3.2. Synthetic L-Polypeptides
Although periodic polypeptides (homopolypeptides) do not exist in nature, knowledge of their structure can contribute to an understanding of the properties of proteins. The -CO-NH-C- main chain is common to all polypeptides and proteins. Also, the different side chains likely influence their contribution properties. Ten homopolypeptides: poly-Gly, poly-L-Ala, poly-DL-Ala, poly-L-Ser, poly-L-Leu, Poly-L-Pro, polyl-L-Lys, poly-L-Tyr, poly-L-Glu and poly-L-gamma-benzyl-glutamate, are outlined below [
18,
20]. Suspension polymerization (10 ml MMA, 0.15 mL initiator, 1 mL water, 0.022–0.055 g polypeptide, at 37 °C for 1 h) was carried out in a conical flask with a stopper. The polymerization product obtained was washed thoroughly with acetone, dried
in vacuo, and then weighed. Samples of polypeptide-MMA-grafted copolymer were treated with 2 mL of glacial acetic acid and 4 mL of 6N hydrochloric acid at 100–110 °C for nine hours. The precipitate was then filtered, washed with water and methanol, and then dissolved in acetone, precipitated with methanol, dried
in vacuo, and weighed. The molecular weight of separated polymers was determined in benzene at 30°C. The infrared spectra of grafted PMMA were identical to that of the original PMMA. (Note: To prevent the hydrolysis of MMA with methacrylic acid, using a glacial acetic acid/HCl solution, methylation of PMMA branches was treated with diazomethane.)
Molecular weight determination of separated polymers: The intrinsic viscosity of the PMMA separated by treatment with glacial acetic acid/HCl was determined in benzene at 30 °C. The number-average molecular weight was calculated using the Equation 2 [
18]:
Characterization of the graft copolymerization was evaluated using the following Equations:
The results are shown in
Table 2. No increase in the percentage graft was observed for poly-Gly, poly-L-Leu, poly-L-Tyr and poly-L-γ-benzyl glutamate. By contrast, the increase in the percentage graft is shown for poly-L-Ala, poly-L-Ser, poly-L-Pro, poly-L-Lys and poly-L-Glu; whereas the number of grafted-PMMA branches declined in the order: poly-L-Ala > poly-L-Ser > poly-DL-Ala > poly-L-Pro > poly-L-Glu > poly-L-Lys.
The discrepancy between the two poly-alanines may be due to the type of helix; i.e., steric hindrance of the side chain between types L and D. The percentage graft for poly-L-tyrosine was zero, possibly due to its radical inhibition of the phenolic O–H group. No grafting was also observed for poly-L-Leu and poly-L-γ-benzyl-glutamate. The weight of MMA-homopolymer formed, the molecular weight, and the number of polymers formed for the polypeptides as a whole were (1.1–1.3) g, (1.1–1.2) × 105 and (10–11) × 10−6 mol, respectively.
Table 2.
Tri-n-butylborane-initiated graft copolymerization of methyl methacrylate with hydrated synthetic poly-L-peptides.
Table 2.
Tri-n-butylborane-initiated graft copolymerization of methyl methacrylate with hydrated synthetic poly-L-peptides.
| | Percent graftt (%) | Mol. wt. of grafted-PMMA | *No.branches | *No. branches | ΔεHOMO of corresponding amino acid a |
|---|
| Polypeptide (pp) | (%) | ×10−5 | (mole/g−pp) × 106 | (mole/mole−pp) × 103 | eV |
|---|
| Poly glycine | 0 | _ | 0 | 0 | 0.014 |
| Poly-L-alanine | 86.1 ± 9.7 | 6.2 ± 0.2 | 1.4 ± 0.1 | 5.9 | 0.184 |
| Poly-DL-alanine | 79 | 19.4 | 0.4 | 1.2 | _ |
| Poly-L-leucine | 0 | _ | 0 | 0 | −0.057 |
| Poly-L-serine | 108.2 ± 43 | 13.5 ± 4.8 | 0.8 ± 0.3 | 4.0 | 0.159 |
| Poly-L-tyrosine | 0 | _ | 0 | 0 | −0.116 |
| Poly-L-lysineb) | 18.0 ± 4.2 | 12.9 ± 0.1 | 0.1 ± 0.1 | 0.3 | −0.385 |
| Poly-L-prolineb) | 49.3 ± 6.7 | 23.2 ± 0.2 | 0.2 ± 0.0 | 2.0 | 0.012 |
| Poly-L-glutamic acid | 25.1 ± 2.6 | 12.8 ± 0.3 | 0.2 ± 0.0 | 0.8 | −0.073 |
| Poly-L-γ- benzyl glutamate | 0 | _ | 0 | 0 | _ |
Currently, computational chemistry has being applied for analyzing the transition structures for the hydroboration of alkenes by borane compounds at the semi-empirical MO calculation level and DFT levels [
26,
27,
28]. It has been reported previously that the relative rates of hydroboration of alkenes are well correlated with their semi-empirical MNDO-calculated HOMO energy level. The higher the HOMO energy of alkenes, the faster the reaction proceeds. This can be accounted for by the enhanced interaction between the HOMO of alkenes and the LUMO of boranes when they are brought together. Formation of the B-C bond occurs predominantly at the position where the atom orbital coefficient in the alkene HOMO is largest [
26,
27]. To explain the present co-catalytic effect of amino acids and the graft copolymerization with polypeptides, the HOMO-LUMO interaction provides a deeper understanding of tri-
n-butylborane-initiated polymerization and copolymerization chemistry. The HOMO energy of amino acids is derived from published data [
29] (
Table 1). Briefly, the initial configurations of amino acids are fully optimized with the semi-empirical AM1 method using MOPAC (FUJITSU MOPAC 2002). The Δε HOMO is determined using the following equation:
As shown in
Table 1, the basic amino acids, Arg, Lys and His, having a large minus Δε HOMO energy, show a large co-catalytic effect, due to their positively charged side chains. The HOMO energy is comparable with the ionization potential (IP) provided by Koopmans
’ theorem, |εHOMO| [
30]. The relationship between the co-catalytic effect (the ratio of the number of polymers formed relative to that of the control) and IP for amino acids has been examined. An acceptable correlation between the co-catalytic effect and IP was observed, with the exception of Cys and His. The equation is as follows:
The higher the IP of amino acids, the higher the co-catalytic effect.
