1. Introduction
Conductive polymers are principally insoluble and infusible, which has been shown by numerous experimental approaches and by thermodynamical considerations [
1]. A “solution” of a conductive polymer would require a single chain having no interaction with a neighbor chain but only interactions with the solvent,
i.e., the chain would be completely surrounded by solvent molecules. Such systems do not and cannot exist.
Therefore, they can only be processed by dispersion. “Dispersion” is a process by which (in the case of solids becoming dispersed in a liquid) agglomerated particles are separated from each other and a new interface, between an inner surface of the liquid dispersion medium and the surface of the particles to be dispersed, is generated. Dispersion is a much more complicated (and less well understood) process than most people believe.
In order to disperse conductive polymers, it must be ascertained what the smallest primary morphological units are that can exhibit all the properties of the macroscopic material. It must also be understood whether these are fibrils, or more or less globular particles? We must know whether they are of micrometer size i.e., 100 nanometers or below?
In a review article [
2], all relevant publications (including proprietary work) have been discussed, for example, the discovery of globular primary particles (around 10 nm in size) which are the primary morphological building unit of all conductive polymers, whether in powder, film or fibrilar form; non-linear phenomena in dispersions (such as rheological properties and sudden conductivity increase at a critical volume concentration); the formation of (so-called) dissipative structures in dispersions; the fact that percolation theory cannot be applied to (polymer matrix based) dispersions of particles having less than 1 µm in size; and, finally, the concise non-equilibrium thermodynamic theory for heterogeneous polymer systems, published in two parts [
3,
4].
Surprisingly, with improved dispersion, not only the conductivity (in contrast to any naive assumption) increased, but also the material crossed the insulator-to-metal boundary to the metallic side [
5]. While the transition from an insulator-type of electron transport to a metal-type—
happening during dispersion—was surprising, observations that conductive polymers could exhibit metallic properties had already been published before. These various observations, partially also confirmed in our laboratories, had been generalized by the “granular metal concept” [
6]. This basically describes the metallic properties in polyaniline, which are understood to be fibrils with more or less straight chains, as emerging in certain limited areas (“islands”), where the chains exhibit a higher degree of order and these “islands” are linked together by less ordered chains, allowing the electrons to move from one “island” to the next.
If this concept is correct, then any dispersion not only would not improve the conductivity, but would in fact deteriorate it (by cutting the chains connecting the “islands”). However, our experiments showed no such deterioration; and on the contrary even finally succeeded in changing from the insulator to the metal side of the IM transition during dispersion. Therefore, the “Organic Nanometal” concept was developed [
1] (p. 513), inspired by the experimental and theoretical background on “mesoscopic metals” (
i.e., conventional metals like Cu, Ag, In,
etc. in nanoscopic form) brought forward by G. Nimtz and coworkers [
7,
8]; which we jointly have been able to show as being very similar to the behavior of polyaniline [
9,
10]. We found the metallic core with a size of 8 nm (assuming globular form), within a total porimary particle size of 9.6 nm, is well in accordance with former conclusions from scanning tunneling microscopy (STM) evaluations in which we deducted a particle size of around 10 nm.
The concept outlines in [
7] that the transport mechanism is purely metallic within the metallic core while the electrons move to the next particle via a barrier (at least 0.8 nm for one particle, in total at least 1.6 nm thick) of a less conductive or even insulating shell around the metallic core, by a mechanism called “tunneling” (sometimes also referred to as “hopping”). Electrons which are tunneling do not need a material type of pathway or file, nor a conductive polymer chain for tunneling through even insulating or empty spaces. As Nimtz has shown later, moreover, electrons do not spend any time within the barrier but arrive at the other side at the same time as they begin tunneling [
11].
At the time the “Organic Nanometal Concept” was conceived, we did not know anything about the conductivity values within the metallic core. We had only been able to measure the macroscopic conductivity which is around 5 S/cm for the raw polymer powder (in pressed pellet form) at the insulator side of the IM transition, and above 100 S/cm up to 600 S/cm for the metallic polyaniline [
12]. Later, in cooperation with two other research groups and using electron spin resonance spectroscopy (EPR), we succeeded to find the intrinsic conductivity within the metallic core to be in the order of 60,000 S/cm [
13]. This is at least 100 times more than we can macroscopically measure.
Yet we still did not understand the structural basis for the conductivity, what structural changes occurred during dispersion within the particle, within the metallic core, and which structural pattern is responsible for higher conductivity compared to the lower (and non-metallic) one. There are numerous papers in the scientific literature proposing structures for polyaniline derived from wide-angle x-ray diffraction studies, for example an overview given in [
14], including a proposal we have contributed [
1] (p. 522), but none have been helpful. The assumption (or “hope”) that there would be a correlation between higher conductivity and higher crystallinity (
i.e., better resolved wide angle x-ray diffraction pattern) did not eventuate. On the contrary, it appears that the peaks’ bandwidths (anyway looking more like spectra of amorphous materials) even get wider with increasing conductivity. The only hint towards a possible mechanism was the finding that the volume of the elementary cell (although the absolute values were, perhaps, not correct) decreases with increasing conductivity. However, at that time, we only investigated a conductivity range over one order of magnitude.
A second indication of what may be happening, and in which direction we should look, came from some first results of a small angle x-ray diffraction (SAXS) [
15]. For the first time, we detected a 3.5 nm small subunit (and confirmed the around 10–15 nm primary particle size), which we assume to represent the diameter of individual polyaniline chains within the primary particle; the chains not being straight but somehow coiled.
