carbon Materials with Increased Electrical Conductivity; USSR: Aghveran, Armenia, 1984; pp. 87–88. [Google Scholar]
- Ai, H.; Koizumi, Y.; Tsuruta, N. Poly(ethynylphenyl)acetylene, Its Copolymer and Composition Thereof. U.S. Patent US-4667006-A, 15 June 1984. [Google Scholar]
- Ai, H.; Koizumi, Y.; Tsuruta, N. Photocurable Compositions of Poly(ethynylphenyl)acetylene, Its Copolymer and Composition Thereof. U.S. Patent US-4767797-A, 30 June 1983. [Google Scholar]
- Lei, Z.; Yang, M.; Chan, X.; Zhang, L. A New Simple Synthesis of Soluble Poly(p-diethynylbenzene) Catalyzed by Cobalt-Phosphine. Polym. J. 1998, 30, 879–882. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Si, J.; Liu, X.; Xu, G.; Ye, P.; Lei, Z.; Zhan, X.; Yang, M. Nonlinear Optical Properties of Some New Conjugated Copolymers. Chinese Phys. Lett. 1998, 15, 189–191. [Google Scholar] [CrossRef]
- Yang, Q.; Si, J.; Wang, Y.; Ye, P. Phase-modulation effects in self-diffraction. Phys. Rev. A 1996, 54, 1702–1708. [Google Scholar] [CrossRef]
- Lavastre, O.; Cabioch, S.; Dixneuf, P.H.; Sedlacek, J.; Vohlidal, J. New Route to Conjugated Polymer Networks: Synthesis of Poly(4-ethynyl)phenylacetylene and Its Transformation into a Conjugated Network. Macromolecules 1999, 32, 4477–4481. [Google Scholar] [CrossRef]
- Vohlídal, J.; Sedláček, J.; Patev, N.; Lavastre, O.; Dixneuf, P.H.; Cabioch, S.; Balcar, H.; Pfleger, J.; Blechta, V. New Substituted Polyacetylenes with Phenyleneethynylene Side Groups [−(C6H4−C⋮C)n−SiiPr3; n = 1, 2]: Synthesis, Characterization, Spectroscopic, and Photoelectric Properties. Macromolecules 1999, 32, 6439–6449. [Google Scholar] [CrossRef]
- Baklouti, M.; Chaabouni, R.; Fontanille, M.; Villenave, J.-J. Polymérisation thermique de la 4-méthoxy-phényl éthynyl cétone, modèle de précurseurs de polymères thermostables. Eur. Polym. J. 1995, 31, 215–221. [Google Scholar] [CrossRef]
- Bondarev, D.; Zedník, J.; Plutnarová, I.; Vohlídal, J.; Sedláček, J. Molecular weight and configurational stability of poly[(fluorophenyl)acetylene]s prepared with metathesis and insertion catalysts. J. Polym. Sci. A Polym. Chem. 2010, 48, 4296–4309. [Google Scholar] [CrossRef]
- Castanon, J.R.; Sano, N.; Shiotsuki, M.; Sanda, F. New Approach to the Polymerization of Disubstituted Acetylenes by Bulky Monophosphine-Ligated Palladium Catalysts. ACS Macro Lett. 2014, 3, 51–54. [Google Scholar] [CrossRef]
- Aso, C.; Kunitake, T.; Saiki, K. Studies on the polymerization of bifunctional monomers. XXI. Polymerization of o-diethinylbenzene. Macromol. Chem. Phys. 1972, 151, 265–274. [Google Scholar] [CrossRef]
- John, J.A.; Tour, J.M. Synthesis of Polyphenylenes and Polynaphthalenes by Thermolysis of Enediynes and Dialkynylbenzenes. J. Am. Chem. Soc. 1994, 116, 5011–5012. [Google Scholar] [CrossRef]
