

DFT and ONIOM Simulation of 1,3-Butadiene Polymerization Catalyzed by Neodymium-Based Ziegler–Natta System


Abstract
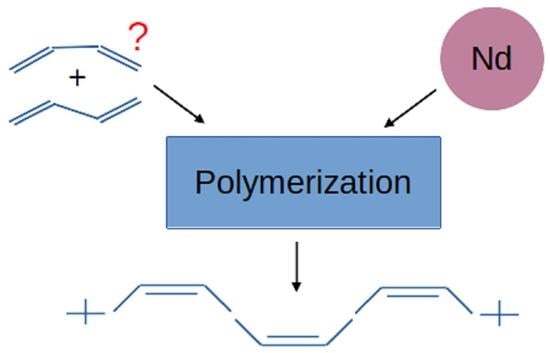
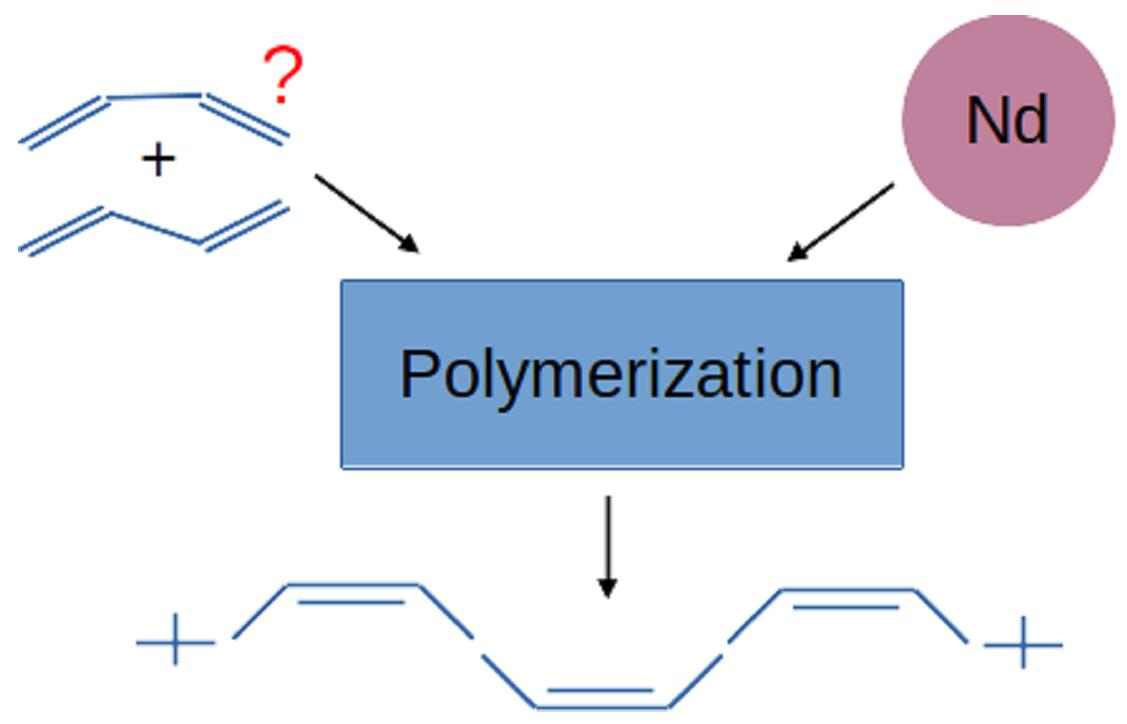
:
1. Introduction
2. Computational Details
3. Results and Discussion
3.1. Structural and Thermochemical Analysis of AS
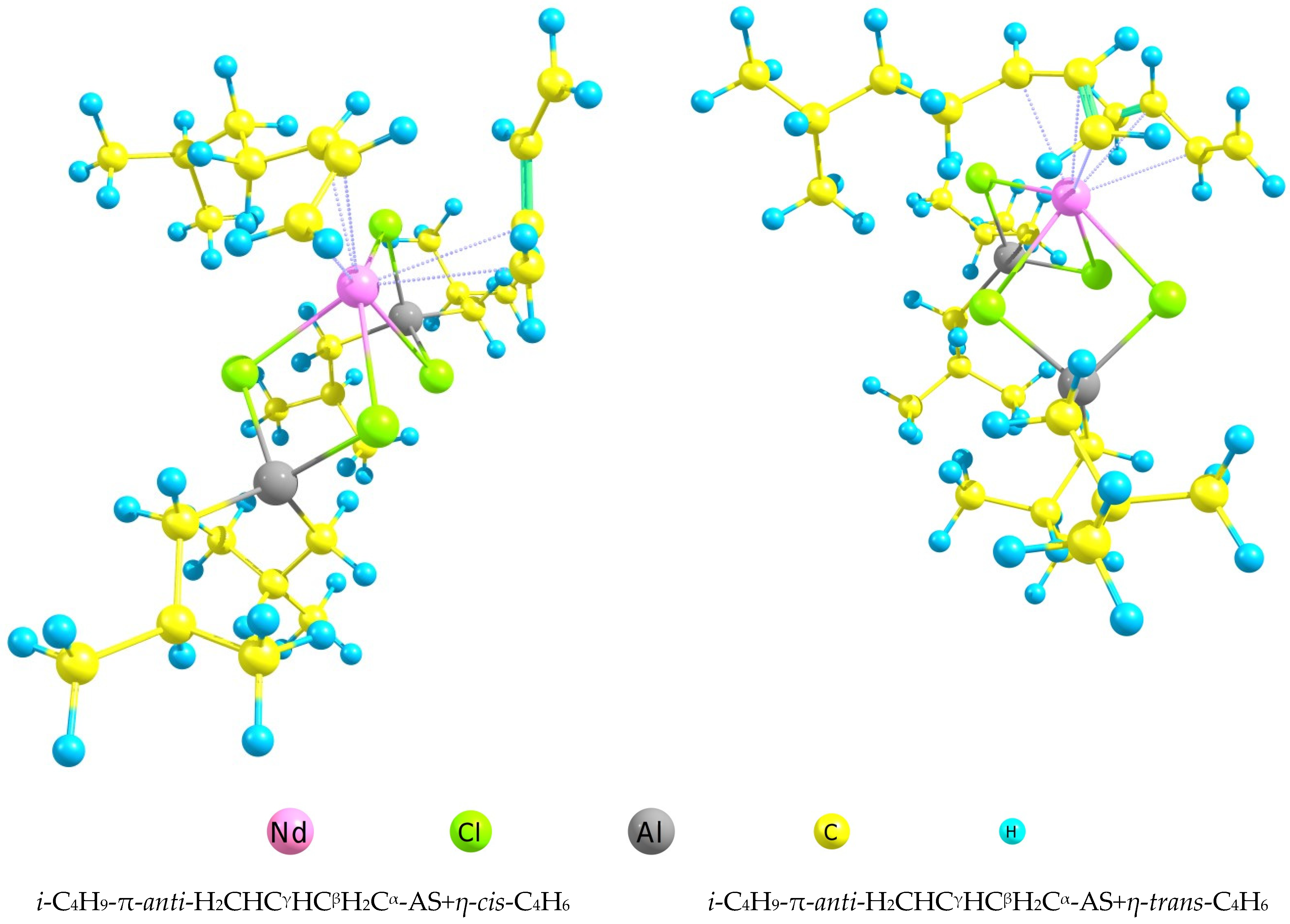
i-C4H9-π-anti-H2CHCγHCβH2Cα-AS+η-trans/cis-C4H6
3.2. Study of Polymerization Mechanism
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ziegler, K.; Holzkamp, E.; Breil, H.; Martin, H. The Mulheim normal pressure polyethylene process. Angew. Chem.-Int. Ed. 1955, 67, 541–547. [Google Scholar] [CrossRef]
- Von Giulio, N.; Corradini, P. “Kristallstruktur des isotaktischen polystyrols”. Die Makromol. Chem. Macromol. Chem. Phys. 1955, 16, 77–80. [Google Scholar] [CrossRef]
- Ziegler, K.; Holzkamp, E.; Breil, H.; Martin, H. Das Mülheimer Normaldruck-Polyäthylen-Verfahren. Angew. Chem. 1956, 68, 721. [Google Scholar] [CrossRef]
- Ziegler, K.; Holzkamp, E.; Breil, H.; Martin, H. Polymerisation von Äthylen und anderen Olefinen. Angew. Chem. 1955, 67, 541–547. [Google Scholar] [CrossRef]
- Natta, C. Stereospecific polymerizations. J. Polym. Sci. 1960, 48, 219–239. [Google Scholar] [CrossRef]
- Natta, G.; Porri, L.; Carbonaro, A.; Stoppa, G. “Polymerization of conjugated diolefins by homogeneous aluminum alkyl-titanium alkoxide catalyst systems. I. Cis-1,4 isotactic poly (1,3-pentadiene)”. Die Makromol. Chem. Macromol. Chem. Phys. 1964, 77, 114–125. [Google Scholar] [CrossRef]
- Natta, G.; Porri, L.; Carbonaro, A. “Polymerization of conjugated diolefins by homogeneous aluminum alkyl-titanium alkoxide catalyst systems. II. 1,2-polybutadiene and 3, 4-polyisoprene”. Die Makromol. Chem. Macromol. Chem. Phys. 1964, 77, 126–138. [Google Scholar] [CrossRef]
- Tinyakova, E.N.; Dolgoplosk, B.A.; Zhuravleva, T.G.; Kovalevskaya, R.N.; Kuren’Gina, T.N. Synthesis of cis and trans polymers of dienes on oxide catalysts and investigation of their structure and properties. J. Polym. Sci. 1961, 52, 159–167. [Google Scholar] [CrossRef]
- Cooper, W.; Vaughan, G. Recent developments in the polymerization of conjugated dienes. Prog. Polym. Sci. 1967, 1, 91–160. [Google Scholar] [CrossRef]
- Tornqvist, E.G.M. The stereo rubbers, William M. Saltman, Wiley-Interscience, New York, 1977, 897 pp., $l49.50. J. Polym. Sci. Polym. Lett. Ed. 1978, 16, 53–55. [Google Scholar] [CrossRef]
- Hsieh, H.L.; Yeh, H.C. Polymerization of butadiene and isoprene with lanthanide catalysts; characterization and properties of homopolymers and copolymers. Rubber Chem. Technol. 1985, 58, 117–145. [Google Scholar] [CrossRef]
- Marina, N.G.; Monakov, Y.B.; Sabirov, Z.M.; Tolstikov, G.A. Lanthanide compounds—Catalysts of stereospecific polymerization of diene monomers. Review. Polym. Sci. USSR 1991, 33, 387–417. [Google Scholar] [CrossRef]