The indication of a volume change along with conductivity increase, and the discovery of a 3.5 nm subunit raised hope of discovering more insights. A better understanding of the structure/conductivity relationship has quite important significance for the goal of increasing the conductivity of conductive polymers and organic metals by one or two more orders of magnitude, towards the intrinsic conductivity.
For a better understanding, at least of polyaniline, also the existence of its complexes with metals needs to be discerned (such metal complexes are, for instance, not possible for poly-ethylenedioxythiophene, PEDOT). Such complexes have hitertho not been published, except as patents [
16,
17]. Here, we will focus on complexes with ions derived from less noble metals than polyaniline (the polyaniline normal potential is between that of Cu and Ag).
The complex with Ag has been discussed in [
18]. This is a complex of a different kind, insofar as it does not involve Ag ions, but Ag metal in nanoscopic form. It is used as a very powerful oxidation prevention and solderability preserving nanolayer for copper pads in printed circuit board manufacturing and assembly [
19].
In the following, the most recent results will be reported and discussed regarding organic metal/conventional metal complexes, their sub-nanoscopic structure and nanoscopic morphology [
20].
2. Results and Discussion
2.1. Polyaniline-Metal complexes
Polyaniline can form complexes with many metal ions by reacting with the base metal, provided its oxidation potential is lower than polyaniline’s potential [
18,
19]. Hence, complexes have been observed forming with Cu
1+, Fe
2+, Zn
2+, In
2+ and others.
When looking too quickly at the reaction of polyaniline with the metal form of Cu, Fe, Zn, or In, respectively, it could easily be concluded that a simple oxidation has taken place, and many experimentalists may have concluded this. However, the result is completely different when simply adding Cu1+, Fe2+, Zn2+ or In2+ ion salts to a water dispersion of polyaniline, compared to the result when allowing a water dispersion of polyaniline to react with Cu, Fe, Zn, or In metal (in granule or powder form), and hence the result of such reaction is much more interesting.
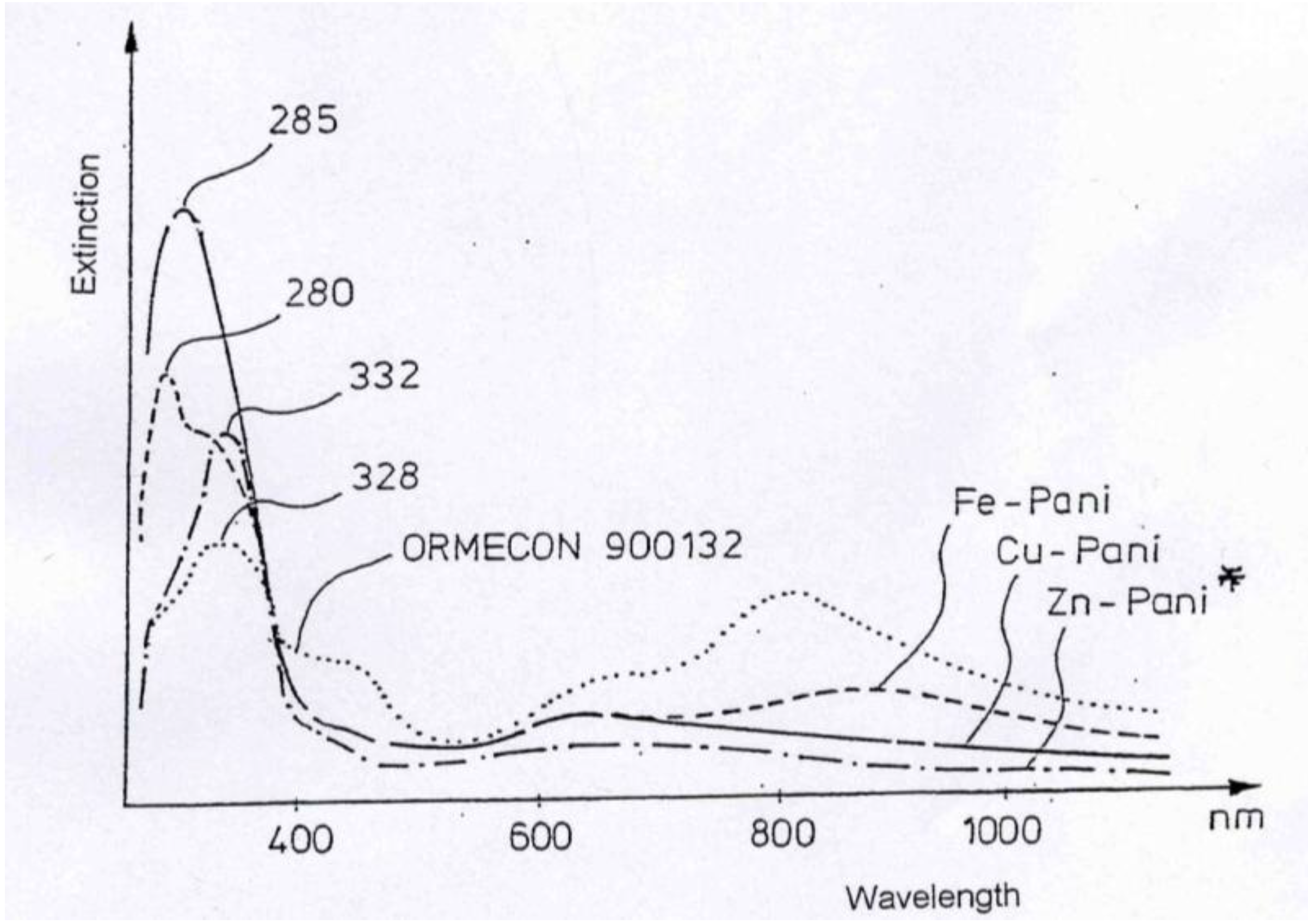
This can easily be seen when comparing the spectra: When mixing the respective metal ions (water solution) with the polyaniline water dispersion, the UV spectra will not change at all, except for some minor dilution effect. However, when following the reaction of polyaniline with the metal (which is a relatively slow reaction taking several hours to complete), the UV spectra gradually change and will ultimately be very different from the starting polyaniline spectrum, as can be seen in
Figure 1,
Figure 2,
Figure 3 and
Figure 4.
