





Thermodynamics of the Glassy Polymer State: Equilibrium and Non-Equilibrium Aspects


Abstract
:1. Introduction
2. The LF Equation-of-State Model and the Glass Transition
2.1. The LF Model
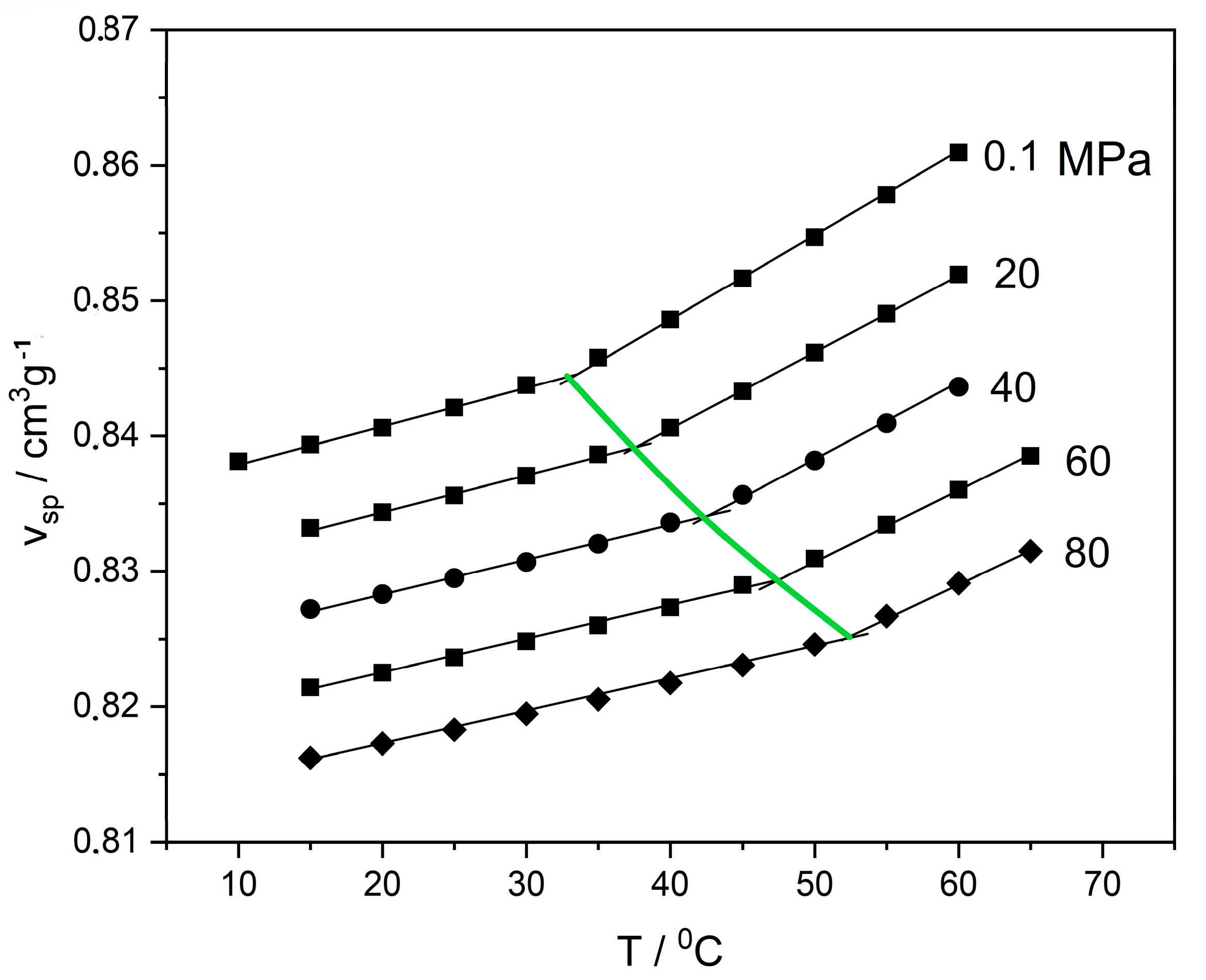
2.2. The Glass Transition
2.3. Glassy Polymer–Compressed Fluid Systems
3. Glass Transition and Hydrogen—Bonding: The LFHB and NRHB Models
NRHB and Retrograde Vitrification
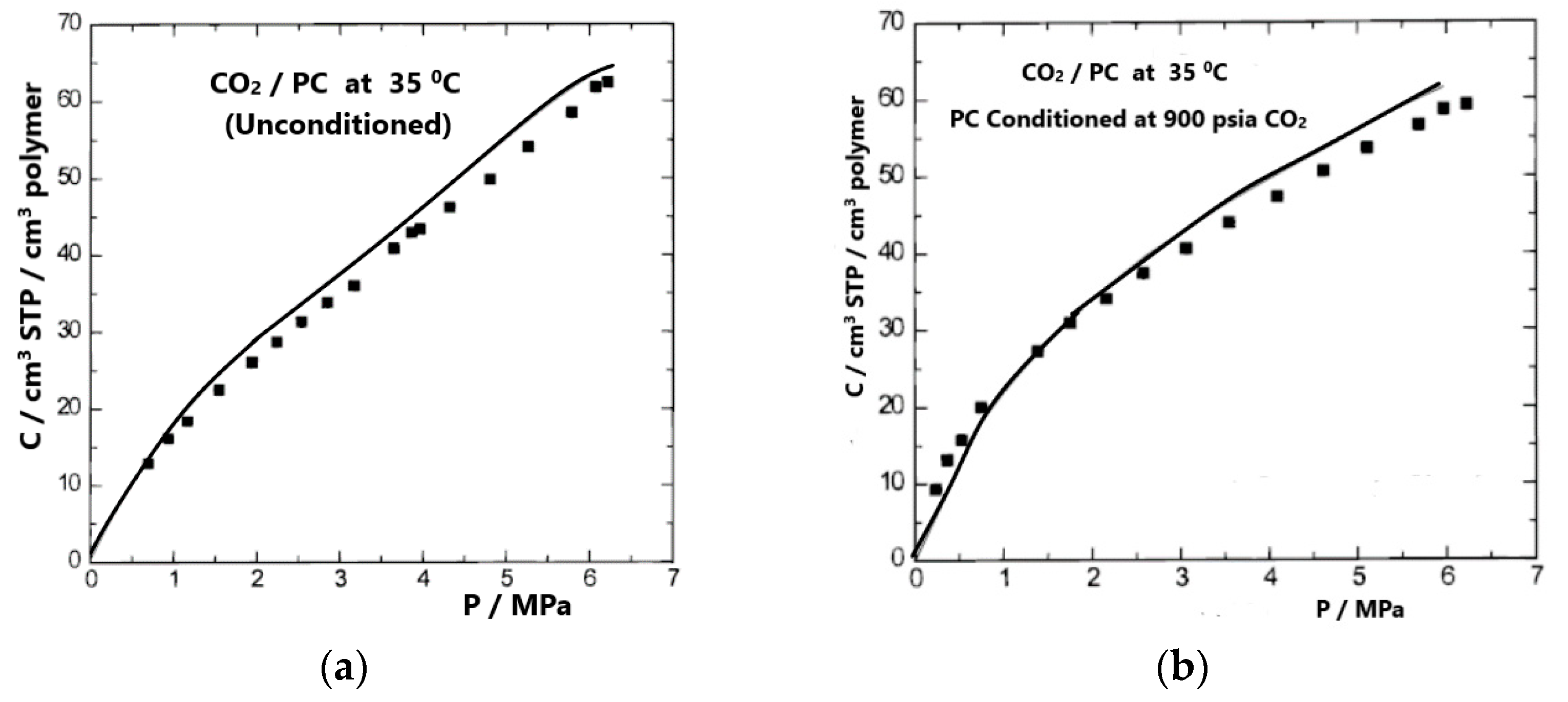
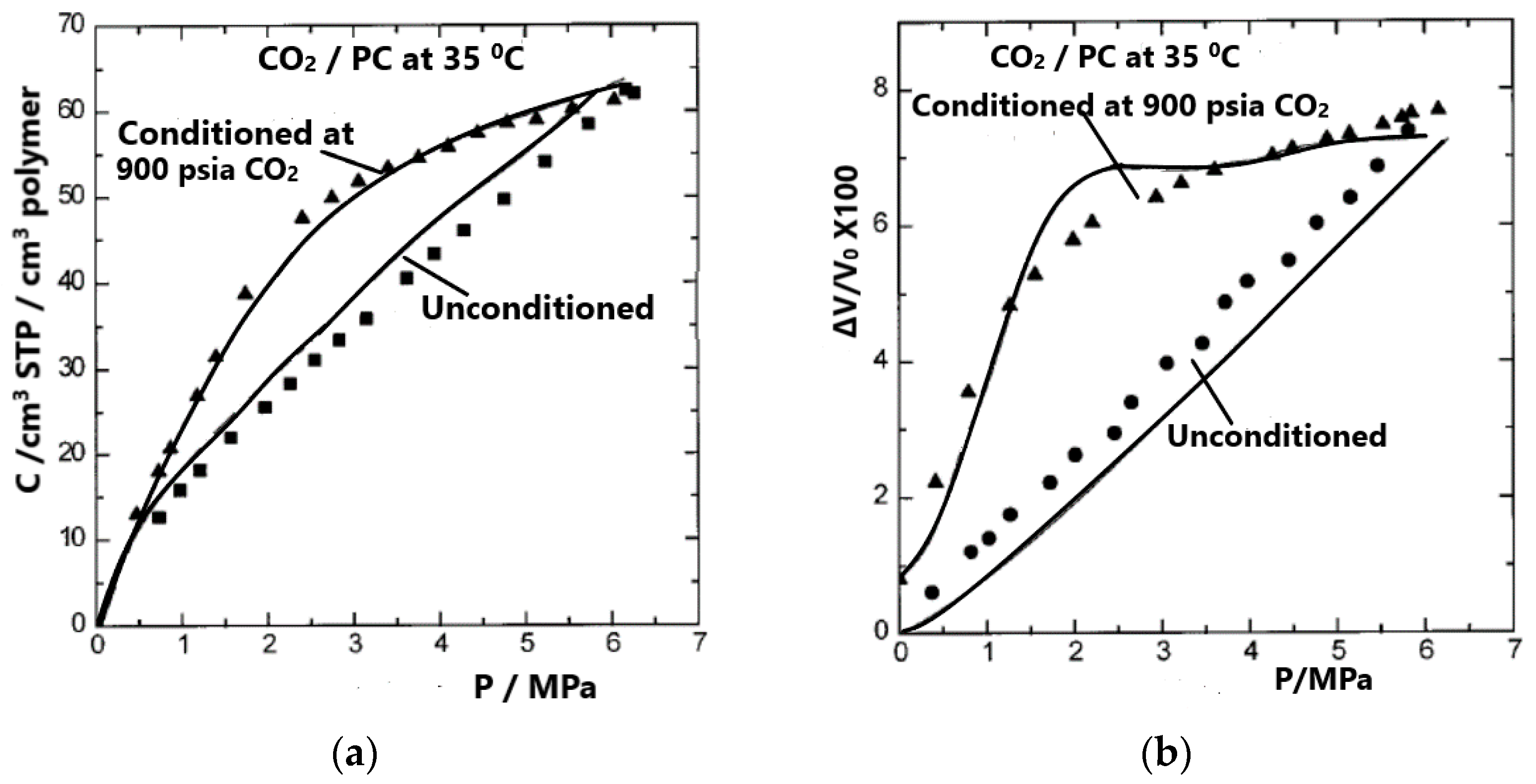
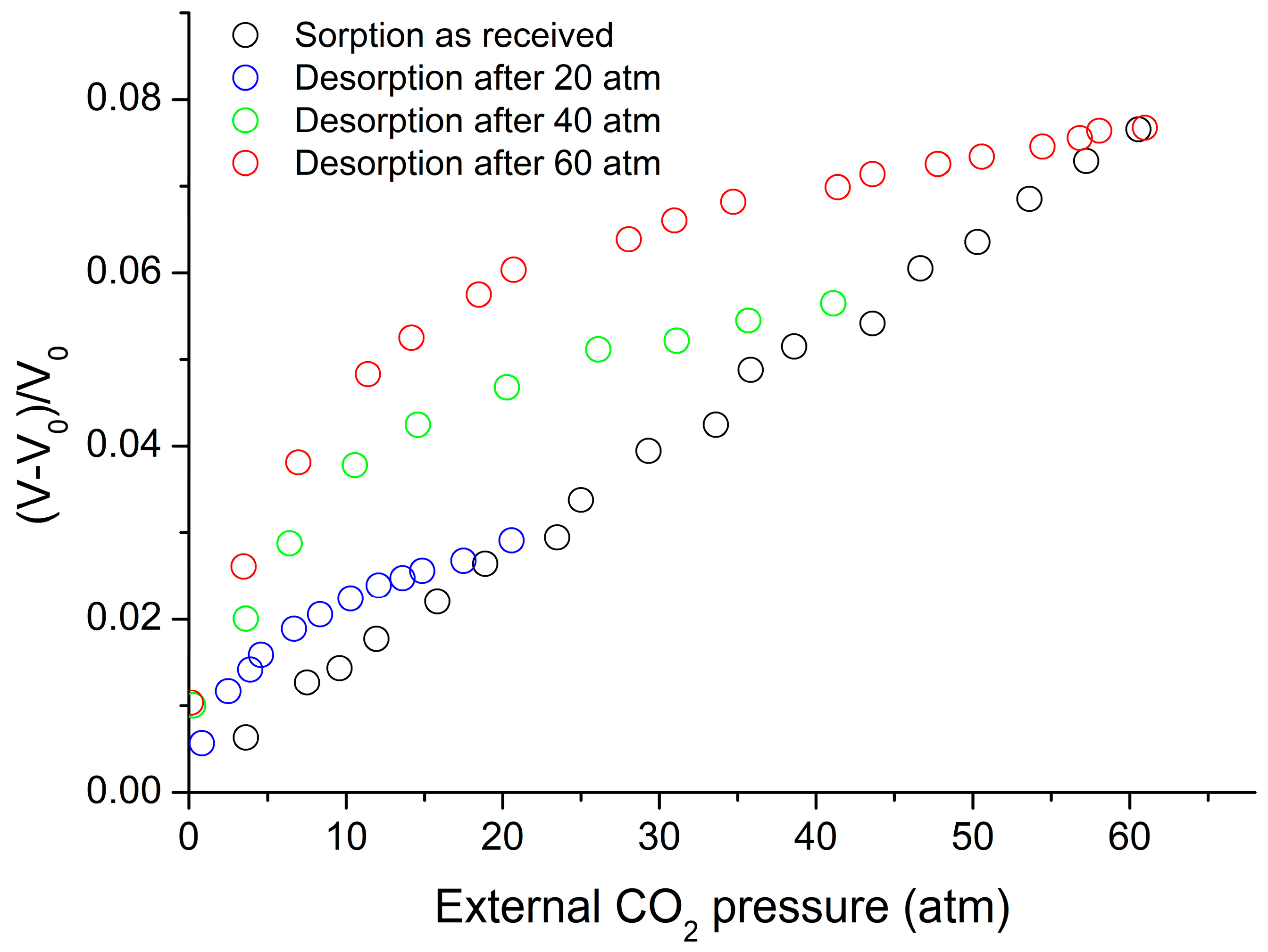
4. Sorption, Solubility, Dilation in Glassy Polymer Systems and the NELF Model
4.1. Definitions of Key Quantities in NELF Model
4.2. NELF Model Calculations
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gee, G. The glassy state in polymers. Contemp. Phys. 1970, 11, 313–334. [Google Scholar] [CrossRef]
- Hutchinson, J.M. Relaxation processes and physical aging. In The Physics of Glassy Polymers, 2nd ed.; Haward, R.N., Young, R.J., Eds.; Springer: Berlin/Heidelberg, Germany, 1997. [Google Scholar]
- Minelli, M.; Sarti, G.C. 110th Anniversary: Gas and Vapor Sorption in Glassy Polymeric Membranes—Critical Review of Different Physical and Mathematical Models. Ind. Eng. Chem. Res. 2020, 59, 341–365. [Google Scholar] [CrossRef]
- Roth, C. Polymer glasses. In Macromolecular Engineering: From Precise Synthesis to Macroscopic Materials and Applications, 2nd ed.; Matyjaszewski, K., Gnanou, Y., Hadjichristidis, N., Muthukumar, M., Eds.; Wiley-VCH GmbH: Berlin, Germany, 2022. [Google Scholar]
- de Gennes, P.G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1979. [Google Scholar]
