
Searching for the Achilles’ Heel of Urethane Linkage—An Energetic Perspective

 , and
, and
Abstract
:1. Introduction
2. Computational Methods
3. Results
3.1. Thermochemical Properties of the Reactants and Products
3.2. Hydrogenation of the Polyurethane Molecule
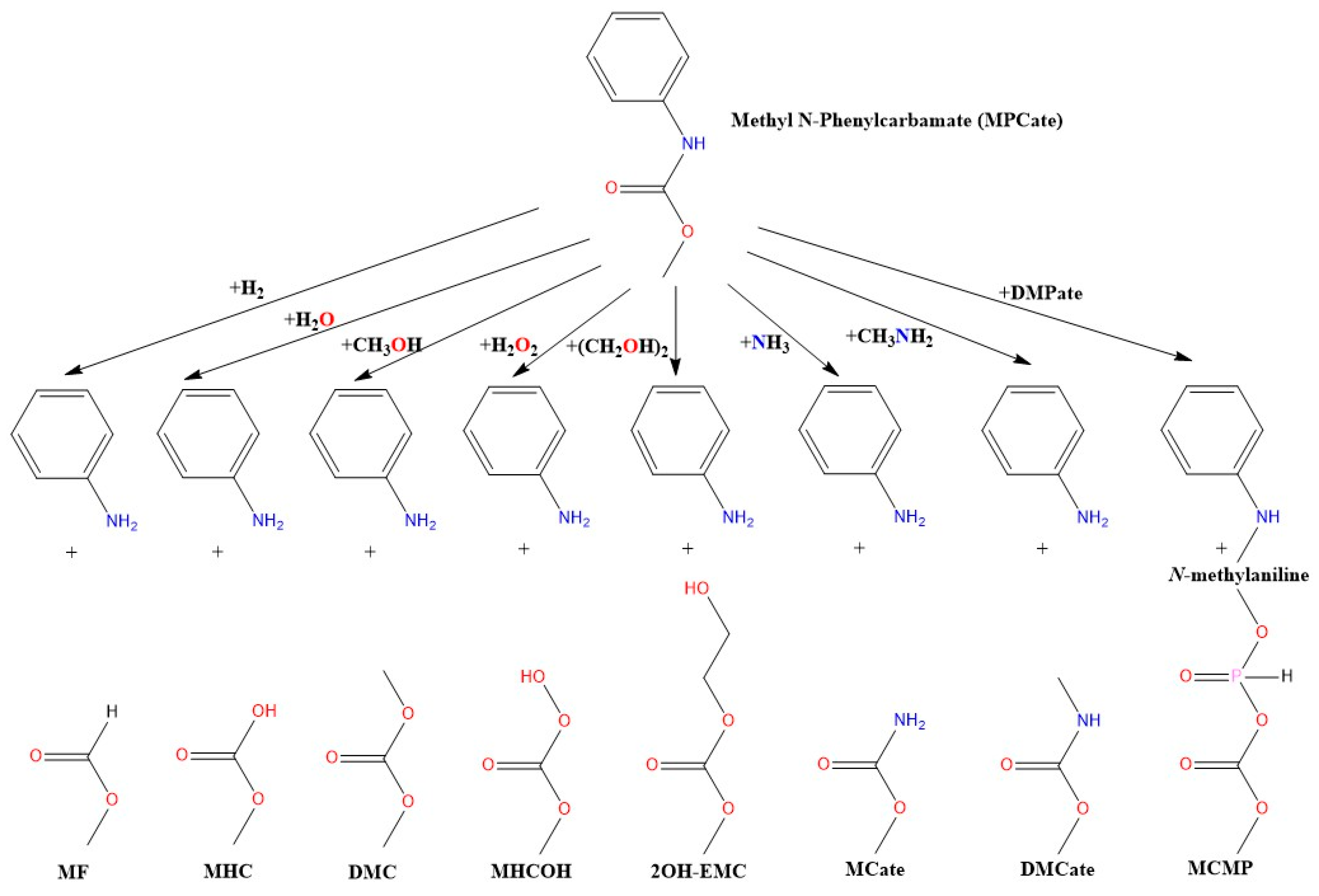
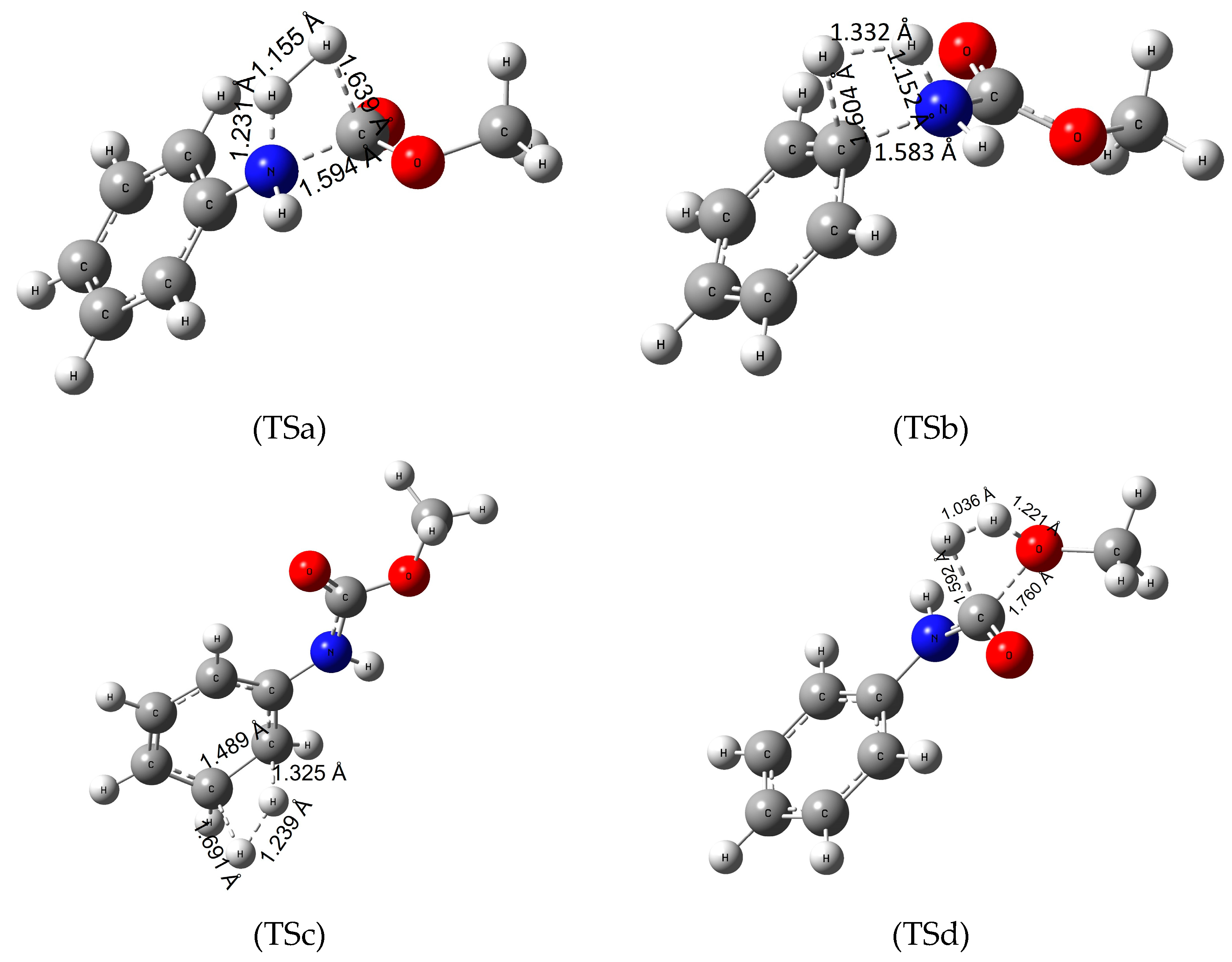
3.3. Reaction Mechanism of Urethane Linkage Termination
3.4. Reaction Mechanism of Urethane Linkage Termination by Water (Hydrolysis)
3.5. Termination Mechanism of the Urethane Linkage by Methanol (Methanolysis)
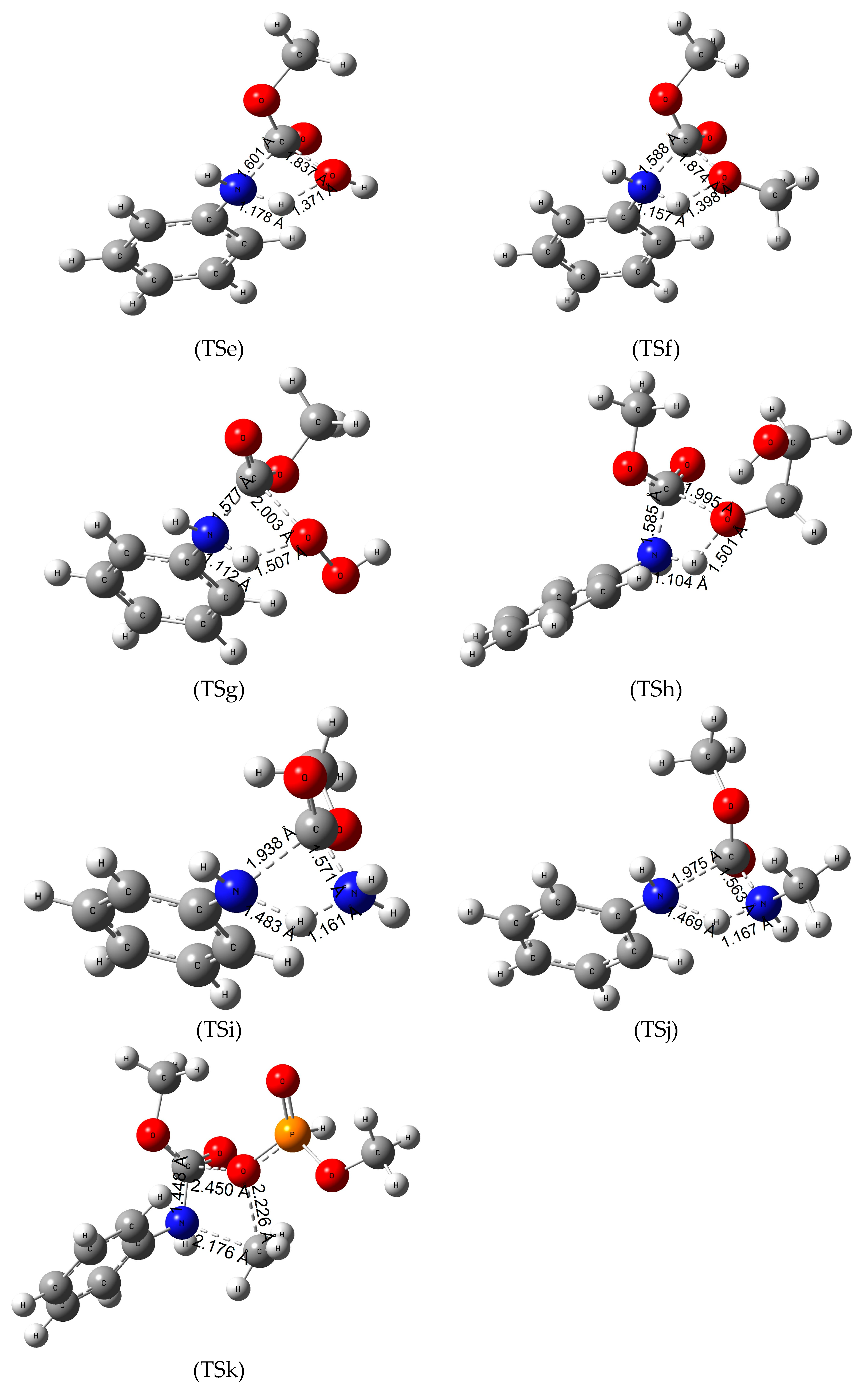
3.6. Reaction Mechanism of Urethane Linkage Termination by Hydrogen Peroxide (H2O2)
3.7. Reaction Mechanism of Urethane Linkage Termination by Ethylene Glycol (EG)
3.8. Reaction Mechanism of Urethane Linkage Termination by Ammonia (Ammonolysis)
3.9. Reaction Mechanism of Urethane Linkage Termination by Methyl Amine (MA)
3.10. Termination of the Urethane Linkage by Dimethyl Phosphite (DMP)
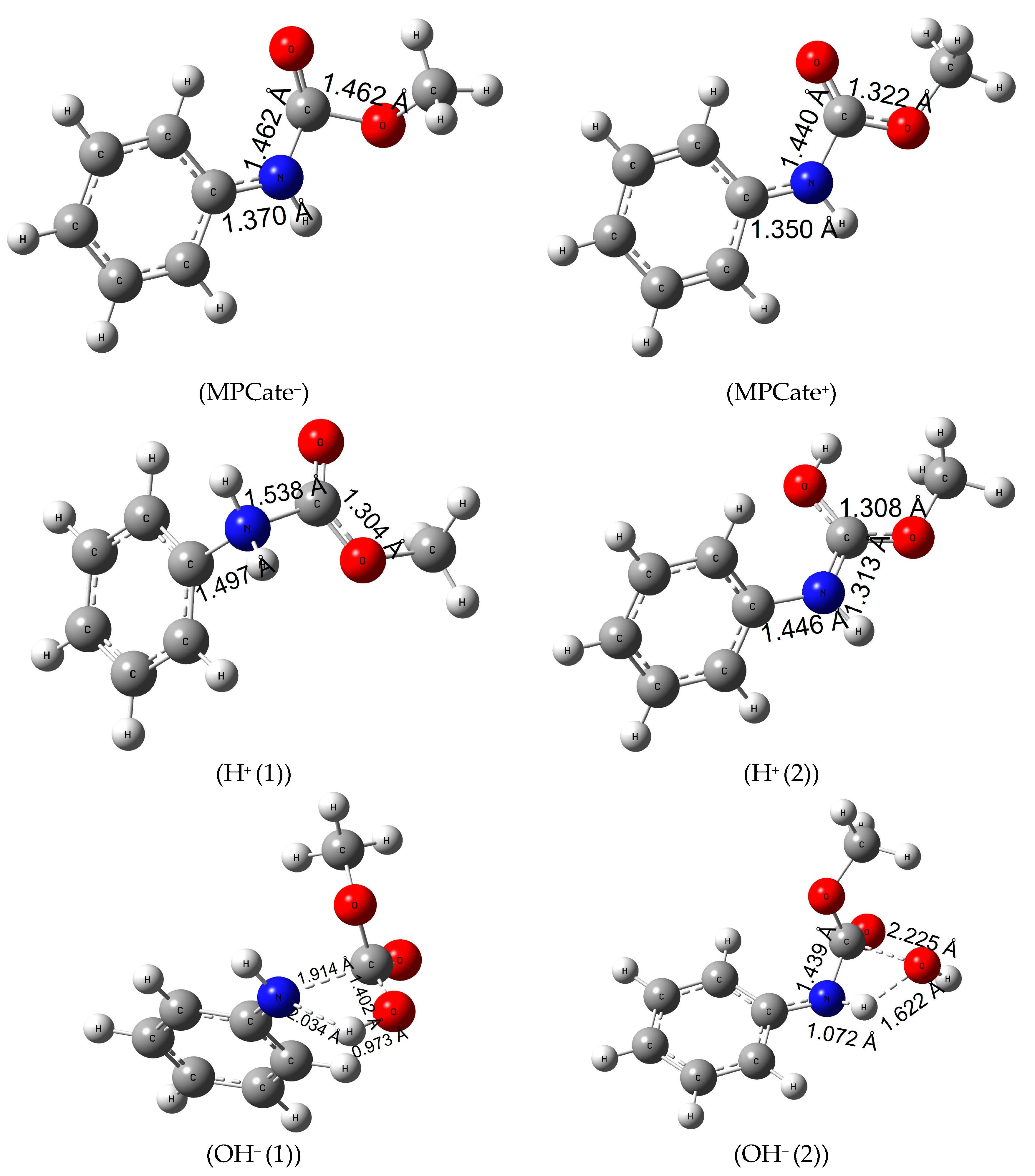
3.11. Reaction Mechanism of Urethane Linkage Termination by Different Ionization Methods
3.12. Experimental Perspectives on Computed Energetics
3.13. Investigation of Reaction Catalysis by Enzymes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kemona, A.; Piotrowska, M. Polyurethane Recycling and Disposal: Methods and Prospects. Polymers 2020, 12, 1752. [Google Scholar] [CrossRef]
- Simón, D.; Borreguero, A.; de Lucas, A.; Rodríguez, J. Recycling of polyurethanes from laboratory to industry, a journey towards the sustainability. Waste Manag. 2018, 76, 147–171. [Google Scholar] [CrossRef]
- Goto, M. Supercritical Water Process for the Chemical Recycling of Waste Plastics. AIP Conf. Proc. 2010, 1251, 169–172. [Google Scholar] [CrossRef]
- Deng, Y.; Dewil, R.; Appels, L.; Ansart, R.; Baeyens, J.; Kang, Q. Reviewing the thermo-chemical recycling of waste polyurethane foam. J. Environ. Manag. 2021, 278, 111527. [Google Scholar] [CrossRef]
- Liang, C.; Gracida-Alvarez, U.R.; Gallant, E.T.; Gillis, P.A.; Marques, Y.A.; Abramo, G.P.; Hawkins, T.R.; Dunn, J.B. Material Flows of Polyurethane in the United States. Environ. Sci. Technol. 2021, 55, 14215–14224. [Google Scholar] [CrossRef]
- Vanbergen, T.; Verlent, I.; De Geeter, J.; Haelterman, B.; Claes, L.; De Vos, D. Recycling of Flexible Polyurethane Foam by Split-Phase Alcoholysis: Identification of Additives and Alcoholyzing Agents to Reach Higher Efficiencies. ChemSusChem 2020, 13, 3835–3843. [Google Scholar] [CrossRef]