Tri-n-butylborane-initiated polymerization of MMA in the presence of amino acids and water in air is controlled by the IP value of the amino acid. Cys with a thiol side chain and His with an imidazole side chain are outliers. It is well known that trialkylboranes react with thiols even at room temperature. Also, tri-n-alkylborane may form a strong complex with His due to its high IP value.
The HOMO in MMA is located on the β-carbon of the double bond, and can be determined by semi-empirical MO calculation PM3 [
31]. Using this method, the HOMO of some polypeptides such as poly-L-Ala and poly-Gly has been calculated. HOMO of poly-L-Ala, with a relatively high percentage graft value, is located at the position of its terminal NH
2. In contrast, HOMO of poly-Gly and that of poly-L-Leu, having a percentage graft value of zero, is located at the position of intramolecular NH (data not shown). Polypeptides with HOMO located in the intramolecular NH group may undergo graft copolymerization only with difficulty because of the steric hindrance of the polypeptide helix, particularly, for long bulky side-chain polypeptides. In this connection, the terminal NH
2 of the end groups in polypeptides may be the most favorable grafting site when HOMO is located in this position. The regioselectivity of HOMO on polypeptides should be examined further.
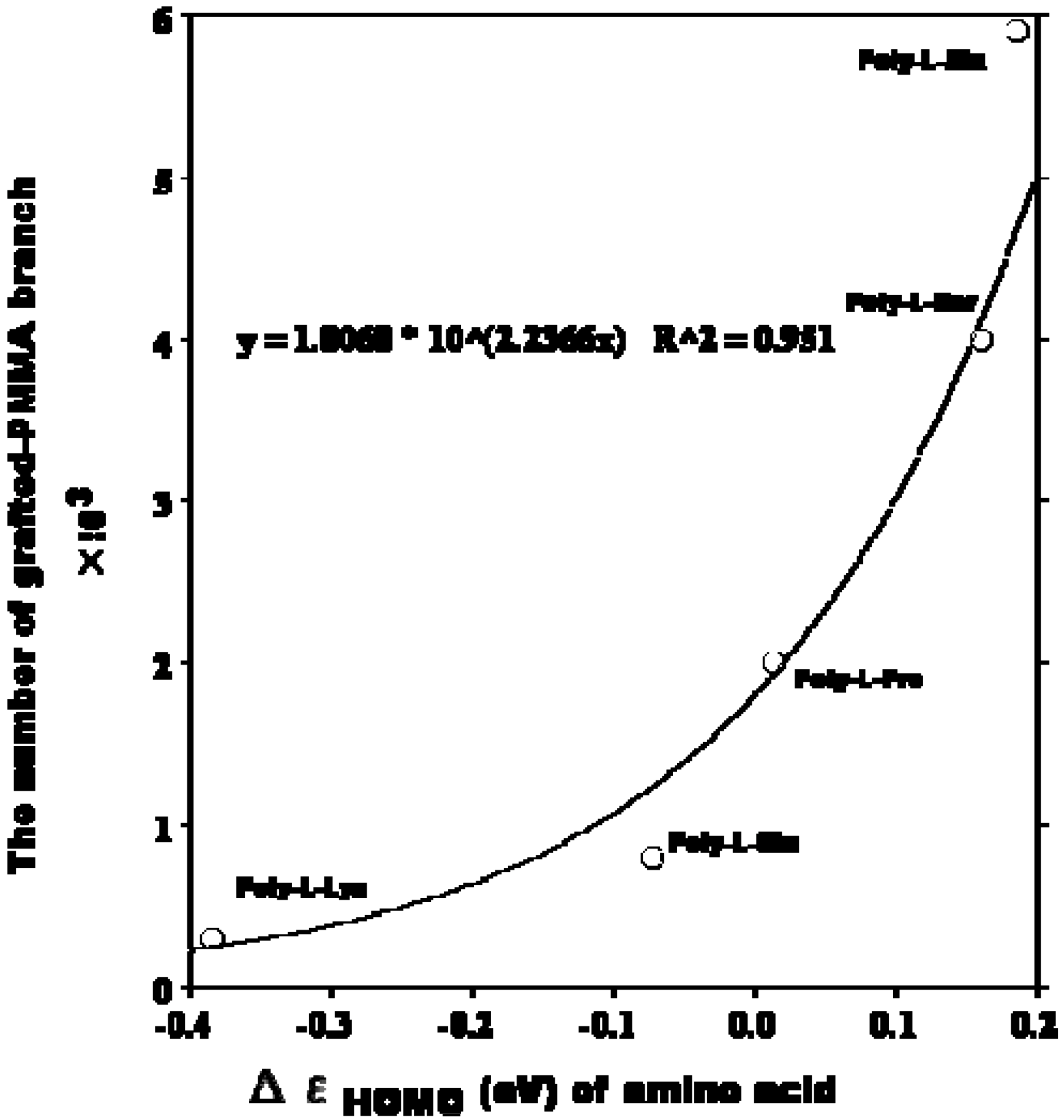
Consideration of the interaction of HOMO of alkenes and LUMO of boranes [
26,
27] could be applied for study of polypeptides. The relationship between the number of grafted-PMMA branches of polypeptides and the Δε HOMO of corresponding amino acids has been examined. A significantly exponential relationship between the two parameters was observed (
Figure 1).
Figure 1.
Relationship between the number of grafted-PMMA branches of poly-L-peptides and the Δε HOMO of the corresponding amino acids.
Figure 1.
Relationship between the number of grafted-PMMA branches of poly-L-peptides and the Δε HOMO of the corresponding amino acids.
