Functionalization of Block Copolymer Vesicle Surfaces



Abstract
: In dilute aqueous solutions certain amphiphilic block copolymers self-assemble into vesicles that enclose a small pool of water with a membrane. Such polymersomes have promising applications ranging from targeted drug-delivery devices, to biosensors, and nanoreactors. Interactions between block copolymer membranes and their surroundings are important factors that determine their potential biomedical applications. Such interactions are influenced predominantly by the membrane surface. We review methods to functionalize block copolymer vesicle surfaces by chemical means with ligands such as antibodies, adhesion moieties, enzymes, carbohydrates and fluorophores. Furthermore, surface-functionalization can be achieved by self-assembly of polymers that carry ligands at their chain ends or in their hydrophilic blocks. While this review focuses on the strategies to functionalize vesicle surfaces, the applications realized by, and envisioned for, such functional polymersomes are also highlighted.1. Introduction
Membranes play a crucial role in several biological and chemical processes. Since Singer and Nicolson introduced the fluid mosaic model of the structure of cell membranes [1], which are viewed as two-dimensional solutions of lipids and globular proteins, polymer chemists and nanoscientists have developed new, artificial membranes that are superior regarding stability and functional versatility, but in most cases are more complex in structure [2,3]. The building blocks of such artificial membranes are, for example, amphiphilic block copolymers composed of hydrophobic and hydrophilic polymer chain blocks. Certain block copolymers self-assemble in an aqueous solution into membranes that, in turn, fuse into their entropically most stable morphology as hollow spheres, called polymer vesicles or polymersomes [4]. It has been shown that biological membrane processes can be successfully mimicked by, for example, reconstituting integral membrane proteins [5], fusing vesicles [6], or entrapping DNA [7]. Furthermore, polymersomes can be designed to be biocompatible [8]. With regard to potential applications of artificial membranes in life science, for instance in drug and gene delivery, cell surface recognition and nanoreactors, the development of so-called smart polymersomes that respond to internal or external stimuli such as temperature, pH, oxidative stress, light, magnetic fields and ultrasound has been highly promoted during the last few years [9-13]. One example of such smart materials are membranes consisting of biohybrid block copolymers that are inherently functional and respond to external stimuli [14]. Especially in the field of drug delivery, such triggers are of interest in order to release a hydrophilic agent at certain disease sites in an organism by changing