- Borda, J.; Pásztor, G.; Zsuga, M. Glycolysis of polyurethane foams and elastomers. Polym. Degrad. Stab. 2000, 68, 419–422. [Google Scholar] [CrossRef]
- Simón, D.; de Lucas, A.; Rodríguez, J.F.; Borreguero, A.M. Flexible polyurethane foams synthesized employing recovered polyols from glycolysis: Physical and structural properties. J. Appl. Polym. Sci. 2017, 134, 45087. [Google Scholar] [CrossRef]
- Gama, N.; Godinho, B.; Marques, G.; Silva, R.; Barros-Timmons, A.; Ferreira, A. Recycling of polyurethane scraps via acidolysis. Chem. Eng. J. 2020, 395, 125102. [Google Scholar] [CrossRef]
- Dai, Z.; Hatano, B.; Kadokawa, J.-I.; Tagaya, H. Effect of diaminotoluene on the decomposition of polyurethane foam waste in superheated water. Polym. Degrad. Stab. 2002, 76, 179–184. [Google Scholar] [CrossRef]
- Bhandari, S.; Gupta, P. Chemical Depolymerization of Polyurethane Foam via Ammonolysis and Aminolysis. In Recycling of Polyurethane Foams; Elsevier: Amsterdam, The Netherlands, 2018; pp. 77–87. [Google Scholar] [CrossRef]
- Mitova, V.; Grancharov, G.; Molero, C.; Borreguero, A.M.; Troev, K.; Rodriguez, J.F. Chemical Degradation of Polymers (Polyurethanes, Polycarbonate and Polyamide) by Esters of H-phosphonic and Phosphoric Acids. J. Macromol. Sci. Part A 2013, 50, 774–795. [Google Scholar] [CrossRef]
- Cabrero-Antonino, J.R.; Adam, R.; Papa, V.; Beller, M. Homogeneous and heterogeneous catalytic reduction of amides and related compounds using molecular hydrogen. Nat. Commun. 2020, 11, 3893. [Google Scholar] [CrossRef]
- Gausas, L.; Kristensen, S.K.; Sun, H.; Ahrens, A.; Donslund, B.S.; Lindhardt, A.T.; Skrydstrup, T. Catalytic Hydrogenation of Polyurethanes to Base Chemicals: From Model Systems to Commercial and End-of-Life Polyurethane Materials. JACS Au 2021, 1, 517–524. [Google Scholar] [CrossRef]
- Johansen, M.B.; Donslund, B.S.; Kristensen, S.K.; Lindhardt, A.T.; Skrydstrup, T. tert-Amyl Alcohol-Mediated Deconstruction of Polyurethane for Polyol and Aniline Recovery. ACS Sustain. Chem. Eng. 2022, 10, 11191–11202. [Google Scholar] [CrossRef]
- Grdadolnik, M.; Drinčić, A.; Oreški, A.; Onder, O.C.; Utroša, P.; Pahovnik, D.; Žagar, E. Insight into Chemical Recycling of Flexible Polyurethane Foams by Acidolysis. ACS Sustain. Chem. Eng. 2022, 10, 1323–1332. [Google Scholar] [CrossRef]