The higher the Δε HOMO becomes, the higher the number of grafted-PMMA branches. However, further studies of this graft mechanism at a higher level of calculation will be necessary.
A small amount of water may play an important role in tri-
n-butylborane-initiated copolymerization of MMA with proteins. The theoretical
ab initio calculation has proposed that a O-H homolysis reaction in the trimethylborone/water complex is endothermic, and its O-H homolysis reaction is much lower than that for pure water: the O-H bond dissociation energy of water (73 kcal/mol) in the borane/water complex is much lower than that of water [
28]. It has been proposed that the trimethylborane/water complex mediates methyl radical generation.
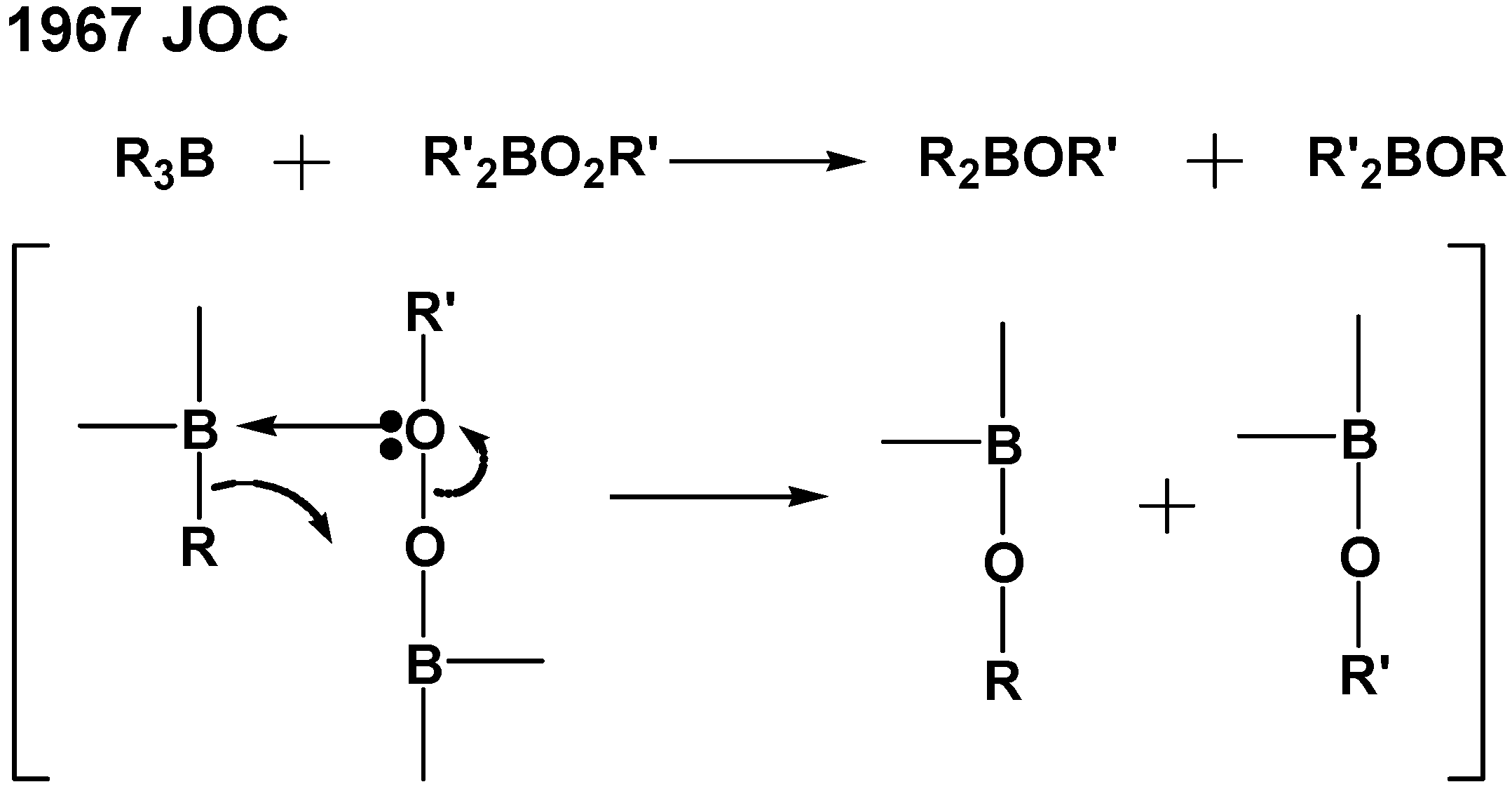
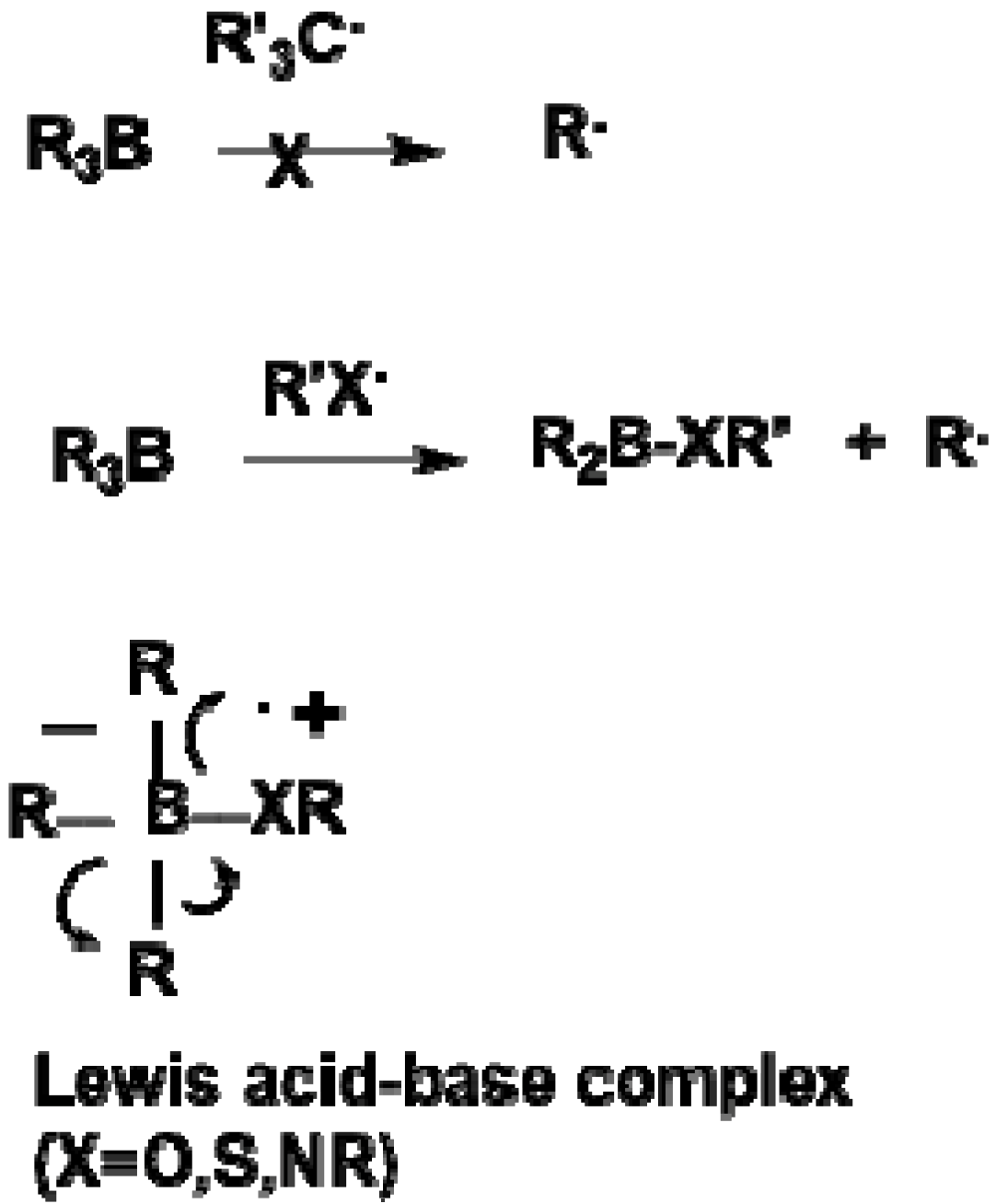
The co-catalytic effect of amino acids on the tri-
n-butylborane-initiated polymerization of MMA in the presence of water is controlled by IP, suggesting that this system proceeds
via a Lewis acid (tri-
n-butylborane)/base (amine) complex, as shown in
Scheme 3. The tri-
n-butylborane/water complex-mediated polymerization system probably belongs to a cation radical polymerization of methyl methacrylate.
Scheme 3.
Reactivity of carbon- and heteroatom-centered radicals with organoboranes [
17].
Scheme 3.
Reactivity of carbon- and heteroatom-centered radicals with organoboranes [
17].
3.2. Copolymerization with Proteins
3.2.1. Silk Fibroin
The results of copolymerization with five types of proteins, rat-tail collagen, wool, gelatin, ovalbumin and silk fibroin, are shown in
Table 3.
The percent graft and graft coefficiency for untreated proteins declines in the order: gelatin > silk fibroin > collagen > ovalbumin> wool. The statistically significant relationship between the percent graft and graft coefficiency is observed (