- Friebe, L.; Nuyken, O.; Obrecht, W. Neodymium-based Ziegler/Natta catalysts and their application in diene polymerization. In Neodymium Based Ziegler Catalysts–Fundamental Chemistry; Springer: Berlin/Heidelberg, Germany, 2006; pp. 1–154. [Google Scholar] [CrossRef]
- Fischbach, A.; Anwander, R. Neodymium Based Ziegler Catalysts Fundamental Chemistry; Nuyken, O., Ed.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 204, pp. 155–281. [Google Scholar] [CrossRef]
- Zhang, Z.; Cui, D.; Wang, B.; Liu, B.; Yang, Y. Polymerization of 1,3-conjugated dienes with rare-earth metal precursors. In Molecular Catalysis of Rare-Earth Elements; Springer: Berlin/Heidelberg, Germany, 2010; pp. 49–108. [Google Scholar] [CrossRef]
- Wang, F.; Liu, H.; Hu, Y.; Zhang, X. Lanthanide complexes mediated coordinative chain transfer polymerization of conjugated dienes. Sci. China Technol. Sci. 2018, 61, 1286–1294. [Google Scholar] [CrossRef]
- Fan, C.; Bai, C.; Cai, H.; Dai, Q.; Zhang, X.; Wang, F. Preparation of high cis-1,4 polyisoprene with narrow molecular weight distribution via coordinative chain transfer polymerization. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 4768–4774. [Google Scholar] [CrossRef]
- Manuiko, G.V.; Salakhov, I.I.; Aminova, G.A.; Akhmetov, I.G.; Dyakonov, G.S.; Bronskaya, V.V.; Demidova, E.V. Mathematical modeling of 1,3-butadiene polymerization over a neodymium-based catalyst in a batch reactor with account taken of the multisite nature of the catalyst and chain transfer to the polymer. Theor. Found. Chem. Eng. 2010, 44, 139–149. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, C.Y.; Hu, Y.M.; Jia, X.Y.; Bai, C.X.; Zhang, X.Q. Reversible coordinative chain transfer polymerization of isoprene and copolymerization with ε-caprolactone by neodymium-based catalyst. Polymer 2012, 53, 6027–6032. [Google Scholar] [CrossRef]
- Wang, F.; Liu, H.; Zheng, W.; Guo, J.; Zhang, C.; Zhao, L.; Zhang, X. Fully-reversible and semi-reversible coordinative chain transfer polymerizations of 1,3-butadiene with neodymium-based catalytic systems. Polymer 2013, 54, 6716–6724. [Google Scholar] [CrossRef]
- Georges, S.; Touré, A.O.; Visseaux, M.; Zinck, P. Coordinative chain transfer copolymerization and terpolymerization of conjugated dienes. Macromolecules 2014, 47, 4538–4547. [Google Scholar] [CrossRef]
- Zheng, W.; Yan, N.; Zhu, Y.; Zhao, W.; Zhang, C.; Zhang, H.; Zhang, X. Highly trans-1,4-stereoselective coordination chain transfer polymerization of 1,3-butadiene and copolymerization with cyclic esters by a neodymium-based catalyst system. Polym. Chem. 2015, 6, 6088–6095. [Google Scholar] [CrossRef]