- John, J.A.; Tour, J.M. Synthesis of polyphenylene derivatives by thermolysis of enediynes and dialkynylaromatic monomers. Tetrahedron 1997, 53, 15515–15534. [Google Scholar] [CrossRef]
- Johnson, J.P.; Bringley, D.A.; Wilson, E.E.; Lewis, K.D.; Beck, L.W.; Matzger, A.J. Comparison of “Polynaphthalenes” Prepared by Two Mechanistically Distinct Routes. J. Am. Chem. Soc. 2003, 125, 14708–14709. [Google Scholar] [CrossRef]
- Kim, D.J.; Kim, K.L.; Cho, H.N.; Kim, C.Y. Synthesis and Properties of Poly (1,2-diethynylbenzene)by Transition Metal Catalysts. Polymer 1994, 18, 297–302. [Google Scholar]
- Caldona, E.B.; Borrego, E.I.; Shelar, K.E.; Mukeba, K.M.; Smith, D.W. Ring-Forming Polymerization toward Perfluorocyclobutyl and Ortho-Diynylarene-Derived Materials: From Synthesis to Practical Applications. Materials 2021, 14, 1486. [Google Scholar] [CrossRef]
- Neenan, T.X.; Whitesides, G.M. Synthesis of high carbon materials from acetylenic precursors. Preparation of aromatic monomers bearing multiple ethynyl groups. J. Org. Chem. 1988, 53, 2489–2496. [Google Scholar] [CrossRef]
- Sergeyev, V.A.; Chernomordik, Y.A.; Korshak, V.V. The effect of the structure of Di- and monoacetylenic monomers, and the order of their addition to the reaction zone on some properties of polyphenylenes. Polym. Sci. USSR 1980, 22, 2130–2135. [Google Scholar] [CrossRef]
- Watson, A.S. Carboxylated Poly(arylacetylenes). U.S. Patent US-3993711-A, 19 March 1975. [Google Scholar]
- Zaldivar, R.J.; Kobayashi, R.W.; Rellick, G.S.; Yang, J.-M. Carborane-catalyzed graphitization in polyarylacetylene-derived carbon-carbon composites. Carbon 1991, 29, 1145–1153. [Google Scholar] [CrossRef]
- Zaldivar, R.J.; Rellick, G.S.; Yang, J.-M. Processing Effects on the Mechanical Behavior of Polyarylacetylene- Derived Carbon-Carbon Composites. SAMPE J. 1992, 27, 32. [Google Scholar]
- Binegar, G.A.; Noblet, J.A.; Zaldivar, R.D.; Sheaffer, P.M.; Rellick, G.S. Effects of Heat Treatment on Microstructure Sand Flexural Properties of Unidirectional Carbon-Carbon Composites; Space Systems Division Air Force Systems Command: Los Angeles, CA, USA, 1989. [Google Scholar]
- Jabloner, H.; Cessna, L.C. Thermo setting polymers of p-diethenylbenzene. Am. Chem. Soc. Polym. Preprints 1976, 17, 169. [Google Scholar]
- Hurwitz, F.I. Ethynylated aromatics as high temperature matrix resins: Hurwitz, F.I. SAMPE Journal Vol 23 No 2 (March/April 1987) pp 49–53. Composites 1988, 19, 418. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, Y. An Overview of Polyarylacetylene as the Resin Matrix for High Temperature Composites. J. Solid Rocket Technol. 2001, 1, 53–59. [Google Scholar]