- McKinney, J.E.; Goldstein, M. PVT relationships for liquid and glassy poly(vinyl acetate). J. Res. Nat. Bur. Std. 1974, 78, 331–353. [Google Scholar] [CrossRef] [PubMed]
- McKinney, J.E.; Simha, R. Thermodynamics of the densification process for polymer glasses. J. Res. Nat. Bur. Std. 1977, 81, 283–297. [Google Scholar] [CrossRef]
- Dixon, D.; Luna-Bárcenas, G.; Johnston, K.P. Microcellular microspheres and microballoons by precipitation with a vapour-liquid compressed fluid antisolvent. Polymer 1994, 35, 3998–4005. [Google Scholar] [CrossRef]
- Tomasko, D.L.; Burley, A.; Feng, L.; Yeh, S.K.; Miyazono, K.; Nirmal-Kumar, S.; Kusaka, I.; Koelling, K. Development of CO2 for polymer foam applications. J. Supercrit. Fluids 2009, 47, 493–499. [Google Scholar] [CrossRef]
- Siripurapu, S.; DeSimone, J.M.; Khan, S.A.; Spontak, R.J. Low temperature Surface Mediated Foaming of Polymer Films. Adv. Mater. 2004, 16, 989–994. [Google Scholar] [CrossRef]
- Zhou, Y.; Tian, Y.; Peng, X. Applications and Challenges of Supercritical Foaming Technology. Polymers 2023, 15, 402. [Google Scholar] [CrossRef]
- Wissinger, R.G.; Paulaitis, E. Glass transitions in polymer/CO2 mixtures at elevated pressures. J. Polym. Sci. Part B Polym. Phys. 1991, 29, 631–633. [Google Scholar] [CrossRef]
- Condo, P.D.; Sanchez, I.C.; Panayiotou, C.G.; Johnston, K.P. Glass transition behavior including retrograde vitrification of polymers with compressed fluid diluents. Macromolecules 1992, 25, 6119–6127. [Google Scholar] [CrossRef]