- Tanaka, R.; Yuuya, K.; Sato, H.; Eberhardt, P.; Nakayama, Y.; Shiono, T. Synthesis of stereodiblock polyisoprene consisting of cis-1,4 and trans-1,4 sequences by using a neodymium catalyst: Change of the stereospecificity triggered by an aluminum compound. Polym. Chem. 2016, 7, 1239–1243. [Google Scholar] [CrossRef] [Green Version]
- Díaz de León Gómez, R.E.; Enríquez-Medrano, F.J.; Maldonado Textle, H.; Mendoza Carrizales, R.; Reyes Acosta, K.; Lopez Gonzalez, H.R.; Romero, J.L.O.; Lugo Uribe, L.E. Synthesis and characterization of high cis-polymyrcene using neodymium-based catalysts. Can. J. Chem. Eng. 2016, 94, 823–832. [Google Scholar] [CrossRef]
- Dai, Q.; Zhang, X.; Hu, Y.; He, J.; Shi, C.; Li, Y.; Bai, C. Regulation of the cis-1,4-and trans-1,4-polybutadiene multiblock copolymers via chain shuttling polymerization using a ternary neodymium organic sulfonate catalyst. Macromolecules 2017, 50, 7887–7894. [Google Scholar] [CrossRef]
- Luo, Y.; Gao, Z.; Chen, J. Rare-earth metal bis (silylamide) complexes supported by ferrocene-substituted amidinate and their performance in cis-1,4 selective polymerization of isoprene. J. Organomet. Chem. 2017, 846, 18–23. [Google Scholar] [CrossRef]
- Hollfelder, C.O.; Jende, L.N.; Diether, D.; Zelger, T.; Stauder, R.; Maichle-Mössmer, C.; Anwander, R. 1,3-Diene Polymerization Mediated by Homoleptic Tetramethylaluminates of the Rare-Earth Metals. Catalysts 2018, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Göttker-Schnetmann, I.; Kenyon, P.; Mecking, S. Coordinative Chain Transfer Polymerization of Butadiene with Functionalized Aluminum Reagents. Angew. Chem. 2019, 131, 17941–17945. [Google Scholar] [CrossRef]
- González-Zapata, J.L.; Enríquez-Medrano, F.J.; González, H.R.L.; Revilla-Vázquez, J.; Carrizales, R.M.; Georgouvelas, D.; Valencia, L.; de León Gómez, R.E.D. Introducing random bio-terpene segments to high cis-polybutadiene: Making elastomeric materials more sustainable. RSC Adv. 2020, 10, 44096–44102. [Google Scholar] [CrossRef]
- Zheng, W.; Yang, Q.; Dong, J.; Wang, F.; Luo, F.; Liu, H.; Zhang, X. Neodymium-based one-precatalyst/dual-cocatalyst system for chain shuttling polymerization to access cis-1,4/trans-1,4 multiblock polybutadienes. Mater. Today Commun. 2021, 27, 102453. [Google Scholar] [CrossRef]
- Wang, H.; Cue, J.M.O.; Calubaquib, E.L.; Kularatne, R.N.; Taslimy, S.; Miller, J.T.; Stefan, M.C. Neodymium catalysts for polymerization of dienes, vinyl monomers, and ε-caprolactone. Polym. Chem. 2021, 12, 6790–6823. [Google Scholar] [CrossRef]
- Cavalcante de Sá, M.C.; Córdova, A.M.T.; Díaz de León Gómez, R.E.; Pinto, J.C. Modeling of Isoprene Solution Coordinative Chain Transfer Polymerization. Macromol. React. Eng. 2021, 15, 2100005. [Google Scholar] [CrossRef]
- Córdova, T.; Enríquez-Medrano, F.J.; Cartagena, E.M.; Villanueva, A.B.; Valencia, L.; Álvarez, E.N.C.; González, R.L.; Díaz-de-León, R. Coordinative Chain Transfer Polymerization of Sustainable Terpene Monomers Using a Neodymium-Based Catalyst System. Polymers 2022, 14, 2907. [Google Scholar] [CrossRef]