r2 = 0.91). However, no significant relationship between the number branches (mole/mole-protein) and the percent graft or graft coefficiency is observed. The percent graft and graft coefficiency for silk fibroin are greater than those for collagen. The number of PMMA branches is 0.1, 0.025, 0.04, 0.02 and 1.3, respectively. Silk fibroin has a one-order greater number of grafting sites than rat-tail collagen. To clarify the good ability of silk fibroin to graft polymerization, it would be important to comprehensively delineate relationships between the chemical structure of proteins (backbone polymer) and grafting sites (No. of branches) or the molecular weight of grafted-PMMA branches. Recently, Cao and Wang [
32] have reported a review of silk-based biomaterials. The structure of silk can be concisely described as follows: Silk fibroin is a giant molecule, of which about two thirds is a crystalline portion and about one third is an amorphous region. The crystalline portion contains repetitive amino acids (-Gly-Ala-Gly-Ala-Gly-Ser-) along its sequence, forming an antiparallel â-sheet, which is formed through hydrogen bonding with the adjacent peptide chain, thus accounting for the stability and mechanical properties of the fiber. The silk fibroins are characterized as natural block copolymers comprising hydrophobic blocks with short side-chain amino acids such as Gly and Ala, and hydrophilic blocks with larger side-chain amino acids, as well as charged amino acids. The two main distinct structures in silk fibroin are silk I and silk II. The structure of silk I contains random-coil and amorphous regions; it is a water-soluble structure. The silk II structural form of the silk fibroins has been characterized as an antiparallel â-sheet structure that is insoluble in several solvents including mild acid and alkali, and several chaotropes. In regenerated silk fibroins, the silk I structure can be easily converted to a â-sheet structure by chemical methods such as treatment with methanol. About 75–80% of silk comprises a Gly-Ala sequence, and is about 5% Ser.
Table 3.
Tri-n-butylborane-initiated graft copolymerization of methyl methacrylates with hydrated rat-tail collagen, wool, gelatin and silk fibroin.
Table 3.