- McHugh, M.A.; Krukonis, V.J. Supercritical Fluid Extraction: Principles and Practice; Butterworths: Boston, MA, USA, 1986. [Google Scholar]
- Pope, D.S.; Koros, W.J. Effect of Various Preexposure Agents on Methane Sorption and Dilation in Tetramethyl Polycarbonate. Macromolecules 1992, 25, 1711–1715. [Google Scholar] [CrossRef]
- Parsons, A.; Heater, K.; Randal, P. Evaluation of Supercritical CO2 Spray Technology as a Cost Effective Approach to Reduction of Solvents in Wood Finishing. In Proceedings of the AIChE Conference, Denver, CO, USA, 14–17August 1994. [Google Scholar]
- Galizia, M.; Stevens, K.A.; Smith, Z.P.; Paul, D.R.; Freeman, B.D. Non-equilibrium lattice fluid modeling of gas solubility in HAB-6FDA polyimide and its thermally rearranged analogs. Macromolecules 2016, 49, 8768–8779. [Google Scholar] [CrossRef]
- Mensitieri, G.; Scherillo, G.; Panayiotou, C.; Musto, P. Towards a predictive thermodynamic description of sorption processes in polymers: The synergy between theoretical EoS models and vibrational spectroscopy. Mater. Sci. Eng. R 2020, 140, 100525. [Google Scholar] [CrossRef]
- Borrmann, D.; Danzer, A.; Sadowski, G. Water Sorption in Glassy Polyvinylpyrrolidone-Based Polymers. Membranes 2022, 12, 434. [Google Scholar] [CrossRef] [PubMed]
- Prinos, J.; Panayiotou, C. Glass-transition temperatures in hydrogen-bonded polymer mixtures. Polymer 1995, 36, 1223–1227. [Google Scholar] [CrossRef]
- Doghieri, F.; Sarti, G.C. Nonequilibrium lattice fluids: A predictive model for the solubility in glassy polymers. Macromolecules 1996, 29, 7885–7896. [Google Scholar] [CrossRef]