- Tereshchenko, K.A.; Ulitin, N.V.; Bedrina, P.S.; Shiyan, D.A.; Lifanov, A.D.; Madzhidov, T.I.; Rusanova, S.N.; Volfson, S.I. Analysis of the Mechanism of Polybutadiene Synthesis in the Presence of the Neodymium Versatate+ Diisobutylaluminum Hydride+ Ethylaluminum Sesquichloride Catalytic System within the Solution of the Inverse Kinetic Problem. Ind. Eng. Chem. Res. 2022, 61, 15961–15969. [Google Scholar] [CrossRef]
- Iovu, H.; Hubca, G.; Simionescu, E.; Badea, E.G.; Dimonie, M. Polymerization of butadiene and isoprene with the NdCl3· 3TBP-TIBA catalyst system. Die Angew. Makromol. Chem. Appl. Macromol. Chem. Phys. 1997, 249, 59–77. [Google Scholar] [CrossRef]
- Srinivasa Rao, G.S.; Upadhyay, V.K.; Jain, R.C. Polymerization of 1,3-butadiene using neodymium chloride tripentanolate–triethyl aluminum catalyst systems. J. Appl. Polym. Sci. 1999, 71, 595–602. [Google Scholar] [CrossRef]
- Ren, C.; Li, G.; Dong, W.; Jiang, L.; Zhang, X.; Wang, F. Soluble neodymium chloride 2-ethylhexanol complex as a highly active catalyst for controlled isoprene polymerization. Polymer 2007, 48, 2470–2474. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, C.; Liu, X.; Gao, K.; Cao, Y.; Zhang, C.; Zhang, X. Methylaluminoxane-activated neodymium chloride tributylphosphate catalyst for isoprene polymerization. J. Appl. Polym. Sci. 2014, 131. [Google Scholar] [CrossRef]
- Kularatne, R.N.; Yang, A.; Nguyen, H.Q.; McCandless, G.T.; Stefan, M.C. Neodymium catalyst for the polymerization of dienes and polar vinyl monomers. Macromol. Rapid Commun. 2017, 38, 1700427. [Google Scholar] [CrossRef]
- Oehme, A.; Gebauer, U.; Gehrke, K.; Beyer, P.; Hartmann, B.; Lechner, M.D. The influence of the catalyst preparation on the homo-and copolymerization of butadiene and isoprene. Macromol. Chem. Phys. 1994, 195, 3773–3781. [Google Scholar] [CrossRef]
- Boisson, C.; Barbotin, F.; Spitz, R. Polymerization of butadiene with a new catalyst based on a neodymium amide precursor. Macromol. Chem. Phys. 1999, 200, 1163–1166. [Google Scholar] [CrossRef]
- Friebe, L.; Nuyken, O.; Windisch, H.; Obrecht, W. Polymerization of 1,3-Butadiene Initiated by Neodymium Versatate/Diisobutylaluminium Hydride/Ethylaluminium Sesquichloride: Kinetics and Conclusions About the Reaction Mechanism. Macromol. Chem. Phys. 2002, 203, 1055–1064. [Google Scholar] [CrossRef]
- Salakhov, I.I.; Akhmetov, I.G.; Kozlov, V.G. Polymerization of butadiene during the action of the catalytic system neodymium versatate-diisobutylaluminum hydride-hexachloro-p-xylene. Polym. Sci. Ser. B 2011, 53, 385–390. [Google Scholar] [CrossRef]