Figure 1.
UV-Vis spectra for polyaniline and its complexes with Cu, Fe and Zn.
Figure 1.
UV-Vis spectra for polyaniline and its complexes with Cu, Fe and Zn.
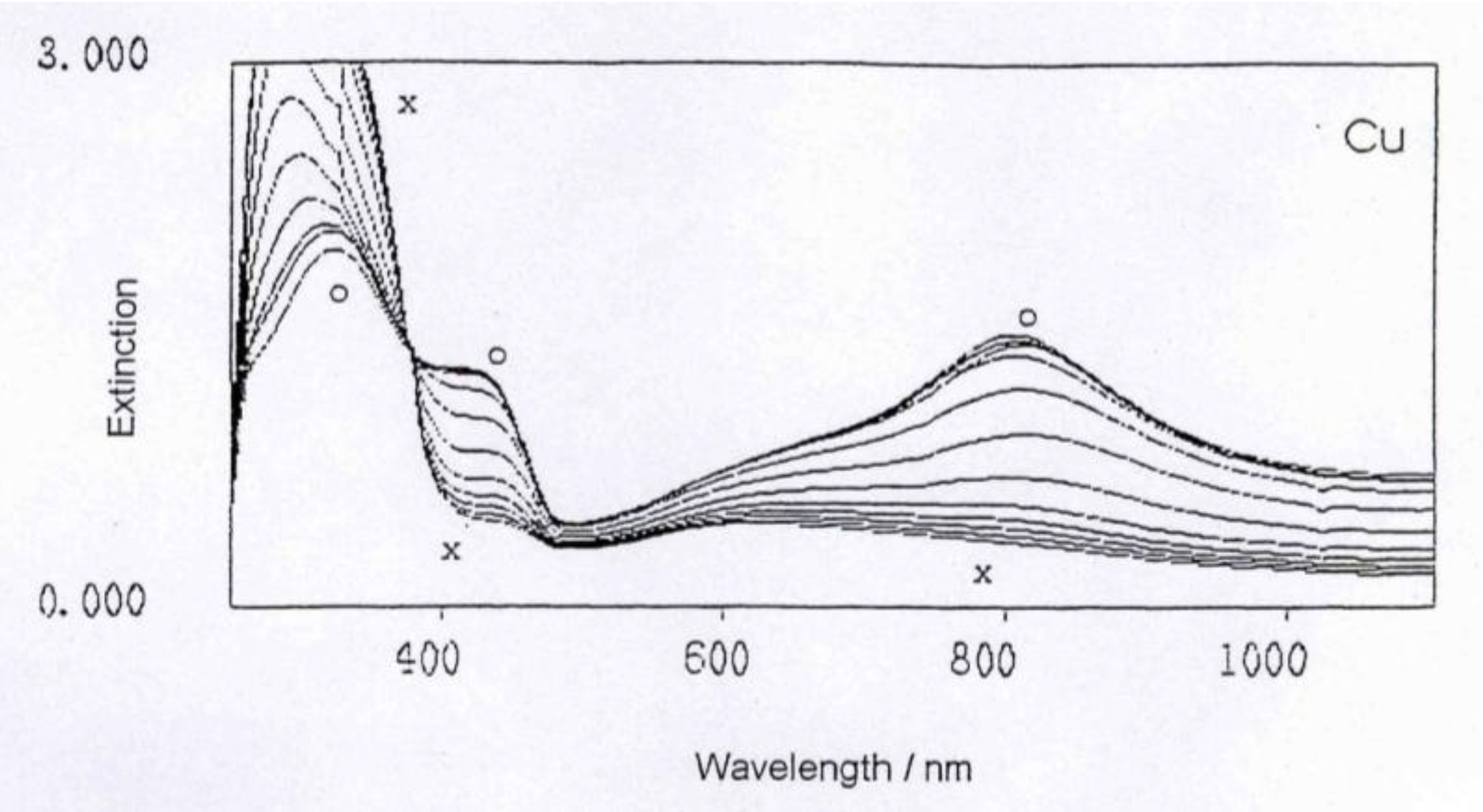
Figure 2.
UV-Vis spectrum evolving during the reaction of polyaniline with Cu metal.
Figure 2.
UV-Vis spectrum evolving during the reaction of polyaniline with Cu metal.