- Baschetti, G.M.; Doghieri, F.; Sarti, G.C. Solubility in glassy polymers: Correlations through the non-equilibrium lattice fluid model. Ind. Eng. Chem. Res. 2001, 40, 3027–3037. [Google Scholar] [CrossRef]
- Sanchez, I.C.; Lacombe, R.H. An elementary molecular theory of classical fluids. Pure fluids. J. Phys. Chem. 1976, 80, 2352–2362. [Google Scholar] [CrossRef]
- Sanchez, I.C.; Lacombe, R.H. Statistical thermodynamics of polymer solutions. Macromolecules 1978, 11, 1145–1156. [Google Scholar] [CrossRef]
- Rodgers, P.A.; Sanchez, I.C. Improvement to the Lattice-Fluid Prediction of Gas Solubilities in Polymer Liquids. J. Polymer Sci. Part B Polymer Phys. 1993, 31, 273–277. [Google Scholar] [CrossRef]
- Sanchez, I.C.; Panayiotou, C. Equations of state thermodynamics of polymer and related solutions. In Models for Thermodynamic and Phase Equilibria Calculations; Sandler, S., Ed.; Marcel Dekker Inc.: New York, NY, USA, 1994. [Google Scholar]
- Flory, P.J. Thermodynamics of high polymer solutions. J. Chem. Phys. 1942, 10, 51–61. [Google Scholar] [CrossRef]
- Flory, P. Polymer Chemistry; Cornel University Press: Ithaca, NY, USA, 1973. [Google Scholar]
- Gibbs, J.H.; Di Marzio, E.A. Nature of the glass transition and the glassy state. J. Chem. Phys. 1958, 28, 373–383. [Google Scholar] [CrossRef]
- Panayiotou, C.; Sanchez, I.C. Hydrogen bonding in fluids: An equation of-state approach. J. Phys. Chem. 1991, 95, 10090–10097. [Google Scholar] [CrossRef]
- Veytsman, B.A. Are Lattice Models Valid for Fluids with Hydrogen Bonds? J. Phys. Chem. 1990, 94, 8499–8500. [Google Scholar] [CrossRef]