- Monakov, Y.B.; Sabirov, Z.M.; Marina, N.G. The nature of active centers and key stages in diene polymerization with lanthanide catalytic systems. Vysokomolekulyarnye Soedineniya. Seriya A Seriya B 1995, 38, 407–417. [Google Scholar]
- Sigaeva, N.N.; Usmanov, T.S.; Budtov, V.P.; Spivak, S.I.; Monakov, Y.B. Catalytic activity distribution of active centers in lanthanide-based diene polymerization. Polym. Sci. Ser. B 2000, 42, 1–5. [Google Scholar]
- Sabirov, Z.M.; Urazbaev, V.N.; Efimov, V.P.; Mullagaliev, I.R.; Monakov, Y.B. On steroespecificity of the effect of Bi-and monometallic active centers in polymerization of butadiene initiated by lanthanide catalysts. In Doklady Physical Chemistry; Pleiades Publishing, Ltd.: New York, NY, USA, 2000; Volume 372, pp. 89–91. [Google Scholar]
- Monakov, Y.B.; Sabirov, Z.M. Basic stages of microstructures formation in polydienes upon lanthanide catalytic systems. Int. J. Polym. Mater. 2001, 50, 57–84. [Google Scholar] [CrossRef]
- Sigaeva, N.N.; Usmanov, T.S.; Budtov, V.P.; Spivak, S.I.; Monakov, Y.B. Determination of a cumulative distribution function of kinetic activity of lanthanide ion-coordination catalytic systems used for diene polymerization. Int. J. Polym. Mater. 2001, 49, 475–489. [Google Scholar] [CrossRef]
- Monakov, B.Y.; Sigaeva, N.N. Active site multiplicity of catalytic systems in polymerization processes. Polym. Sci. Ser. C 2001, 43, 61–80. [Google Scholar]
- Monakov, Y.B.; Sabirov, Z.M.; Duvakina, N.V.; Glukhov, E.A.; Ponomareva, O.A. Nature of active centers in diene copolymerization initiated by Ziegler-Natta catalysts: The role of a non-transition-metal organic compound. Polym. Sci. Ser. A 2001, 43, 91–95. [Google Scholar]
- Sigaeva, N.N.; Usmanov, T.S.; Budtov, V.P.; Monakov, Y.B. Effect of organoaluminum compound on kinetic nonuniformity and structure of active centers of neodymium catalytic systems in butadiene polymerization. Russ. J. Appl. Chem. 2001, 74, 1141–1146. [Google Scholar] [CrossRef]
- Monakov, Y.B.; Sabirov, Z.M.; Urazbaev, V.N.; Efimov, V.P. Relationship between the stereospecificity of lanthanide catalysts and the structures of active sites and dienes, the nature of a cocatalyst, and preparation conditions. Kinet. Catal. 2001, 42, 310–316. [Google Scholar] [CrossRef]
- Monakov, Y.B.; Sabirov, Z.M.; Urazbaev, V.N.; Efimov, V.P. Diene polymerization initiated by NdCl3. 3TBP-based catalytic systems. Multiplicity of active centers and their structure and stereospecificity distributions. Polym. Science. Ser. A 2002, 44, 228–231. [Google Scholar]
- Sigaeva, N.N.; Usmanov, T.S.; Budtov, V.P.; Spivak, S.I.; Zaikov, G.E.; Monakov, Y.B. The influence of the nature of organoaluminum compound on kinetic heterogeneity of active sites in lanthanide-based diene polymerization. J. Appl. Polym. Sci. 2003, 87, 358–368. [Google Scholar] [CrossRef]