Tri-n-butylborane-initiated graft copolymerization of methyl methacrylates with hydrated rat-tail collagen, wool, gelatin and silk fibroin.
| Protein | Percent graft | Mol. wt. of grafted-PMMA branches | *No. branches | *No. branches | Graft efficiency | Total conversion of monomer |
|---|
| | (%) | ×10-5 | (mole/g-protein) ×106 | (mole/mole-protein)d) | (%) | (%) |
|---|
| Collagen | 75.7 ± 6.7 | 18.2 ± 1.9 | 0.4 ± 0.1 | 0.1 | 2.6 ± 0.2 | 16.8 ± 2.7 |
| Collagena | 139.1 ± 17.7 | 20.1 ± 2.1 | 0.7 ± 0.2 | 0.2 | 2.5 ± 0.1 | _ |
| Wool | 11.5 | 3.1 | 0.5 | 0.025 | 0.8 | 9.3 |
| Gelatin | 130.8 ± 35.9 | 19.9 ± 3.4 | 0.7 ± 0.3 | 0.04 | 3.8 ± 0.6 | 19.6 ± 1.8 |
| Ovalbumin | 44.6 ± 7.6 | 12.4 ± 1.6 | 0.3 ± 0.0 | 0.02 | 1.8 ± 0.4 | 15.9 ± 2.1 |
| Silk fibroin | 85.5 ± 21.4 | 2.3 ± 0.7 | 3.6 ± 0.2 | 1.3 | 3.6 ± 0.9 | 11.8 ± 0.1 |
| Silk fibroina) | 25.7 ± 5.9 | 1.4 ± 0.1 | 1.8 ± 0.5 | 0.7 | 1.7 ± 0.4 | 10 ± 1 |
| Silk fibroinb) | 26 ± 3.6 | 1.4 ± 0.3 | 1.8 ± 0.3 | 0.7 | _ | _ |
| Silk fibroinc) | 249.4 | 6.4 | 3.9 | 1.4 | _ | _ |
The average molecular weight of grafted-PMMA branches of silk fibroin is approximately 2 × 105, being one order less than that of collagen, gelatin and hydrophilic poly-L-peptides (Lys, Pro, Glut). In this system, the average molecular weight of the homopolymer is approximately 1.1 × 105–1.2 × 105, indicating that the molecular weight of the grafted PMMA is similar to that of a homopolymer. This finding suggests that grafting of MMA onto silk fibroin probably occurs at the surface of the protein. 2-Tolylisocyanate and formic acid have chemically modified the functional groups of the amino acids in silk fibroin. The percentage grafting and the number of PMMA branches of the treated-silk fibroin are reduced to approximately one half of the corresponding value for the original silk fibroin, indicating that hydrophilic blocks with larger side-chain amino acids, particularly charged amino acids, may be involved in the grafting sites of silk fibroin. Interestingly, even when modifiers have masked silk fibroin, the graft copolymerization results in a lower percentage of grafting. It is assumed from this that hydrophobic blocks with short side-chain amino acids, such as Gly and Ala, may be involved in the graft copolymerization. Also, silk fibroin treated with formic acid vapor for 72 h shows a large percentage grafting of 249%, being approximately 10-fold greater than that of isocyanate- or acetic acetic anhydride-treated, or original, silk fibroin. Formic acid vapor induces swelling of the backbone polymers. However, the number of its PMMA branches is similar to that of the control.
Tsukada
et al. [
11] have examined the infrared spectrum of poly(MMA)-grafted silk fibers using tri-
n-butylborane, and demonstrated overlapping absorption bands of silk fiber with a β-sheet structure and those of the grafted MMA polymer. Also, a grafted silk fiber with a graft yield of more than 140% was shown to be composed of two endothermic peaks at 321 °C and 396 °C on the DSC curve, attributable to the thermal decomposition of silk fiber and grafted poly-MMA chain, respectively. Also, Tsukada
et al. [
33] have examined MMA-grafted silk fibers obtained by using potassium persulfate (KPS) and tri-
n-butylborane, which were evaluated in terms of the gel permeation chromatography (GPC) elution pattern of the poly-MMA chains. A bimodal molecular weight distribution, with a heavy and a light component on the grafted silk fiber, was observed. The average molecular weight of the heavy component ranged from 48.5 to 200 kDa for poly-MMA copolymerized by the KPS reaction system, and from 336 to 816 kDa for the poly-MMA copolymerized by the tri-
n-butylborane reaction system. The light component had an average molecular weight lower than 1,000 Da, being similar in all the samples examined. Scanning electron microscopy (SEM) demonstrated the presence of MMA oligomers formed on the fiber surface during grafting. Taken together with the finding of our previous study, it seems that the masked portion of silk fibroin treated with modifiers is probably compatible with hydrophilic blocks that include larger side-chain amino acids/charged amino acids. The grafted-PMMA branches on the hydrophilic block may be responsible for the high molecular weight. Interestingly, the average number of branches does not exceed two. Poly
-L-Ala and poly-Gly have been used as a model polypeptide of silk fibroin because silk contains predominantly Ala and Gly residues. The graft copolymerization of MMA with polypeptides occurs for poly-L-Ala but not for poly-Gly. Also, the characterization of graft copolymerization with poly-L-Ala is likely similar to that of silk. However, the most favorable grafting sites on silk fibroin remain unclear because the number of grafted-PMMA branches is very small.
3.2.2. Collagens
Collagen One (Type I) is contained in skin, tendon, vasculature, ligaments, organs, bone (the main component) and teeth (dentin, cementum). The collagen content of dentin is about 20%, and that of other components (mainly minerals) is about 80%, comprising hydroxyapatite (Ca
10(PO
4)
6(OH)
2) with some phosphate. The sequence of collagen often follows the pattern Gry-Pro-X or Gly-X-Hyp, where X may be any of various other amino acid residues. Pro or hydroxyPro constitutes about 1/6 of the total sequence, Gly accounting for 1/3 and Ala for about 1/10 [
34]. Also, gelatin is an irreversibly hydrolyzed form of collagen, with a similar amino acid composition, being mainly Gly21%, Pro 12%, hydroxyPro12%, Glu 10% and Ala 9% [
34].