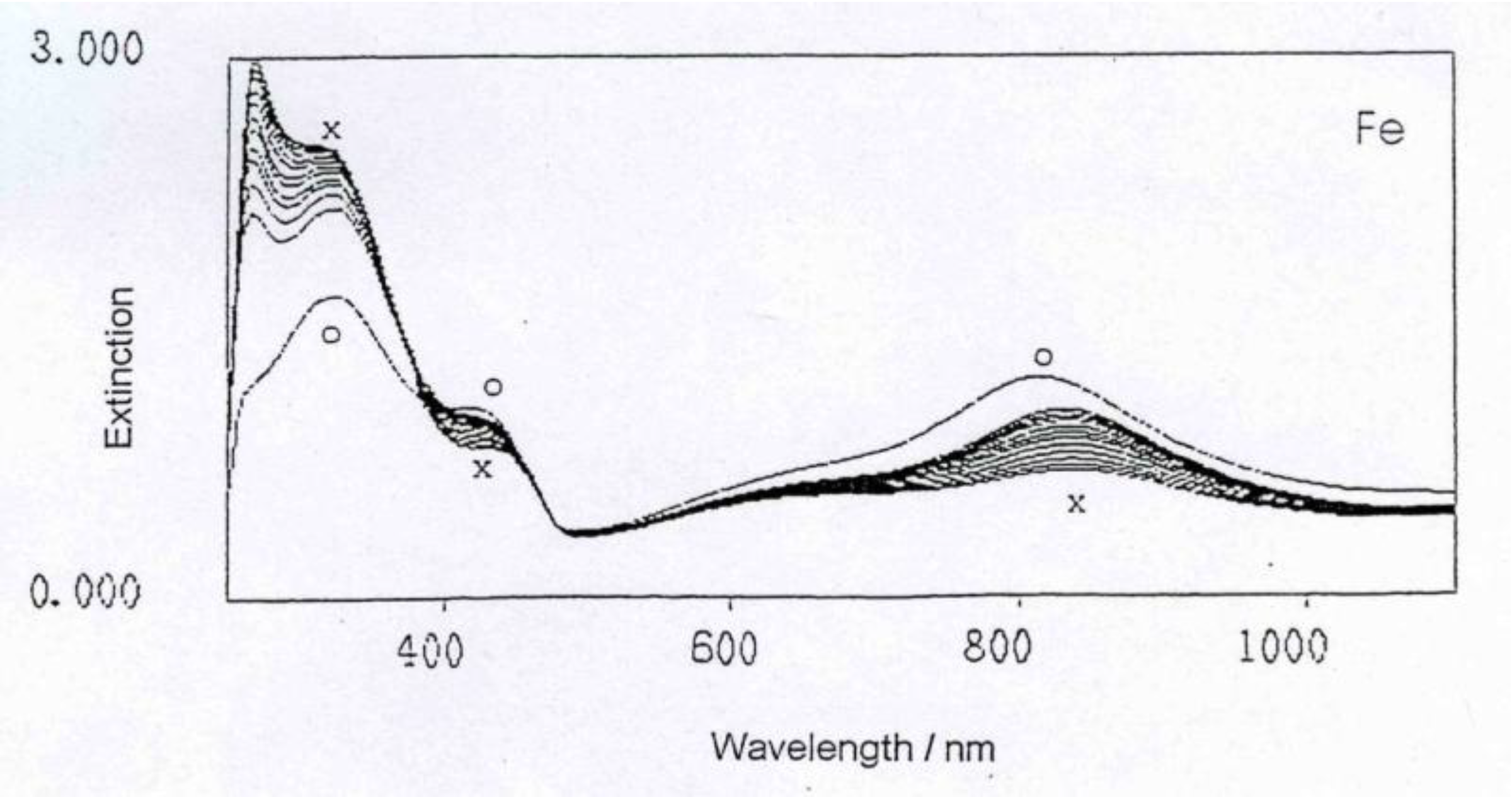
Figure 3.
UV-Vis spectrum evolving during the reaction of polyaniline with Fe metal.
Figure 3.
UV-Vis spectrum evolving during the reaction of polyaniline with Fe metal.
Figure 4.
UV-Vis spectrum of polyaniline (pure) and polyanilne/In complex; the curve with the lowest absorption around 800 nm representing the complex after reaction completed.
Figure 4.
UV-Vis spectrum of polyaniline (pure) and polyanilne/In complex; the curve with the lowest absorption around 800 nm representing the complex after reaction completed.
The general feature of all spectra is that the absorption strength around 800 nm decreases with proceeding complex formation, also the absorption strength around 450 nm decreases, with an isosbestic point appearing around 390 nm, and the absorption in the UV region increases. These effects are the most impressive in the formation of the polyaniline-Cu complex. This is not so surprising as the amount of Cu1+ in this complex is 1 per about 12 aniline units, while in the case of the other ions (Fe2+, Zn2+, In2+) the relation is roughly 1:24, as was found by elemental analysis.
These spectra clearly show that new compositions have been formed, true polyaniline/metal ion complexes.
The conclusion that we have drawn (in the case of Cu) with Cu
1+ comes from two different experiments: In printed circuit board finishing with the immersion tin process ORMECON CSN, where a water dispersion of polyaniline is used as a Cu surface preparation pretreatment called “predip” (trade name OMP 7000), the Cu complex plays a key role. Here, in contrast to other immersion tin processes not using such an organic metal predip, the relation between Cu release and Sn deposition is almost exactly 2:1 [
21] which corresponds to a net reaction equation such as:
For competitive tin deposition processes, a relation of roughly 1.5 to 1.0:1 has been observed which indicates a mixture of Cu2+ and Cu1+ or, in extreme cases only, Cu2+ to be released.
In addition, x-ray photoelectron spectroscopy (XPS) spectra of the Cu after treatment with the organic metal (in comparison to untreated Cu) clearly show the Cu
1+ oxidation stage [
1] (p. 535).
The conclusion that in the case of iron the Fe(II) oxidation state is involved, has been drawn based on earlier work in connection with studies of the reaction mechanism leading to the passivation of iron and steel and its subsequent enhanced corrosion protection which was found to proceed via the Fe
2+ ion [
22].