- Urazbaev, V.N.; Efimov, V.P.; Sabirov, Z.M.; Monakov, Y.B. Structure of active centers, their stereospecificity distribution, and multiplicity in diene polymerization initiated by NdCl3-based catalytic systems. J. Appl. Polym. Sci. 2003, 89, 601–603. [Google Scholar] [CrossRef]
- Tobisch, S. Theoretical investigation of the mechanism of cis−trans regulation for the allylnickel(II)-catalyzed 1,4 polymerization of butadiene. Acc. Chem. Res. 2002, 35, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Tobisch, S. Mechanism of the chain termination of the allylnickel(II)-catalyzed polymerization of 1,3-butadiene. A density functional investigation for the cationic [NiII(RC3H4)(cis-C4H6)L]+ active catalyst. Macromolecules 2003, 36, 6235–6244. [Google Scholar] [CrossRef]
- Tobisch, S. Mechanistic insight into the selective trans-1,4-polymerization of butadiene by terpyridine–iron(II) complexes—A computational study. Can. J. Chem. 2009, 87, 1392–1405. [Google Scholar] [CrossRef]
- Lin, F.; Wang, M.; Pan, Y.; Tang, T.; Cui, D.; Liu, B. Sequence and regularity controlled coordination copolymerization of butadiene and styrene: Strategy and mechanism. Macromolecules 2017, 50, 849–856. [Google Scholar] [CrossRef]
- Bahri-Laleh, N.; Hanifpour, A.; Mirmohammadi, S.A.; Poater, A.; Nekoomanesh-Haghighi, M.; Talarico, G.; Cavallo, L. Computational modeling of heterogeneous Ziegler-Natta catalysts for olefins polymerization. Prog. Polym. Sci. 2018, 84, 89–114. [Google Scholar] [CrossRef]
- Guo, Z.A.; Xian, J.Y.; Rong, L.R.; Qin, H.; Jie, Z. Theoretical study of metal ion impact on geometric and electronic properties of terbutaline compounds. Mon. Für Chem.-Chem. Mon. 2019, 150, 1355–1364. [Google Scholar] [CrossRef]
- Nsiri, H.; Belaid, I.; Larini, P.; Thuilliez, J.; Boisson, C.; Perrin, L. Ethylene–Butadiene Copolymerization by Neodymocene Complexes: A Ligand Structure/Activity/Polymer Microstructure Relationship Based on DFT Calculations. ACS Catal. 2016, 6, 1028–1036. [Google Scholar] [CrossRef]
- Romano, E.; Budzelaar, P.H.; De Rosa, C.; Talarico, G. Unconventional Stereoerror Formation Mechanisms in Nonmetallocene Propene Polymerization Systems Revealed by DFT Calculations. J. Phys. Chem. A 2022, 126, 6203–6209. [Google Scholar] [CrossRef]
- Manuyko, G.V.; Aminova, G.A.; Dyakonov, G.S.; Ahmetov, I.G.; Bronskaya, V.V. Kinetic heterogeneity of neodymium catalyst systems modified with methylaluminoxane. Theor. Found. Chem. Eng. 2015, 49, 246–251. [Google Scholar] [CrossRef]
- Busygin, V.M.; Gil’manov, K.K.; Gil’mutdinov, N.R.; Akhmetov, I.G.; Salakhov, I.I.; Akhmetova, D.R.; Vagizov, A.M. Method of Producing Polymers and Copolymers of Conjugated Dienes (Versions). RU Patent 2422468, 27 June 2011. [Google Scholar]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 8, e1327. [Google Scholar] [CrossRef]