- Liu, J.; He, J.; Xue, R.; Xu, B.; Qian, X.; Xin, F.; Blank, L.M.; Zhou, J.; Wei, R.; Dong, W.; et al. Biodegradation and up-cycling of polyurethanes: Progress, challenges, and prospects. Biotechnol. Adv. 2021, 48, 107730. [Google Scholar] [CrossRef]
- Chaffin, K.A.; Chen, X.; McNamara, L.; Bates, F.S.; Hillmyer, M.A. Polyether Urethane Hydrolytic Stability after Exposure to Deoxygenated Water. Macromolecules 2014, 47, 5220–5226. [Google Scholar] [CrossRef]
- Liu, L.; Zhu, Z.; Wu, Y. Depolymerization kinetics for thermoplastic polyurethane elastomer degradation in subcritical methanol. Polym. Degrad. Stab. 2017, 140, 126–135. [Google Scholar] [CrossRef]
- Liu, L.; Tang, L.; Wu, Y.; Ni, Y.; Zhu, Z. Degradation process investigation of thermoplastic polyurethane elastomer in supercritical methanol. Polym. Degrad. Stab. 2013, 98, 2520–2528. [Google Scholar] [CrossRef]
- Zamani, S.; van der Voort, S.H.E.; Lange, J.-P.; Kersten, S.R.A.; Ruiz, M.P. Polyurethane Recycling: Thermal Decomposition of 1,3-Diphenyl Urea to Isocyanates. Polymers 2023, 15, 2522. [Google Scholar] [CrossRef]
- Olazabal, I.; González, A.; Vallejos, S.; Rivilla, I.; Jehanno, C.; Sardon, H. Upgrading Polyurethanes into Functional Ureas through the Asymmetric Chemical Deconstruction of Carbamates. ACS Sustain. Chem. Eng. 2023, 11, 332–342. [Google Scholar] [CrossRef]
- Akutsu-Shigeno, Y.; Adachi, Y.; Yamada, C.; Toyoshima, K.; Nomura, N.; Uchiyama, H.; Nakajima-Kambe, T. Isolation of a bacterium that degrades urethane compounds and characterization of its urethane hydrolase. Appl. Microbiol. Biotechnol. 2006, 70, 422–429. [Google Scholar] [CrossRef]
- Tournier, V.; Duquesne, S.; Guillamot, F.; Cramail, H.; Taton, D.; Marty, A.; André, I. Enzymes’ Power for Plastics Degradation. Chem. Rev. 2023, 123, 5612–5701. [Google Scholar] [CrossRef]
- Boros, R.Z.; Koós, T.; Wafaa, C.; Nehéz, K.; Farkas, L.; Viskolcz, B.; Szőri, M. A theoretical study on the phosgenation of methylene diphenyl diamine (MDA). Chem. Phys. Lett. 2018, 706, 568–576. [Google Scholar] [CrossRef]
- Boros, R.Z.; Farkas, L.; Nehéz, K.; Viskolcz, B.; Szőri, M. An Ab Initio Investigation of the 4,4′-Methlylene Diphenyl Diamine (4,4′-MDA) Formation from the Reaction of Aniline with Formaldehyde. Polymers 2019, 11, 398. [Google Scholar] [CrossRef]
- Thangaraj, R.; Fiser, B.; Qiu, X.; Li, C.; Viskolcz, B.; Szőri, M. An Ab Initio Investigation on Relevant Oligomerization Reactions of Toluene Diisocyanate (TDI). Polymers 2022, 14, 4183. [Google Scholar] [CrossRef]