- Hergenrother, P.M. The Use, Design, Synthesis, and Properties of High Performance/High Temperature Polymers: An Overview. High Perform. Polym. 2003, 15, 3–45. [Google Scholar] [CrossRef]
- Fu, H.J.; Huang, Y.D.; Liu, L. Influence of fibre surface oxidation treatment on mechanical interfacial properties of carbon fibre/polyarylacetylene composites. Mater. Sci. Technol. 2004, 20, 1655–1660. [Google Scholar] [CrossRef]
- Cao, J.; Ding, X.; Zhuang, Y.; Jiao, Y. Studies of Polyarylacetylene. Mater. Rev. 2001, 8, 64–66. [Google Scholar]
- Häußler, M.; Dong, H.; Lam, J.W.Y.; Zheng, R.; Qin, A.; Tang, B.-Z. Hyperbranched conjugative macromolecules constructed from triple-bond building blocks. Chin. J. Polym. Sci. 2005, 23, 567. [Google Scholar] [CrossRef]
- Grundy, M.; Ye, Z. Cross-linked polymers of diethynylbenzene and phenylacetylene as new polymer precursors for high-yield synthesis of high-performance nanoporous activated carbons for supercapacitors, hydrogen storage, and CO2 capture. J. Mater. Chem. A 2014, 2, 20316–20330. [Google Scholar] [CrossRef]
- Zhang, B.; Zhou, Q.; Wang, Y.; Song, N.; Ni, L. Synthesis of ordered mesoporous carbon using m-Diethynylbenzene as a new precursor. Mater. Lett. 2017, 189, 317–320. [Google Scholar] [CrossRef]
- Bakker, R.; Weijers, P.; Spee, D.A.; van Steenbergen, M.J.; van der Werf, C.H.M.; Rath, J.K.; Schropp, R.E.I. Synthesis of poly(meta-diethynyl benzene) with initiated chemical vapour deposition. Thin Solid Films 2011, 519, 4418–4420. [Google Scholar] [CrossRef]
- Sergeyev, V.A.; Chernomordik, Y.A.; Slonimskii, G.L.; Askadskii, A.A.; Tolchinskii, Y.I.; Sokolova, Y.B.; Shebanova, M.P.; Korshak, V.V. Synthesis and properties of polyphenylenes based on 4,4′-diethynyldiphenyl and 4,4′-diethynyldiphenyl oxide. Polym. Sci. USSR 1980, 22, 2402–2409. [Google Scholar] [CrossRef]
- Ried, W. Athinierungsrekationen. In Neuere Methoden der Praparativen Organischen Chemie; Verlag Chemie: Weinheim, German, 1966; Volume 4, p. 88. [Google Scholar]
- Rohde, O.; Wegner, G. On the Polymerization of Crystalline 1,4-Diethynylnaphthalene, 1. The Structure of the Photopolymer. Makromol. Chem. 1978, 179, 1999–2011. [Google Scholar] [CrossRef]
- Rohde, O.; Wegner, G. On the Polymerization of Crystalline 1,4-Diethynylnaphthalene, 2 The Reaction Mechanism. Makromol. Chem. 1978, 179, 2013–2029. [Google Scholar] [CrossRef]
- Cho, H.N.; Kim, S.H.; Kim, C.Y. Synthesis and characterization of poly(1,8-diethynylnaphthalene). Polym. Bull. 1995, 34, 125–132. [Google Scholar] [CrossRef]
- Ratto, J.J.; Dynes, P.J.; Hamermesh, C.L. The synthesis and thermal polymerization of 4,4′-diethynylphenyl ether. J. Polym. Sci. Polym. Chem. Ed. 1980, 18, 1035–1046. [Google Scholar] [CrossRef]