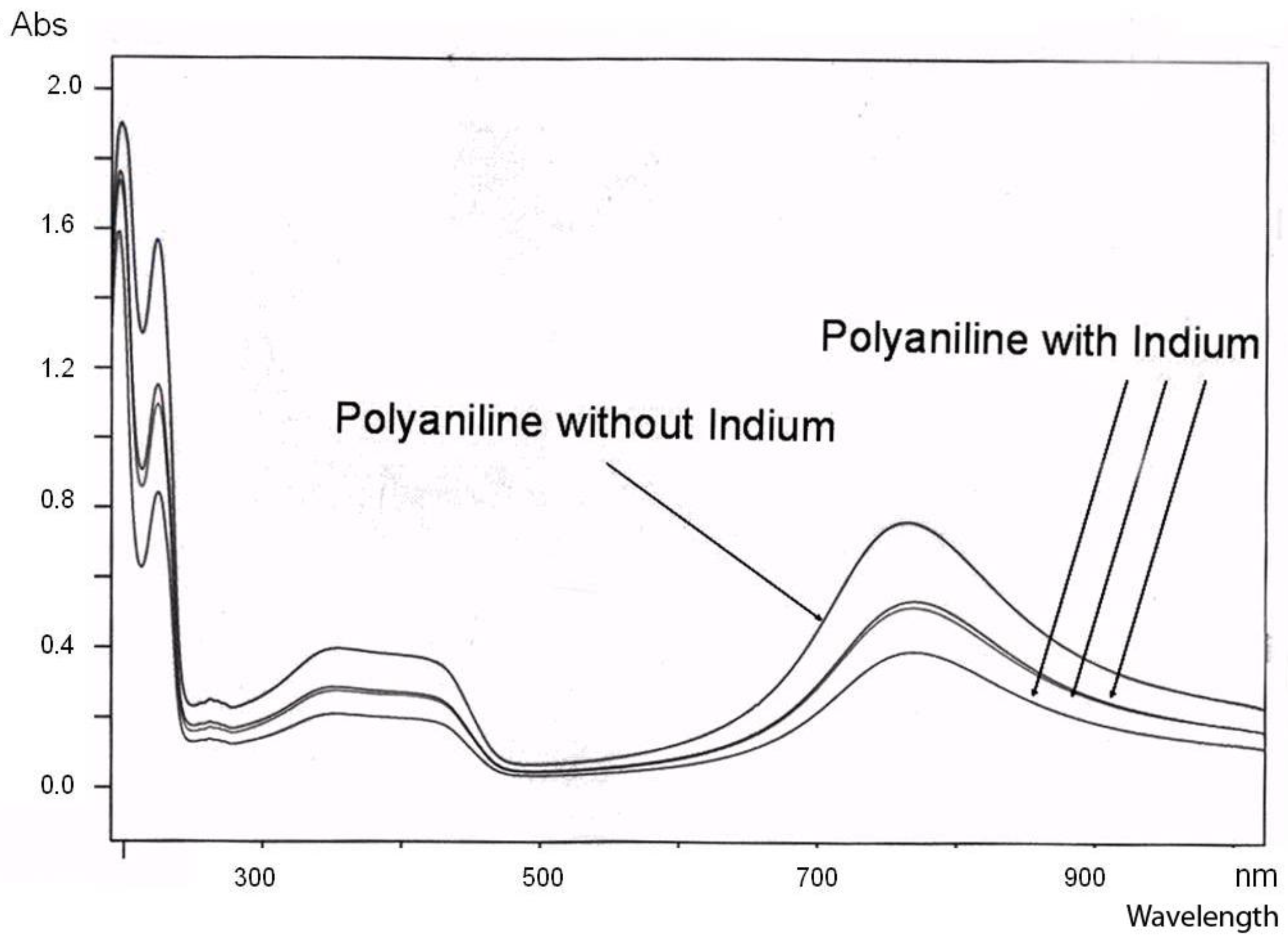
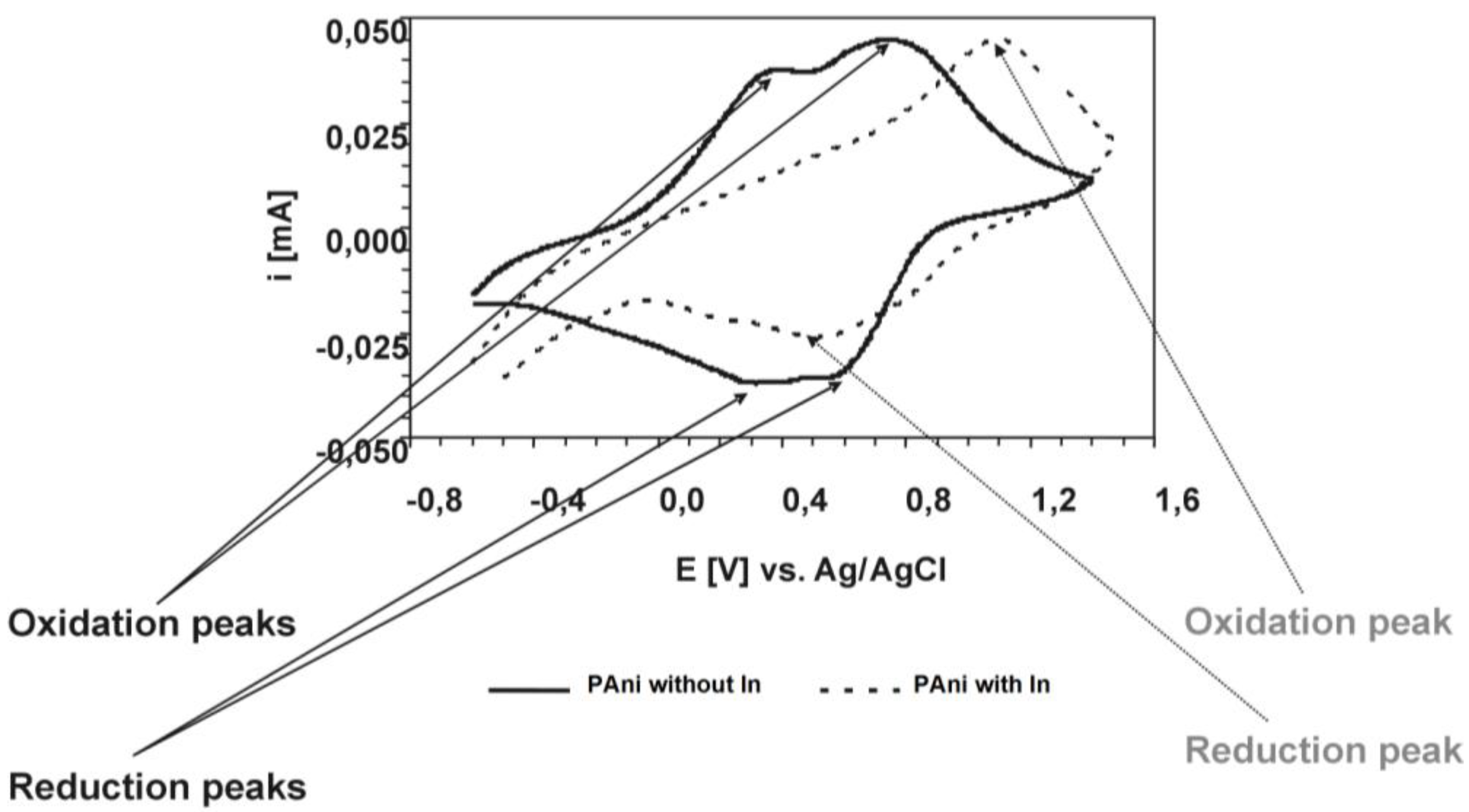
For the polyaniline/indium complex, we additionally performed cyclovoltammetric studies [
23]. While the pure polyaniline shows two oxidation and, correspondingly, two reduction peaks, the polyaniline-In complex only shows one single oxidation peak (at a significantly higher voltage) and one corresponding reduction peak (
Figure 5).
Figure 5.
Cyclovoltammogram of Polyaniline dispersions with and without indium. The figure shows the 5th cyclovoltammogram (CV) scan on PAni layers (PAni with and without In, respectively) deposited from dispersion onto Indium/Tin oxide (ITO); CVs performed in propylenecarbonate/0.1 M TEABF4 at 25 mV.
Figure 5.