- Thangaraj, R.; Horváth, T.; Boros, R.Z.; Viskolcz, B.; Szőri, M. A Theoretical Study on the Phosgenation of 2,4-Toluenediamine (2,4-TDA). Polymers 2022, 14, 2254. [Google Scholar] [CrossRef]
- Lenzi, V.; Crema, A.; Pyrlin, S.; Marques, L. Current State and Perspectives of Simulation and Modeling of Aliphatic Isocyanates and Polyisocyanates. Polymers 2022, 14, 1642. [Google Scholar] [CrossRef]
- Cheikh, W.; Rózsa, Z.B.; López, C.O.C.; Mizsey, P.; Viskolcz, B.; Szőri, M.; Fejes, Z. Urethane Formation with an Excess of Isocyanate or Alcohol: Experimental and Ab Initio Study. Polymers 2019, 11, 1543. [Google Scholar] [CrossRef]
- Gertig, C.; Erdkamp, E.; Ernst, A.; Hemprich, C.; Kröger, L.C.; Langanke, J.; Bardow, A.; Leonhard, K. Reaction Mechanisms and Rate Constants of Auto-Catalytic Urethane Formation and Cleavage Reactions. ChemistryOpen 2021, 10, 534–544. [Google Scholar] [CrossRef]
- Waleed, H.Q.; Hadjadj, R.; Viskolcz, B.; Fiser, B. Effect of morpholine, and 4-methylmorpholine on urethane formation: A computational study. Sci. Rep. 2023, 13, 17950. [Google Scholar] [CrossRef]
- Waleed, H.Q.; Csécsi, M.; Konyhás, V.; Boros, Z.R.; Viskolcz, B.; Fejes, Z.; Fiser, B. Aliphatic tertiary amine catalysed urethane formation—A combined experimental and theoretical study. Phys. Chem. Chem. Phys. 2022, 24, 20538–20545. [Google Scholar] [CrossRef]
- Waleed, H.Q.; Pecsmány, D.; Csécsi, M.; Farkas, L.; Viskolcz, B.; Fejes, Z.; Fiser, B. Experimental and Theoretical Study of Cyclic Amine Catalysed Urethane Formation. Polymers 2022, 14, 2859. [Google Scholar] [CrossRef]
- Baboul, A.G.; Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Gaussian-3 theory using density functional geometries and zero-point energies. J. Chem. Phys. 1999, 110, 7650–7657. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.P.G.A.; Petersson, G.A.; Nakatsuji, H.J.R.A.; et al. Gaussian 16, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version 6.1; Semichem Inc.: Shawnee, KS, USA, 2019. [Google Scholar]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on the Generalized Born Approximation with Asymmetric Descreening. J. Chem. Theory Comput. 2009, 5, 2447–2464. [Google Scholar] [CrossRef]
- Nicolaides, A.; Rauk, A.; Glukhovtsev, M.N.; Radom, L. Heats of Formation from G2, G2(MP2), and G2(MP2,SVP) Total Energies. J. Phys. Chem. 1996, 100, 17460–17464. [Google Scholar] [CrossRef]