- Becke, A. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Stoychev, G.L.; Auer, A.A.; Neese, F. Automatic generation of auxiliary basis sets. J. Chem. Theory Comput. 2017, 13, 554–562. [Google Scholar] [CrossRef]
- Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted abinitio pseudopotentials for the rare earth elements. J. Chem. Phys. 1989, 90, 1730–1734. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cammi, R.; Mennucci, B.; Tomasi, J. Fast evaluation of geometries and properties of excited molecules in solution: A Tamm-Dancoff model with application to 4-dimethylaminobenzonitrile. J. Phys. Chem. A 2000, 104, 5631–5637. [Google Scholar] [CrossRef]
- Dapprich, S.; Komáromi, I.; Byun, K.S.; Morokuma, K.; Frisch, M.J. A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. J. Mol. Struct. THEOCHEM 1999, 461, 1–21. [Google Scholar] [CrossRef]
- Grimme, S.; Bannwarth, C.; Shushkov, P. A robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent interactions of large molecular systems parametrized for all spd-block elements (Z = 1–86). J. Chem. Theory Comput. 2017, 13, 1989–2009. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB—An accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehlert, S.; Stahn, M.; Spicher, S.; Grimme, S. Robust and efficient implicit solvation model for fast semiempirical methods. J. Chem. Theory Comput. 2021, 17, 4250–4261. [Google Scholar] [CrossRef]
- Anno, T. Out-of-Plane Vibrations of a Conjugated Hydrocarbon: Trans-Butadiene. J. Chem. Phys. 1958, 28, 944–949. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AS+η-cis-C4H6 | AS+η-trans-C4H6 | |
|---|---|---|
| R(Nd-Cl), Å | 2.85 | 2.87 |
| R(Nd-C(i-C4H9)), Å | 2.42 | 2.39 |
| R(Nd-C1(C4H6)), Å | 3.28 | 3.36 |
| R(Nd-C2(C4H6)), Å | 3.03 | 3.08 |
| R(Nd-C3(C4H6)), Å | 3.09 | 3.14 |
| R(Nd-C4(C4H6)), Å | 3.30 | 3.51 |
| R(Al-Cl), Å | 2.31 | 2.31 |
| R(C(C4H6)-C(i-C4H9), Å | 3.22 | 3.64 |
| ∠Cl-Nd-Cl, degrees | 74.0, 84.0 | 73.0, 85.0 |
| ∠Cl-Al-Cl, degrees | 95.7 | 94.9 |
| ∠Nd-Cl-Al, degrees | 94.0 | 94.0 |
| ∠Nd-Cl-Cl-Al, degrees | 171.0 | 164.0 |
| ∠Cl-Cl-Nd-Cl, degrees | 87.0 | 87.0 |
| i-C4H9-π-anti-H2CHCγHCβH2Cα-AS | i-C4H9-π-syn-H2CHCγHCβH2Cα-AS | |
|---|---|---|
| R(Nd-Cl), Å | 2.82 | 2.82 |
| R(Nd-Cα(C4H6)), Å | 2.62 | 2.50 |
| R(Nd-Cβ(C4H6)), Å | 2.67 | 2.71 |
| R(Nd-Cγ(C4H6)), Å | 2.64 | 2.84 |
| R(Nd-Cδ(C4H6)), Å | 2.96 | 3.95 |
| R(Al-Cl), Å | 2.32 | 2.32 |
| R(C(C4H9)-C(i-C4H9), Å | 1.54 | 1.54 |
| ∠Cl-Nd-Cl, degrees | 74.0, 91.0 | 74.0, 83.0 |
| ∠Cl-Al-Cl, degrees | 95.0 | 93.0 |
| ∠Nd-Cl-Al, degrees | 93.0 | 94.0, 91.0 |
| ∠Nd-Cl-Cl-Al, degrees | 172.0 | 176.0, 146.0 |
| ∠Cl-Cl-Nd-Cl, degrees | 76.0 | 77.0 |
| i-C4H9-π-anti-H2CHCγHCβH2Cα-AS+η-cis-C4H6 | i-C4H9-π-anti-H2CHCγHCβH2Cα-AS+η-trans-C4H6 | |
|---|---|---|
| R(Nd-Cl), Å | 2.87 | 2.90 |
| R(Nd-C1(C4H6)), Å | 3.07 | 3.42 |
| R(Nd-C2(C4H6)), Å | 3.25 | 3.10 |