Cyclovoltammogram of Polyaniline dispersions with and without indium. The figure shows the 5th cyclovoltammogram (CV) scan on PAni layers (PAni with and without In, respectively) deposited from dispersion onto Indium/Tin oxide (ITO); CVs performed in propylenecarbonate/0.1 M TEABF4 at 25 mV.
To conclude, it can be stated that polyaniline forms metal ion complexes via a reaction with the base metal, however with a low coordination number (1:12 in the case of Cu1+, 1:24 in the case of Me2+). The exact structure of these complexes is not known. Three of these complexes are already commercially used: The Cu complex in printed circuit board finishing; the Fe complex in corrosion protection; and the In complex in hole injection layers for organic light emitting diodes (OLEDs).
2.2. X-Ray Diffraction Studies
For additional insight, we performed x-ray diffraction studies, but this time down to the range of 2θ = 2°. For this study, we used polyaniline taken from different stages of the multi-step dispersion process.
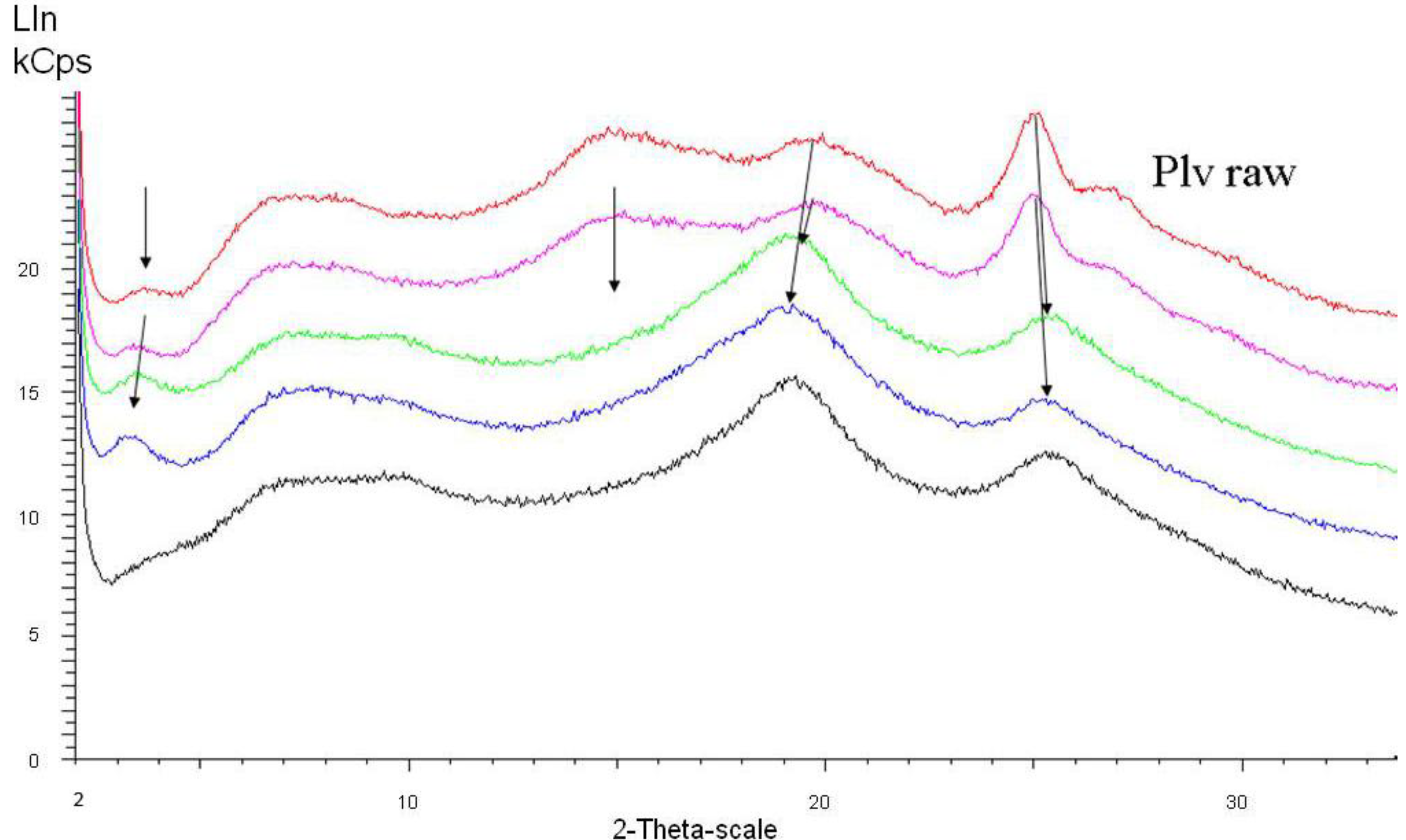
Figure 6 shows spectra of the raw (dry) polyaniline powder taken directly after polymerization and samples from different stages of the first dispersion step (in total, for commercial products, three subsequent dispersion steps are used [
24]). The starting conductivity (raw powder) is 5 S/cm, the maximum conductivity after the first dispersion step is 30 S/cm.
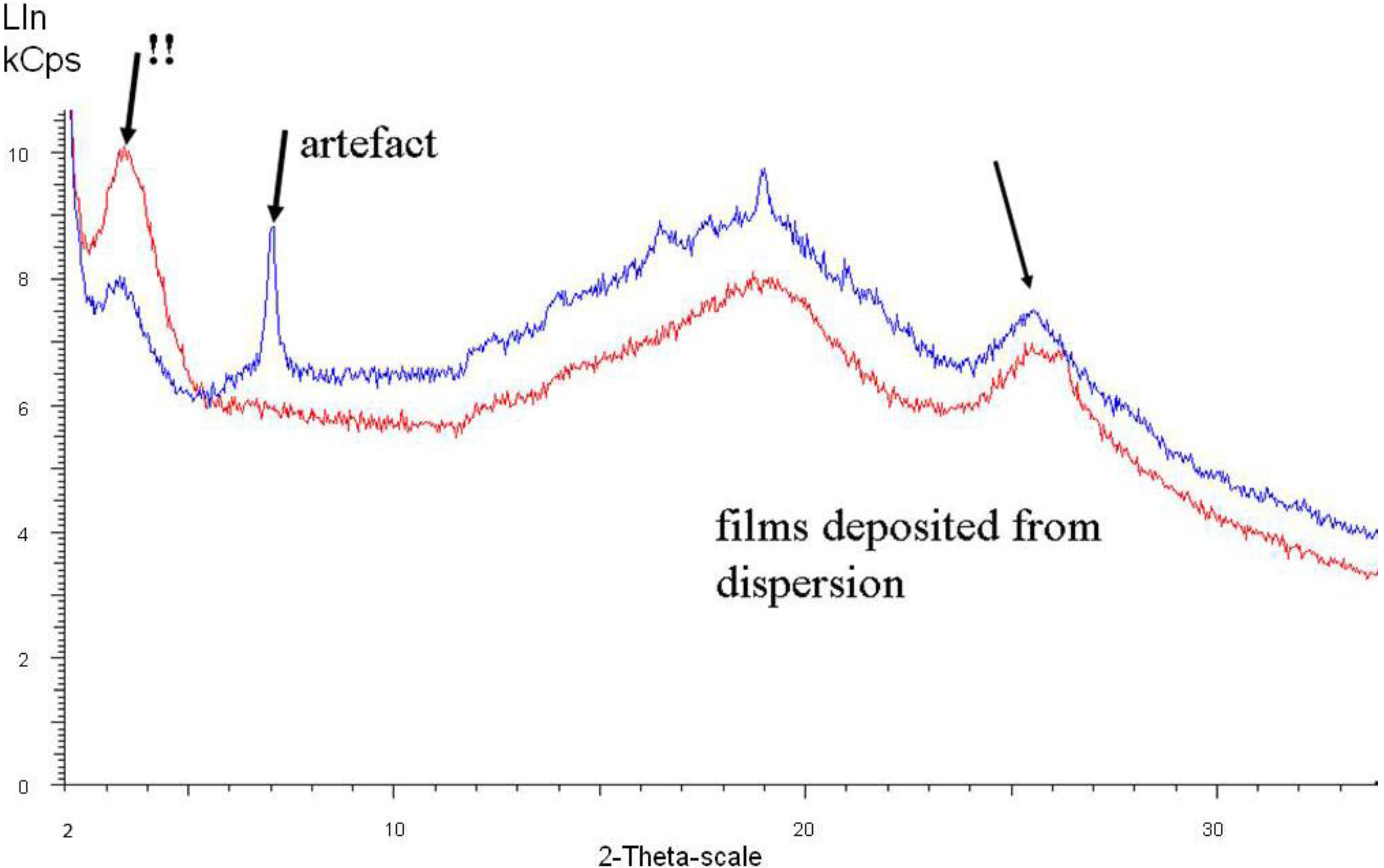
Figure 7 shows spectra of two samples taken from films which have been deposited after the third dispersion step.