- Johnson, R.D., III; Russell, D. NIST Computational Chemistry Comparison and Benchmark Database, NIST Standard Reference Database 101. Available online: http://cccbdb.nist.gov/ (accessed on 8 May 2023).
- Ruscic, B.; Bross, D.H. Active Thermochemical Tables (ATcT), Version 1.124: Argonne National Laboratory. Available online: http://atct.anl.gov/ (accessed on 1 March 2023).
- Goos, E.; Burcat, A.; Ruscic, B. Extended Third Millennium Ideal Gas and Condensed Phase Thermochemical Database for Combustion with Updates from Active Thermochemical Tables. Available online: http://garfield.chem.elte.hu/Burcat/hf.doc (accessed on 8 May 2023).
- NIST National Institute of Standards and Technology. NIST Chemistry Webbook, SRD 69, Group Additivity Based Estimates. Available online: https://webbook.nist.gov/chemistry/grp-add/ (accessed on 3 April 2023).
- Zhou, Y.; Wu, J.; Lemmon, E.W. Thermodynamic Properties of Dimethyl Carbonate. J. Phys. Chem. Ref. Data 2011, 40, 043106. [Google Scholar] [CrossRef]
- Pedley, J.B.; Naylor, R.D.; Kirby, S.P. Thermochemical Data of Organic Compounds, 2nd ed.; Springer Science and Business Media LLC: Dordrecht, The Netherlands, 1986. [Google Scholar] [CrossRef]
- Hemmaplardh, B.; King, A.D. Solubility of methanol in compressed nitrogen, argon, methane, ethylene, ethane, carbon dioxide, and nitrous oxide. Evidence for association of carbon dioxide with methanol in the gas phase. J. Phys. Chem. 1972, 76, 2170–2175. [Google Scholar] [CrossRef]
- Meijs, G.F.; McCarthy, S.J.; Rizzardo, E.; Chen, Y.; Chatelier, R.C.; Brandwood, A.; Schindhelm, K. Degradation of medical-grade polyurethane elastomers: The effect of hydrogen peroxide in vitro. J. Biomed. Mater. Res. 1993, 27, 345–356. [Google Scholar] [CrossRef]
- Vakil, A.U.; Petryk, N.M.; Du, C.; Howes, B.; Stinfort, D.; Serinelli, S.; Gitto, L.; Ramezani, M.; Beaman, H.T.; Monroe, M.B.B. In vitro and in vivo degradation correlations for polyurethane foams with tunable degradation rates. J. Biomed. Mater. Res. Part A 2023, 111, 580–595. [Google Scholar] [CrossRef]
- O’Neil, M. The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals, 15th ed.; Royal Society of Chemistry: Cambridge, UK, 2013. [Google Scholar]
- Troev, K.; Atanasov, V.; Tsevi, R.; Grancharov, G.; Tsekova, A. Chemical degradation of polyurethanes. Degradation of microporous polyurethane elastomer by dimethyl phosphonate. Polym. Degrad. Stab. 2000, 67, 159–165. [Google Scholar] [CrossRef]