As shown in
Table 4, the number of grafted-PMMA branches of gelatin is approximately double that of rat-tail collagen, possibly due to the higher cross-linking in collagen than that in gelatin. Comparing collagen treated with acetic anhydride/pyridine modifiers with the original form, this treatment enhances the number of grafted-PMMA branches in collagen. Collagen comprises a polar region and an amorphous region, similar to silk fibroin. A highly basic block is present in the triple helix near the the carboxyl terminus [
35]. Even if this block is masked by treatment with modifiers, the percentage grafting is not reduced, but conversely enhanced, possibly due to the modifier’s swelling effect on collagens. The average molecular weight of the grafted-PMMA branches of collagen and gelatin is similar (approximately 20 × 10
5). That of ovalbumin is 12 × 10
5, being half that of collagen. Ovalbumin is a monomeric phosphoprotein with 385 amino acid residues. The N-terminal protein is acetylated and contains four cysteine and one cysteine residue [
36]. Hydrophobic interactions and disulfide bond formation between ovalbumin molecules is involved in aggregation and surface gelatinization [
37]. This structure of ovalbumin may be responsible for the relatively large molecular weight of the grafted-PMMA branches. By contrast, wool, a fiber protein, shows characteristics of graft copolymerization similar to those of silk fiber protein. Each of the amino acid residues is distributed almost equally [
38].
On the other hand, the molecular weight of the homopolymer for all tested proteins (collagen, gelatin, silk fibroin, ovalbumin and wool) is similar, being 1.1 × 10
−5 –1.2 × 10
5. Also, that of the homopolymer derived from modifier-treated silk fibroin and collagen is 1.1×10
5. A great difference in average molecular weight between the grafted-PMMA and the homopolymer is observed for collagen, gelatin and hydrophilic polypeptides. The considerably high molecular weight of the grafted-PMMA branches of these backbone polymers may be associated with the so-called “gel effect”,
i.e., a markedly increased molecular weight and entanglements in the bulk [
39]. Indeed, it has been found that grafted PMMA is in part converted to an insoluble form even after intensive acetone extraction, possibly due to the entanglements of resin polymer in the bulky hydrophilic backbone polymer. These results may be attributed to the heterogeneity of the system.
Graft copolymerization of MMA onto chrome-tanned pig skin has been reported to occur upon irradiation with
60Co-rays, and proof of grafting has been obtained through the detection of amino acid end groups in the isolated grafts by reaction with ninhydrin [
40]. It is assumed that the N-terminal groups of amino acid ends are involved in radical reactions in biological systems. Gelatin has NH
2-Lys as a predominant end group. Ser, Thr, Ala, Asp and Glu residues also occur [
34]. Furthermore, the end group of collagen has been estimated from enzymatically hydrolyzed collagen products (polypeptides), Gly being the main terminal amino acid (NH
2-Gly-Pro----), with much smaller amounts of other amino acids (Ala, Glu, Aspa) [
34]. The N-terminal groups of silk fibroin comprise 92% Gly [
41]. The terminal end groups of the amino acid residues of many proteins commonly comprise Gly. Our results suggest that any NH
2-Gly in the end group of a protein may not be involved in grafting because of the zero percentage grafting of polyglycine. If Gly residues are mainly related to graft copolymerization, a large number of grafted-PMMA branches would be observed for the indicated proteins. N-terminal groups of amino acid residues,
i.e., Ala, Ser and Asp with a high Δε HOMO may become grafting site candidates. However, further study of this issue may be necessary.
3.2.3. Effect of Initiator Concentration on Graft Copolymerization
The percentage grafting, the molecular weight of grafted-PMMA branches, the number of grafted-PMMA branches, grafting efficiency (%) and the total conversion of monomer (%) with varying initiator concentration are shown in
Table 4.
Table 4.
Effect of the concentration of tri-n-butylborane on the graft copolymerization of MMA onto hydrated bovine dentin collagen.
Table 4.
Effect of the concentration of tri-n-butylborane on the graft copolymerization of MMA onto hydrated bovine dentin collagen.
| Tri-n-butylborane | Percent graft | Mol. wt. of grafted-PMMA branch | No. of grafted-PMMA branch×106 | Graft efficiency | Total conversion of monomer |
|---|
| ×102 mol/liter | (%) | ×10−5 | mol/g-protein | (%) | (%) |
|---|
| 3 | 9.7 | 8.5 | 0.1 | 3.0 | 4.3 |
| 4 | 40.0 | 53.2 | 0.2 | 5.4 | 10.0 |
| 11 | 49.5 | 38.9 | 0.1 | 3.5 | 18.6 |
| 19 | 81.8 | 49.8 | 0.2 | 5.0 | 22.5 |
| 25 | 71.9 | 40.0 | 0.2 | 3.8 | 25.0 |
| 37 | 5.8 | 8.5 | 0.1 | 0.2 | 32.0 |
| 47 | 0.8 | _ | _ | 0.03 | 34.7 |
To optimize the reaction conditions for obtaining the maximum percentage grafting, the concentrations of initiator were varied. A significant parabolic relationship between the percentage grafting or the molecular weight of grafted PMMA branches and the square root of the initiator concentration (C
1/2) yielded equations 9 and 10:
There is an acceptable relationship for the percentage grafting versus the molecular weight of grafted PMMA (r2 = 9.45, p < 0.01).
Above 37 mol, the percentage grafting and the grafting efficiency decrease markedly.
By contrast, the total conversion of monomer (%) is linearly related to the square root of the initiator concentration, and expressed as the following equation:
Termination occurs in free radical polymerization by one of two mechanisms: coupling or disproportionation. Either mechanism involves a reaction between two growing chain ends. Comparing Equation 9 with Equation 11, the termination mechanisms of homopolymers in MMA solution and the growing PMMA radical in the backbone polymers apparently differ. The growing PMMA radical derived from cation radical polymerization mediated by the tri-
n-butylborane/water complex (
i.e., tri-
n-butylborane as a Lewis acid) may be terminated by the terminal N-amino acid residues end group of backbone polymers such as collagen and gelatin. This is also assumed from the considerably small value of E
a for tri-
n-butylborane (4 kcal/mol) [
15]. It is well known that polymerization initiators with a small E
a preferably undergo ionic polymerization in water. Since the number of grafted-PMMA branches of collagen is considerably small (0.1), graft copolymerization of MMA onto collagen by tri-
n-butylborane might be a pseudo-graft copolymerization.