- Panayiotou, C.; Tsivintzelis, I.; Economou, I.G. Nonrandom Hydrogen-Bonding Model of Fluids and their Mixtures. 2. Multicomponent Mixtures. Ind. Eng. Chem. Res. 2007, 46, 2628–2636. [Google Scholar] [CrossRef]
- Panayiotou, C.; Acree, W.E.; Zuburtikudis, I. COSMO-RS and LSER models of solution thermodynamics: Towards a COSMO-LSER equation of state model of fluids. J. Mol. Liq. 2023, 390, 122992. [Google Scholar] [CrossRef]
- Scherillo, G.; Loianno, V.; Pierleoni, D.; Esposito, R.; Brasiello, A.; Minelli, M.; Doghieri, F.; Mensitieri, G. Modeling Retrograde Vitrification in the Polystyrene−Toluene System. J. Phys. Chem. B 2018, 122, 3015–3022. [Google Scholar] [CrossRef]
- Pierleoni, D.; Minelli, M.; Scherillo, G.; Mensitieri, G.; Loianno, V.; Bonavolontà, F.; Doghieri, F. Analysis of polystyrene-toluene system through ‘dynamic’ sorption tests: Glass transitions and retrograde vitrification. J. Phys. Chem. B 2017, 121, 9969–9981. [Google Scholar] [CrossRef]
- Boudouris, D.; Panayiotou, C. On the solubility of gas molecules in glassy polymers and the non-equilibrium approach. Macromolecules 1998, 31, 7915–7920. [Google Scholar] [CrossRef]
- Fleming, G.K.; Koros, W.J. Dilation of polymers by sorption of carbon dioxide at elevated pressures. 1. Silicone rubber and unconditioned polycarbonate. Macromolecules 1986, 19, 2285. [Google Scholar] [CrossRef]
- Fleming, G.K.; Koros, W.J. Carbon dioxide conditioning effects on sorption and volume dilation behavior for bisphenol A-polycarbonate. Macromolecules 1990, 23, 1353–1360. [Google Scholar] [CrossRef]