| R(Nd-C3(C4H6)), Å | 3.96 | 3.15 |
| R(Nd-C4(C4H6)), Å | 4.83 | 3.51 |
| R(Nd-Cα(C4H6)), Å | 2.68 | 2.67 |
| R(Nd-Cβ(C4H6)), Å | 2.67 | 2.68 |
| R(Nd-Cγ(C4H6)), Å | 2.60 | 2.64 |
| R(Nd-Cδ(C4H6)), Å | 2.97 | 3.06 |
| R(Al-Cl), Å | 2.30 | 2.30 |
| R(C1(C4H6)-Cδ(C4H6), Å | 3.93 | 3.58 |
| ∠Cl-Nd-Cl, degrees | 74.0, 105.0 | 73.0, 87.0 |
| ∠Cl-Al-Cl, degrees | 95.0 | 95.0 |
| ∠Nd-Cl-Al, degrees | 93.6 | 95.0 |
| ∠Nd-Cl-Cl-Al, degrees | 168.0, 150.0 | 175.0, 165.0 |
| ∠Cl-Cl-Nd-Cl, degrees | 77.0 | 79.0 |
| Initial Structure | ΔG≠ (trans–cis) | ΔG (trans–cis) | ΔG≠ (Polymer) | ΔG≠ (Total) | ΔG (Polymer) | Product |
|---|---|---|---|---|---|---|
| I stage | ||||||
| i-C4H9-AS-T | 43.0 | 6.6 | 54.3 | 60.9 | −51.7 | i-C4H9-C-AS |
| i-C4H9-AS-T | 67.0 | 67.0 | −55.1 | i-C4H9-T-AS | ||
| II stage | ||||||
| i-C4H9-C-AS-T | 41 | 19.4 | 42.3 | 61.7 | −38.5 | i-C4H9-CC-AS |
| i-C4H9-C-AS-T | - | - | 75.0 | 75.0 | −32.9 | i-C4H9-CT-AS |
| i-C4H9-T-AS-T | 41.9 | 29.2 | 38.8 | 68.0 | −46.1 | i-C4H9-TC-AS |
| i-C4H9-T-AS-T | - | - | 72.5 | 72.5 | −39.2 | i-C4H9-TT-AS |
| III stage | ||||||
| i-C4H9-CC-AS-T | 24.6 | 14.6 | 51.7 | 66.3 | −35.0 | i-C4H9-CCC-AS |
| i-C4H9-CC-AS-T | - | - | 102.0 | 102.0 | −52.8 | i-C4H9-CCT-AS |
| i-C4H9-TT-AS-T | - | - | 77.5 | 77.5 | −40.4 | i-C4H9-TTT-AS |
| IV stage | ||||||
| i-C4H9-CCC-AS-T | 24.7 | 6.3 | 54.2 | 60.5 | −60.4 | i-C4H9-CCCC-AS |
| i-C4H9-CCC-AS-T | - | - | 80.1 | 80.1 | −51.6 | i-C4H9-CCCT-AS |
| i-C4H9-TTT-AS-T | - | - | 72.4 | 72.4 | −35.7 | i-C4H9-TTTT-AS |
| V stage | ||||||
| i-C4H9-CCCC-AS-T | 24.8 | 11.3 | 50.2 | 61.5 | −45.0 | i-C4H9-CCCCC-AS |
| i-C4H9-CCCC-AS-T | - | - | 77.3 | 77.3 | −57.2 | i-C4H9-CCCCT-AS |
| i-C4H9-TTTT-AS-T | - | - | 72.4 | 72.4 | −35.7 | i-C4H9-TTTTT-AS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masliy, A.N.; Akhmetov, I.G.; Kuznetsov, A.M.; Davletbaeva, I.M. DFT and ONIOM Simulation of 1,3-Butadiene Polymerization Catalyzed by Neodymium-Based Ziegler–Natta System. Polymers 2023, 15, 1166. https://doi.org/10.3390/polym15051166
Masliy AN, Akhmetov IG, Kuznetsov AM, Davletbaeva IM. DFT and ONIOM Simulation of 1,3-Butadiene Polymerization Catalyzed by Neodymium-Based Ziegler–Natta System. Polymers. 2023; 15(5):1166. https://doi.org/10.3390/polym15051166
Chicago/Turabian StyleMasliy, Alexey N., Ildar G. Akhmetov, Andrey M. Kuznetsov, and Ilsiya M. Davletbaeva. 2023. "DFT and ONIOM Simulation of 1,3-Butadiene Polymerization Catalyzed by Neodymium-Based Ziegler–Natta System" Polymers 15, no. 5: 1166. https://doi.org/10.3390/polym15051166
APA StyleMasliy, A. N., Akhmetov, I. G., Kuznetsov, A. M., & Davletbaeva, I. M. (2023). DFT and ONIOM Simulation of 1,3-Butadiene Polymerization Catalyzed by Neodymium-Based Ziegler–Natta System. Polymers, 15(5), 1166. https://doi.org/10.3390/polym15051166