The effect of time (0–120 min) on graft copolymerization with bovine dentin collagen has been examined [bovine dentin collagen, 0.050 g; MMA, 10 mL; initiator, 0.15 mL; water, 0.2 mL; 37 °C] (data not shown). The percentage grafting increases with reaction time, the relationship being significantly linear (r2 = 0.99). The percent grafting continues to increase even after 120 min. The tri-n-butylborane/water complex-initiated copolymerization starts without an induction period, and polymerization continues for a relatively long time.
3.2.4. Copolymerization with Various Collagens
Table 5 shows the graft copolymerization of MMA with various hydrated collagens.
The number of grafted-PMMA branches is comparably small, whereas the molecular weight of grafted-PMMA branches is very high. Premature collagen (calf dermis, deciduous dentin) shows a greater number of branches than the corresponding mature form, particularly that of calf. This may be due to cross-linking in mature collagen. The number of branches of calf dermis collagen is similar to that of gelatin, and the number of grafted-MMA branches of dentin powder shows a value similar that of soft tissue type-one collagen.
There have been some reports of graft copolymerization of methacrylates with hydrated collagen by use of ceric ammonium nitrate [
42] and potassium persulfate [
43] as an initiator. There is an increase in the percentage grafting of collagen with such initiators. The activation energy (E
a) of graft copolymerization for acryl amide and methyl methacrylate onto cellulose by ceric ammonium nitrate is low (
circa 3.4 kcal/mol), being within a temperature range of 20 °C to 45 °C [
42]. By contrast, the overall E
a of aqueous polymerization of acryl amide by potassium persulfate/2-mercaptoethanol is
circa 32.1 kcal/mol) [
43]. The E
a of ceric ammonium nitrate is similar to that of tri-
n-butylborane, whereas that of the potassium persulfate/2-mercaptoethanol redox system is 8-fold greater than that of tri-
n-butylborane. As described in 3.2.1, the molecular weight of the heavy component of MMA-grafted silk fiber with the tri-n-butylborane reaction system is about 4-fold greater than that with the potassium persulfate reaction system [
33]. This indicates that the tri-
n-butylborane system may be applicable for enhancing the molecular weight of MMA-grafted branches. The characterization of copolymerization by the tri-
n-butylborane reaction system probably promotes the adhesive strength of this resin system to tooth collagen due to the high percentage grafting with a large molecular weight, even if the MMA-grafted chains are considerably small.
Table 5.
Tri-n-butylborane-initiated graft copolymerization of methyl methacrylates with hydrated various collagens (A) and bovine dentin powder (B).
Table 5.
Tri-n-butylborane-initiated graft copolymerization of methyl methacrylates with hydrated various collagens (A) and bovine dentin powder (B).
| A | | | |
| | Percent graft | Mol. wt. of grafted-PMMA branch | No. of grated-PMMA branch (mole/g-collagen) |
| Type one collagen | (%) | ×10−5 | ×106 |
| Calf dermis | 48.8 | 7.6 | 0.6 |
| Matur bovine dermis | 4 | 6.5 | 0.1 |
| Bovine achilles tendon | 7 | 9.6 | 0.1 |
| Bovine peridontal ligament | 12.6 | 7.5 | 0.2 |
| Bovine femoral bone | 8.9 | 10.5 | 0.1 |
| Bovine Dermisa) | 15.2 | 10.9 | 0.1 |
| Dentin (human permanent teeth) | 4.3 | 6 | 0.1 |
| Dentin (human permanent teeth) | 13.5 | 9.3 | 0.2 |
| Dentin (human decidous teeth) | 10 | 6.8 | 0.2 |
| B | | | |
| | Percent graft | Mol. wt. of grafted-PMMA branch | No. of grafted-PMMA branch (mole/g-collagen) |
| Bovine dentin powder | (%) | ×10−5 | ×106 |
| Dentin (mesh-size 200-325)b) | 0 | _ | _ |
| Dentin(mesh-size 200-325) | 27.6 | 19.5 | 0.2 |
| Dentin (mesh-size 180-200)c) | 11.7 | 7 | 0.3 |
| Dentin (mesh-size 180-200) | 8.6 | 7.3 | 0.1 |
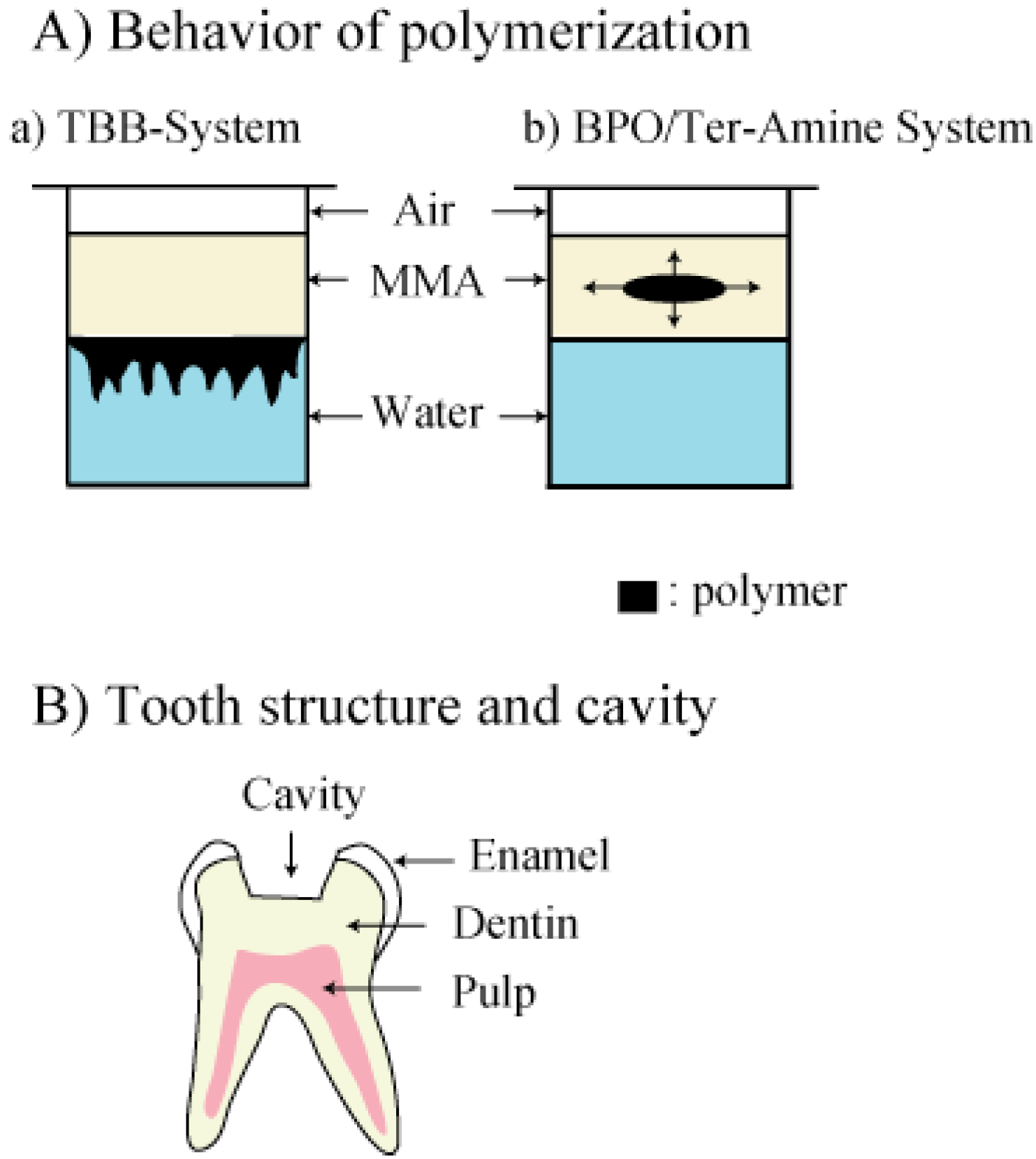
To elucidate the good adaptation of tri-
n-butylborane-initiated PMMA resins to the cavity prepared after caries treatments, Masuhara [
3] previously reported the beneficial effect of the polymerization of MMA initiated by tri-