- Olabisi, O.; Simha, R. Pressure-volume-temperature studies of amorphous and crystallizable polymers. I. Experimental. Macromolecules 1975, 8, 206–210. [Google Scholar] [CrossRef]
- Zoller, P. A Study of the Pressure-Volume-Temperature Relationships of Four Related Amorphous Polymers: Polycarbonate, Polyarylate, Phenoxy, and Polysulfone. J. Polym. Sci. Polym. Phys. Ed. 1982, 20, 1453. [Google Scholar] [CrossRef]
- Zoller, P.; Walsh, D.J. Standard Pressure Volume Temperature Data for Polymers; Technomic Publishing AG: Basel, Switzerland, 1995. [Google Scholar]
- Vargaftik, N.B. Handbook of Physical Properties of Liquids and Gases, 2nd ed.; Hemisphere Publishing: Washington, DC, USA, 1975. [Google Scholar]
- Scherillo, G.; Sanguigno, L.; Galizia, M.; Lavorgna, M.; Musto, P.; Mensitieri, G. Non-equilibrium compressible lattice theories accounting for hydrogen bonding interactions: Modelling water sorption thermodynamics in fluorinated polyimides. Fluid Phase Equilibr. 2012, 334, 166. [Google Scholar] [CrossRef]
- De Angelis, M.G.; Sarti, G.C. Solubility of Gases and Liquids in Glassy Polymers. Ann. Rev. Chem. Biomol. Eng. 2011, 2, 97–120. [Google Scholar] [CrossRef] [PubMed]
- Sarti, G.C.; De Angelis, M.G. Calculation of the Solubility of Liquid Solutes in Glassy Polymers. AIChE J. 2012, 58, 292–301. [Google Scholar] [CrossRef]
- Panayiotou, C.; Vera, J.H. Thermodynamics or r-mer Fluids and their Mixtures. Polymer J. 1982, 14, 681–694. [Google Scholar] [CrossRef]
- Panayiotou, C.; Stefanis, E.; Tsivintzelis, I.; Pantoula, M.; Economou, I. Nonrandom Hydrogen-Bonding Model of Fluids and Their Mixtures 1. Pure Fluids. Ind. Eng. Chem. Res. 2004, 43, 6592–6606. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LFHB | NRHB | ||||||