- Sun, Y.; Chmielewski, A.G. Applications of Ionizing Radiation in Materials Processing; Institute of Nuclear Chemistry and Technology: Warsaw, Poland, 2017; Volume 1. [Google Scholar]
- Nakajima-Kambe, T.; Shigeno-Akutsu, Y.; Nomura, N.; Onuma, F.; Nakahara, T. Microbial degradation of polyurethane, polyester polyurethanes and polyether polyurethanes. Appl. Microbiol. Biotechnol. 1999, 51, 134–140. [Google Scholar] [CrossRef]
- Skleničková, K.; Abbrent, S.; Halecký, M.; Kočí, V.; Beneš, H. Biodegradability and ecotoxicity of polyurethane foams: A review. Crit. Rev. Environ. Sci. Technol. 2020, 52, 157–202. [Google Scholar] [CrossRef]
- Howard, G.T. Biodegradation of polyurethane: A review. Int. Biodeterior. Biodegrad. 2002, 49, 245–252. [Google Scholar] [CrossRef]
- Zou, X.; Jiang, X.; Wen, Y.; Wu, S.; Nadege, K.; Ninette, I.; Zhang, H.; Jin, Q.; Wang, X. Enzymatic synthesis of structured lipids enriched with conjugated linoleic acid and butyric acid: Strategy consideration and parameter optimization. Bioprocess Biosyst. Eng. 2020, 43, 273–282. [Google Scholar] [CrossRef]
- Müller, V.; Burger, Y. HDCR-katalysierte Hydrogenierung von CO2 zum H2-Carrier Ameisensäure. Biospektrum 2022, 28, 763–766. [Google Scholar] [CrossRef]
- Malthouse, J.P.G. Kinetic Studies of the Effect of pH on the Trypsin-Catalyzed Hydrolysis of N-α-benzyloxycarbonyl-l-lysine-p-nitroanilide: Mechanism of Trypsin Catalysis. ACS Omega 2020, 5, 4915–4923. [Google Scholar] [CrossRef]
- Canto, V.P.D.; Thompson, C.E.; Netz, P.A. Polyurethanases: Three-dimensional structures and molecular dynamics simulations of enzymes that degrade polyurethane. J. Mol. Graph. Model. 2019, 89, 82–95. [Google Scholar] [CrossRef]
- Nie, K.; Xie, F.; Wang, F.; Tan, T. Lipase catalyzed methanolysis to produce biodiesel: Optimization of the biodiesel production. J. Mol. Catal. B Enzym. 2006, 43, 142–147. [Google Scholar] [CrossRef]
- Talon, R.; Leroy, S. Fermented Foods: Fermented Meat Products and the Role of Starter Cultures. In Encyclopedia of Food Microbiology, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 870–874. [Google Scholar] [CrossRef]
- Heshof, R.; De Graaff, L.H.; Villaverde, J.J.; Silvestre, A.J.; Haarmann, T.; Dalsgaard, T.K.; Buchert, J. Industrial potential of lipoxygenases. Crit. Rev. Biotechnol. 2016, 36, 665–674. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | ∆f,298.15KH0 (g) (kJ/mol) | Method | Ref. |