As can be seen in
Figure 6 and
Figure 7, the originally dominant peak around 2θ = 25° is weakening (= lower degree of crystallinity) and shifted to higher angle values (= denser packing, as previously observed and commented on in the introduction section), while the peak around 2θ = 20° slightly increases in intensity and shifts to lower angle values.
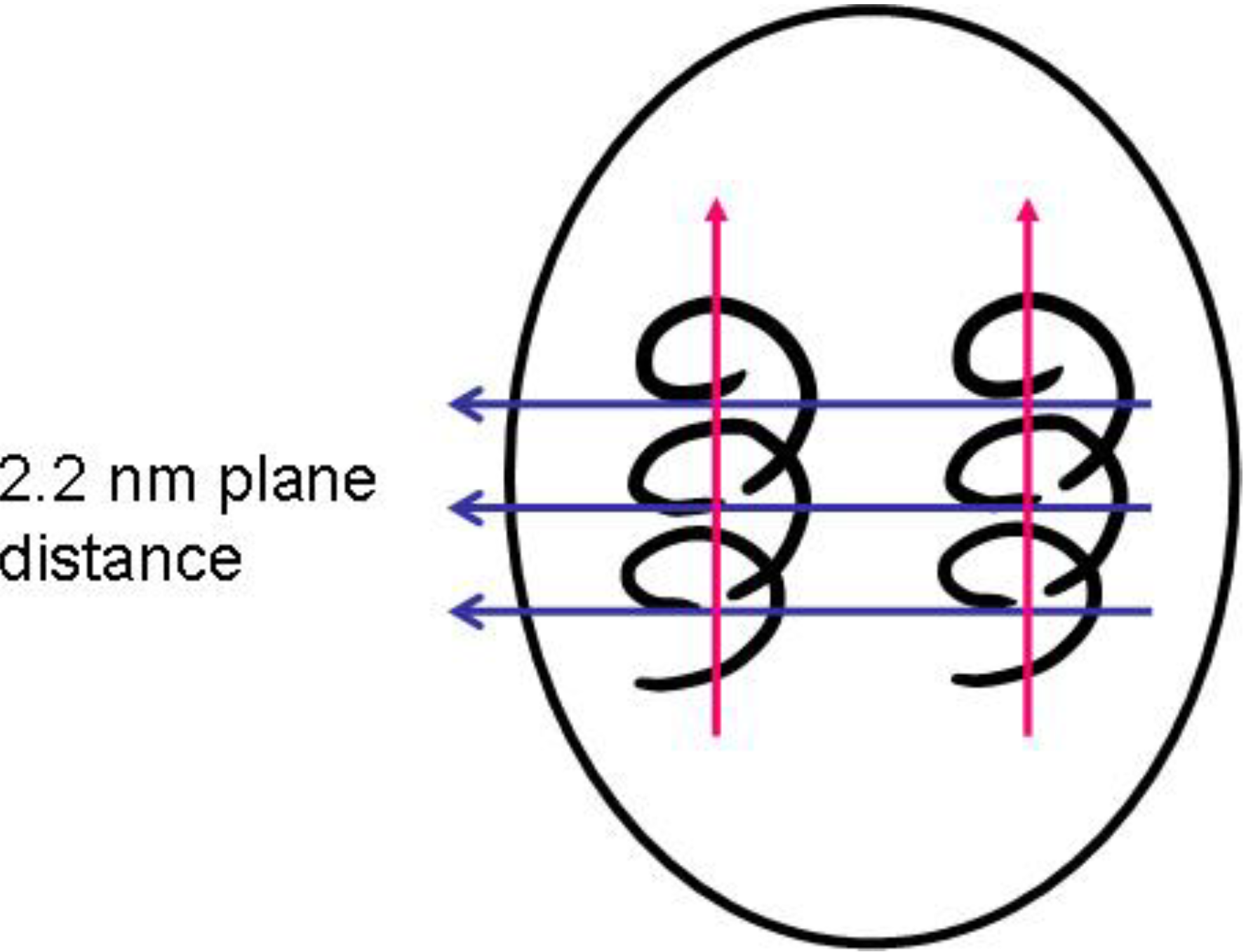
With increasing conductivity, especially detectable in
Figure 7, a peak at around 2θ = 3.5° (originally very weak) emerges and becomes the most prominent one. This 2θ value represents a diffraction plane distance of around 2.2 nm, hence smaller than the 3.5 nm subunits found in the SAXS experiments. This means that within the 3.5 nm subunit, some order is emerging which is characterized by a diffraction plane pattern with 2.2 nm distance.
Figure 6.
X-ray diffraction of polyaniline starting with the raw powder (red curve), following the progress of dispersion in the first dispersion step (from purple over green to blue curve, the latter representing the final stage of the first dispersion step); the black curve represents the same material as the blue curve after extracting all dispersion additives.
Figure 6.
X-ray diffraction of polyaniline starting with the raw powder (red curve), following the progress of dispersion in the first dispersion step (from purple over green to blue curve, the latter representing the final stage of the first dispersion step); the black curve represents the same material as the blue curve after extracting all dispersion additives.
Figure 7.
X-ray diffraction of polyaniline films deposited after completed dispersion steps. The blue curve represents a film having 50 S/cm, the red curve 100 S/cm conductivity.
Figure 7.
X-ray diffraction of polyaniline films deposited after completed dispersion steps. The blue curve represents a film having 50 S/cm, the red curve 100 S/cm conductivity.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