|---|---|---|---|---|---|---|---|
| T * (K) | P * (MPa) | ρ * (g cm−3) | εh * (J mol−1) | εs * (J mol−1 K−1) | vsp,0 * (cm3 g−1) | s | |
| PC | 755 | 534 | 1.275 | 7973.4 | −2.8371 | 0.77968 | 0.728 |
| CO2 | 300 | 630 | 1.515 | 3468.4 | −4.5855 | 0.79641 | 0.909 |
| LFHB | NRHB | ||||||
|---|---|---|---|---|---|---|---|
| T * (K) | P * (MPa) | ρ * (g cm−3) | εh * (J mol−1) | εs * (J mol−1 K−1) | vsp,0 * (cm3 g−1) | s | |
| 6FDA_6FpDA | 750.1 | 476.5 | 1.806 | 5471.1 | 3.8652 | 0.5174 | 0.7757 |
| H2O | 484.1 | 452.7 | 1.0647 | 5336.5 | −6.506 | 0.9703 | 0.8610 |
| LFHB | NRHB | |||||
|---|---|---|---|---|---|---|
| E11 (J mol−1) | S11 (J mol−1 K−1) | E11 (J mol−1) | S11 (J mol−1 K−1) | |||
| H2O | −18,424 | −19.83 | −16,100 | −14.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panayiotou, C. Thermodynamics of the Glassy Polymer State: Equilibrium and Non-Equilibrium Aspects. Polymers 2024, 16, 298. https://doi.org/10.3390/polym16020298
Panayiotou C. Thermodynamics of the Glassy Polymer State: Equilibrium and Non-Equilibrium Aspects. Polymers. 2024; 16(2):298. https://doi.org/10.3390/polym16020298
Chicago/Turabian StylePanayiotou, Costas. 2024. "Thermodynamics of the Glassy Polymer State: Equilibrium and Non-Equilibrium Aspects" Polymers 16, no. 2: 298. https://doi.org/10.3390/polym16020298
APA StylePanayiotou, C. (2024). Thermodynamics of the Glassy Polymer State: Equilibrium and Non-Equilibrium Aspects. Polymers, 16(2), 298. https://doi.org/10.3390/polym16020298