|---|---|---|---|
| (carboperoxyoxy)methane (C2H4O4) | −471.1 | AS | |
| −494.5 | GA | ||
| 2-hydroxyethyl methyl carbonate (C4H8O4) | −752.9 | AS | |
| −753.5 | GA | ||
| ammonia (NH3) | −42.4 | AS | |
| −45.6 | Burcat | ||
| aniline (C6H7N) | 85.6 | AS | |
| 87.0 | Burcat | ||
| dimethyl carbonate (C3H6O3) | −569.3 | AS | [46] |
| −571.0 | lit. | ||
| dimethyl phosphite (C2H7O3P) | −758.3 | AS | |
| ethylene-glycol (C2H6O2) | −387.9 | AS | |
| −389.4 | Burcat | ||
| formanilide (C7H7NO) | −64.8 | AS | |
| −55.2 | GA | ||
| H2O | −240.5 | AS | |
| −241.8 | Burcat | ||
| hydrogen-peroxide (H2O2) | −131.0 | AS | |
| −135.9 | Burcat | ||
| methanol (CH3OH) | −199.4 | AS | |
| −200.9 | Burcat | ||
| methyl carbonic methyl phosphonic anhydride (MCMPA-C3H7O5P) | −1113.6 | AS | |
| methyl methylcarbamate (C3H7NO2) | −402.7 | AS | |
| −367.7 | GA | ||
| methyl phenyl urethane (C8H9NO2) | −286.4 | AS | [47] |
| −186.7 (cr) | lit. | ||
| methylamine (CH3NH2) | −17.5 | AS | |
| −20.9 | Burcat | ||
| methylcarbamate (C2H5NO2) | −406.3 | GA | |
| −412.0 | Burcat | ||
| methylformate (C2H4O2) | −358.8 | AS | |
| −360.0 | Ruisic | ||
| methyl-hydrogencarbonate (C2H4O3) | −589.6 | AS | [48] |
| −607 | lit. | ||
| methyl-phenylamine (C7H9N) | 92.4 | AS | |
| 86.6 | GA | ||
| OH− | −138.5 | AS | |
| −139.0 | Ruisic |
| Reactant | ΔR;298.15KG0 (kJ/mol) [ΔTS;298.15KG0 (kJ/mol)] | ΔR;398.15KG (kJ/mol) [ΔTS;398.15KG (kJ/mol)] | ||||
|---|---|---|---|---|---|---|
| Gas Phase (ε = 1) | Aniline (ε = 6.8) | Water (ε = 78.4) | Gas Phase (ε = 1) | Aniline (ε = 6.8) | Water (ε = 78.4) | |
| H2 (TSa) | 5.2 [339.6] | 5.2 [311.0] | −6.4 [290.4] | 7.3 [325.0] | 0.9 [298.8] | −6.5 [281.6] |
| H2 (TSb) | −48.9 [424.9] | −56.0 [378.1] | −63.8 [362.0] | −47.7 [431.7] | −56.9 [384.8] | −64.8 [367.7] |
| H2 (TSc) | 41.2 [452.2] | 35.0 [373.9] | 28.3 [329.1] | 48.8 [459.4] | 41.5 [379.6] | 33.8 [334.3] |
| H2 (TSd) | 57.9 [393.2] | 40.9 [382.2] | 35.5 [370.7] | 5.4 [346.1] | −11.7 [335.1] | −15.5 [323.4] |
| Reactant | ΔR;298.15KG0 (kJ/mol) [ΔTS;298.15KG0 (kJ/mol)] | ΔR;398.15KG0 (kJ/mol) [ΔTS;398.15KG0 (kJ/mol)] | ||||
|---|---|---|---|---|---|---|
| Gas Phase (ε = 1) | Aniline (ε = 6.8) | Water (ε = 78.4) | Gas Phase (ε = 1) | Aniline (ε = 6.8) | Water (ε = 78.4) | |
| H2 (TSa) | 5.2 [339.6] | 5.2 [311.0] | −6.4 [290.4] | 7.3 [325.0] | 0.9 [298.8] | −6.5 [281.6] |