- Liu, J.; Zheng, R.; Tang, Y.; Häussler, M.; Lam, J.W.Y.; Qin, A.; Ye, M.; Hong, Y.; Gao, P.; Tang, B.Z. Hyperbranched Poly(silylenephenylenes) from Polycyclotrimerization of A2 -Type Diyne Monomers: Synthesis, Characterization, Structural Modeling, Thermal Stability, and Fluorescent Patterning. Macromolecules 2007, 40, 7473–7486. [Google Scholar] [CrossRef]
- Wang, Z.; Shi, Y.; Wang, J.; Li, L.; Wu, H.; Yao, B.; Sun, J.Z.; Qin, A.; Tang, B.Z. Indium-catalyzed polycyclotrimerization of diynes: A facile route to prepare regioregular hyperbranched polyarylenes. Polym. Chem. 2014, 5, 5890–5894. [Google Scholar] [CrossRef]













































{kind=link}
{kind=link}
{kind=link}
{kind=link}
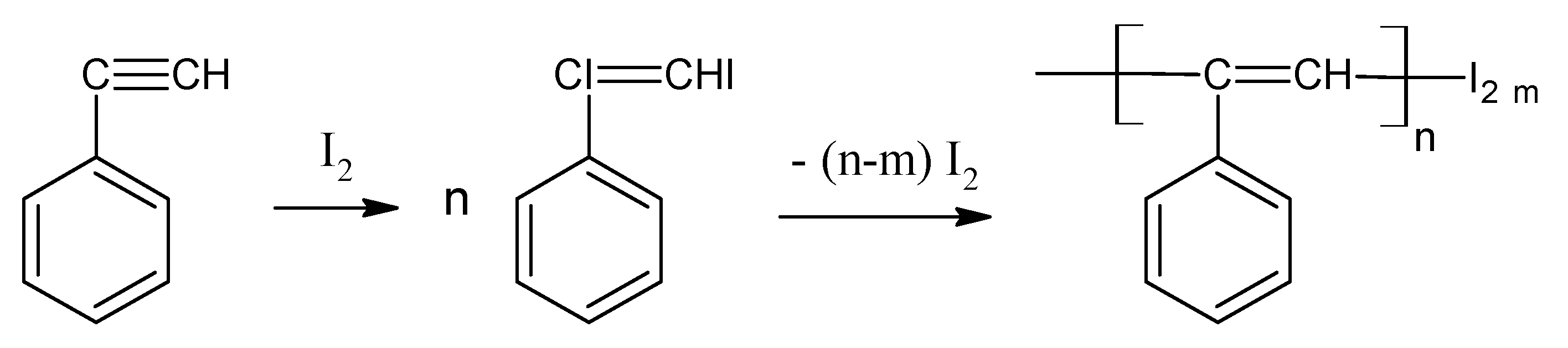{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Solvent | T, °C | τ, min | Y, % |
|---|---|---|---|---|
| PDEBC-2 | Benzene | 82 | 10 | 59 |
| PDEBC-4 | Toluene | 110 | 10 | 72 |
| PDEBC-5 | Toluene | 82 | 120 | 8 |
| Solvent | Reaction Time τ, min | PDEB Yield, % | |||
|---|---|---|---|---|---|
| Soluble Fraction | Insoluble Fraction | ||||
| HMPA | 3 | 47 | - | 1300/10.3 | 5/1 |
| HMPA | 20 | 48 | - | 1340/10.6 | 5/1 |
| HMPA | 40 | 49 | - | 1370/10.9 | 5.3/1 |
| DMSO | 1 | 5 | - | 1800/14 | 7/1 |
| DMSO | 20 | 58 | 23 | 3160/25 | 7/1 |
| DMSO | 60 | 63 | 26 | 3730/29.6 | 8/1 |
| Samples | Time, h | Yield, % | Solubility in THF a | |||
|---|---|---|---|---|---|---|
| 1 | 2 | 30.1 | ~100 | 10,200 | 2.67 | 5/1 |
| 2 | 5 | 53.6 | 90 | 11,500 | 2.85 | - |
| 3 | 10 | 73.6 | 80 | 13,300 | 3.20 | 5.1/1 |
| 4 | 15 | 75.7 | 80 | 17,500 | 3.31 | 5.6/1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Misin, V.M.; Maltseva, I.E.; Kazakov, M.E.; Volkov, V.A. The Polymers of Diethynylarenes—Is Selective Polymerization at One Acetylene Bond Possible? A Review. Polymers 2023, 15, 1105. https://doi.org/10.3390/polym15051105
Misin VM, Maltseva IE, Kazakov ME, Volkov VA. The Polymers of Diethynylarenes—Is Selective Polymerization at One Acetylene Bond Possible? A Review. Polymers. 2023; 15(5):1105. https://doi.org/10.3390/polym15051105
Chicago/Turabian StyleMisin, Vyacheslav M., Irina E. Maltseva, Mark E. Kazakov, and Vladimir A. Volkov. 2023. "The Polymers of Diethynylarenes—Is Selective Polymerization at One Acetylene Bond Possible? A Review" Polymers 15, no. 5: 1105. https://doi.org/10.3390/polym15051105
APA StyleMisin, V. M., Maltseva, I. E., Kazakov, M. E., & Volkov, V. A. (2023). The Polymers of Diethynylarenes—Is Selective Polymerization at One Acetylene Bond Possible? A Review. Polymers, 15(5), 1105. https://doi.org/10.3390/polym15051105