n-butylborane on tooth structures. Results are shown in
Figure 2.
These simple experimental results suggested that polymerization of MMA initiated by the TBB system may occur preferably at moist dentin surface with collagens because native collagen fibers contain water intrinsically and consequently, a good cavity adaptation between dentin and the resin may be obtained [
3] (
Figure 2B). Indeed, the good adhesive ability of TBB-system resins provided sufficient protection of pulp [
4,
5]. Note: Dentin collagen has a proportion of
circa 20% of dentin, whereas enamel contains no collagen and other proteins are <<1%. Currently, in adhesive dentistry, tooth surfaces are treated by acids for a few seconds to enhance the adhesive strength of resin to tooth. Tooth enamel surfaces are decalcified by this etching treatment and most of etched hard-tissues are a well-known brushite (dicalcium phosphate dihydrate, CaHPO
4 2H
2O) which is derived from hydroxyapatite of enamel structure. The formed brushite layer (porous layer with about 50 μm width) works as an anchor site for adhesive materials, in addition to the marked increase of enamel surface area due to etching treatments. Since brushite has a better affinity to water than that of hydroxyapatite, TBB-initiated resins for enamel bonding systems with a coupling agent were more effective, compared with BPO/
tert-amine’ ones [
3].
Figure 2.
The distinct difference of polymerization behavior of methyl methacrylate (MMA) at the surface of water between (a) tri
-n-butylborane (TBB) and (b) benzoyl peroxide (BPO)/
tert-amine systems (retouched figure taken from the Ref. [
3] with permission.) (A) and tooth structure and cavity (B). A given amount of MMA and water were added to sample tubes. The mixture solution was partitioned into the two phases of water and MMA; the upper phase, MMA and the lower phase, water. Next, to initiate polymerization, TBB and BPO/
tert-amine initiators were added to sample tubes, respectively. a) TBB-initiated polymerization began at the surface of water. Resin forming occurred in water. b) BPO/
tert-amine-initiated polymerization occurred in the central part of MMA solution and accelerated circumferentially. Also, no polymerization was influenced by water. Polymerization shrinkage occurred toward the center of the resin body in a tube.
Figure 2.
The distinct difference of polymerization behavior of methyl methacrylate (MMA) at the surface of water between (a) tri
-n-butylborane (TBB) and (b) benzoyl peroxide (BPO)/
tert-amine systems (retouched figure taken from the Ref. [
3] with permission.) (A) and tooth structure and cavity (B). A given amount of MMA and water were added to sample tubes. The mixture solution was partitioned into the two phases of water and MMA; the upper phase, MMA and the lower phase, water. Next, to initiate polymerization, TBB and BPO/
tert-amine initiators were added to sample tubes, respectively. a) TBB-initiated polymerization began at the surface of water. Resin forming occurred in water. b) BPO/
tert-amine-initiated polymerization occurred in the central part of MMA solution and accelerated circumferentially. Also, no polymerization was influenced by water. Polymerization shrinkage occurred toward the center of the resin body in a tube.
Ikemura
et al. [
44] recently wrote a review titled “Our development of dental adhesives”. They reported as follows: Based on the general principle of adhesion that is “good wettability and good hardening”, our research strategy has focused on the dual effects of radical polymerization initiators (to make good hardening) and adhesive monomers (to form good wettability) on adhesion.
Currently, selfcuring PMMA resins initiated by partly oxidized tri-
n-butylborane have undoubtedly achieved success in delivering outstanding bonding performance [
44]. Note: Partly oxidized tri-
n-butylborane is used commercially, since tri-
n-butylborane is unstable in air. Many researchers seek more stable polymerization initiators with active initiation like tri-
n-butylborane.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