| H2O (TSe) | 2.8 [235.5] | 2.8 [220.5] | 8.5 [205.7] | 3.3 [239.3] | 8.4 [224.9] | 9.0 [209.9] |
| CH3OH (TSf) | 5.7 [219.3] | 9.8 [205.5] | 11.1 [192.4] | 4.5 [223.6] | 8.9 [210.6] | 9.5 [196.9] |
| H2O2 (TSg) | 1.4 [193.0] | 1.4 [171.9] | 13.4 [164.1] | 3.2 [199.2] | −3.6 [172.3] | 13.2 [169.0] |
| C2H6O2 (TSh) | 21.2 [221.7] | 21.2 [204.5] | 16.7 [192.8] | 20.9 [228.2] | 23.3 [209.7] | 14.7 [197.2] |
| NH3 (TSi) | 3.3 [250.2] | 1.2 [225.5] | 1.4 [216.9] | 3.9 [254.8] | 1.0 [230.0] | 2.7 [222.0] |
| CH3NH2 (TSj) | −12.1 [229.7] | −14.1 [204.2] | −22.8 [193.1] | −14.8 [233.6] | −15.8 [208.4] | −25.3 [196.9] |
| C2H7O3P (TSk) | 16.5 [318.0] | 13.5 [291.0] | 22.4 [277.4] | 17.3 [323.3] | 12.2 [292.2] | 23.8 [280.8] |
| Reactant | ΔR;298.15KG0 (kJ/mol) [ΔTS;298.15KG0 (kJ/mol)] | ΔR;398.15KG0 (kJ/mol) [ΔTS;398.15KG0 (kJ/mol)] | ||||
|---|---|---|---|---|---|---|
| Gas Phase (ε = 1) | Aniline (ε = 6.8) | Water (ε = 78.4) | Gas Phase (ε = 1) | Aniline (ε = 6.8) | Water (ε = 78.4) | |
| MPCate− | 95.0 | −71.5 | −116.3 | 92.9 | −72.5 | −117.5 |
| MPCate+ | 789.4 | 616.9 | 594.5 | 788.8 | 616.7 | 594.5 |
| H+ (1) | −833.1 | 67.5 | 38.7 | −835.1 | 66.3 | 37.2 |
| H+ (2) | −943.0 | 138.2 | 152.3 | −849.5 | 66.6 | 36.2 |
| OH− (1) | 108.5 [140.3] | 89.3 [144.7] | 65.9 [139.7] | 112.4 [143.9] | 94.0 [150.3] | 69.4 [143.1] |
| OH− (2) | 16.0 [176.0] | 17.7 [148.8] | 6.7 [114.8] | 16.8 [180.1] | 19.2 [153.9] | 6.7 [118.2] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horváth, T.; Kecskés, K.; Jordán Csábrádiné, A.; Szőri-Dorogházi, E.; Viskolcz, B.; Szőri, M. Searching for the Achilles’ Heel of Urethane Linkage—An Energetic Perspective. Polymers 2024, 16, 1126. https://doi.org/10.3390/polym16081126
Horváth T, Kecskés K, Jordán Csábrádiné A, Szőri-Dorogházi E, Viskolcz B, Szőri M. Searching for the Achilles’ Heel of Urethane Linkage—An Energetic Perspective. Polymers. 2024; 16(8):1126. https://doi.org/10.3390/polym16081126
Chicago/Turabian StyleHorváth, Tamás, Karina Kecskés, Anikó Jordán Csábrádiné, Emma Szőri-Dorogházi, Béla Viskolcz, and Milán Szőri. 2024. "Searching for the Achilles’ Heel of Urethane Linkage—An Energetic Perspective" Polymers 16, no. 8: 1126. https://doi.org/10.3390/polym16081126
APA StyleHorváth, T., Kecskés, K., Jordán Csábrádiné, A., Szőri-Dorogházi, E., Viskolcz, B., & Szőri, M. (2024). Searching for the Achilles’ Heel of Urethane Linkage—An Energetic Perspective. Polymers, 16(8), 1126. https://doi.org/10.3390/polym16081126