the permeability or even the morphology of the vesicle membrane [10]. For efficient therapeutic applications, another essential issue is the targeting of specific sites, such as diseased cells or tissue, by an active receptor-ligand mechanism [15]. This targeting can be realized by attaching or introducing functional, in the majority of cases biological, ligands, such as sugars, aptamers, peptides and proteins, vitamins and antibodies [16] to the membrane surface. The specific immobilization of polymersomes to solid substrates and surfaces is also of high importance in other fields of application—in sensors and for surface modifications [17-19].
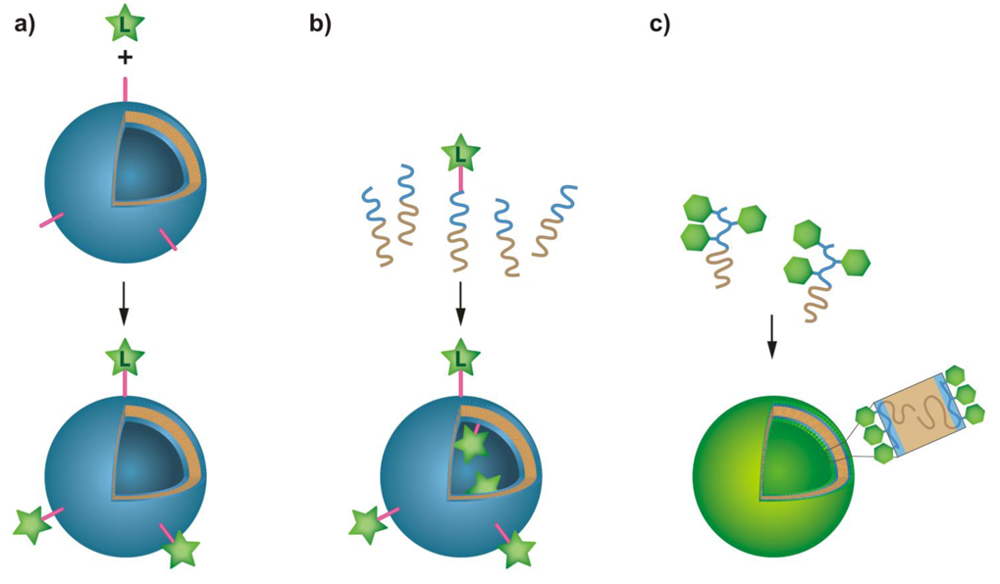
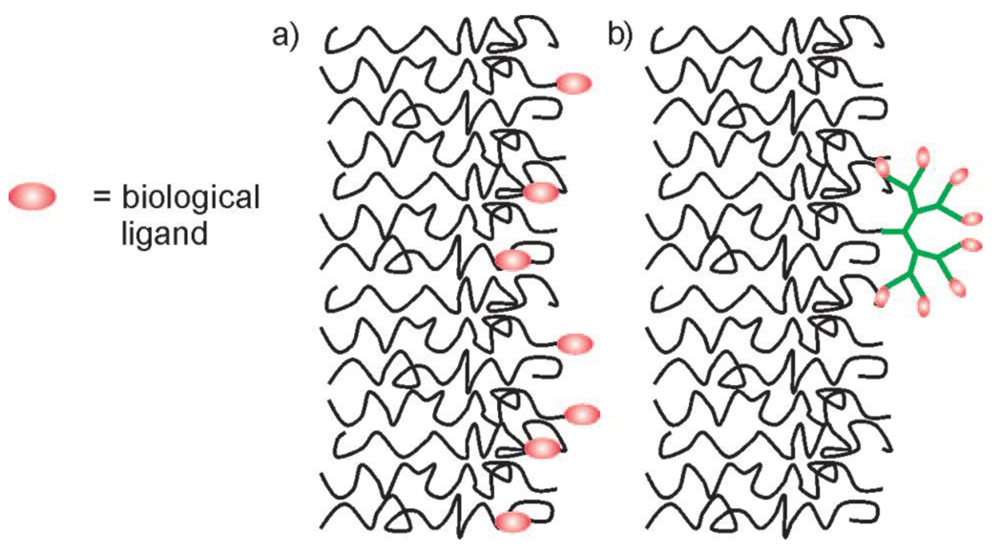
Various ways exist to generate a wide range of functionalities on the surface of the hydrophilic leaflets of amphipilic block copolymer membranes (Figure 1). In this review, we highlight the state of the art on how to functionalize block copolymer vesicle surfaces and show their applications in biomedicine, surface- and nanoscience. For this purpose, we focus on four different concepts of membrane modification: First, we discuss the conjugation of ligands to preformed vesicles, where the conjugation chemistries in aqueous solution will be highlighted. In the second part, we examine the formation of membranes from end-functionalized block copolymers. Then, we outline the formation of membranes from biohybrid block copolymers consisting of synthetic polymers and biologically relevant sugars, peptides, proteins and oligonucleotides. Finally, we show that membrane proteins that were reconstituted in block copolymer membranes, can act as specific recognition and attachment sites.
2. Functionalization of Block Copolymer Membrane Surfaces
2.1. Conjugation to Preformed Polymersomes
Polymersomes that have assembled from amphiphilic block copolymers have proven to be useful tools as drug delivery systems [8,20-22], nanoreactors and sensors [13,17,23-28]. The attachment of targeting ligands or enzymes to the polymersomes, as well as the immobilization of polymer vesicles on surfaces, is of crucial importance in most of the previously mentioned applications. In this chapter, we will review the chemical methods that have been used for conjugation of a variety of ligands to the membrane surfaces of preformed polymersomes (Figure 1(a)). The main requirements of such conjugation chemistries are their viability in an aqueous environment, the need to avoid cross-linking between polymersomes and between ligands, as well as stability and irreversibility of the resulting bond. Furthermore, it is desirable that the bonds be detectable and quantifiable in a simple, non-destructive manner. The state of the art in terms of common conjugation chemistry that fulfills all or some of these requirements is summarized in Table 1.
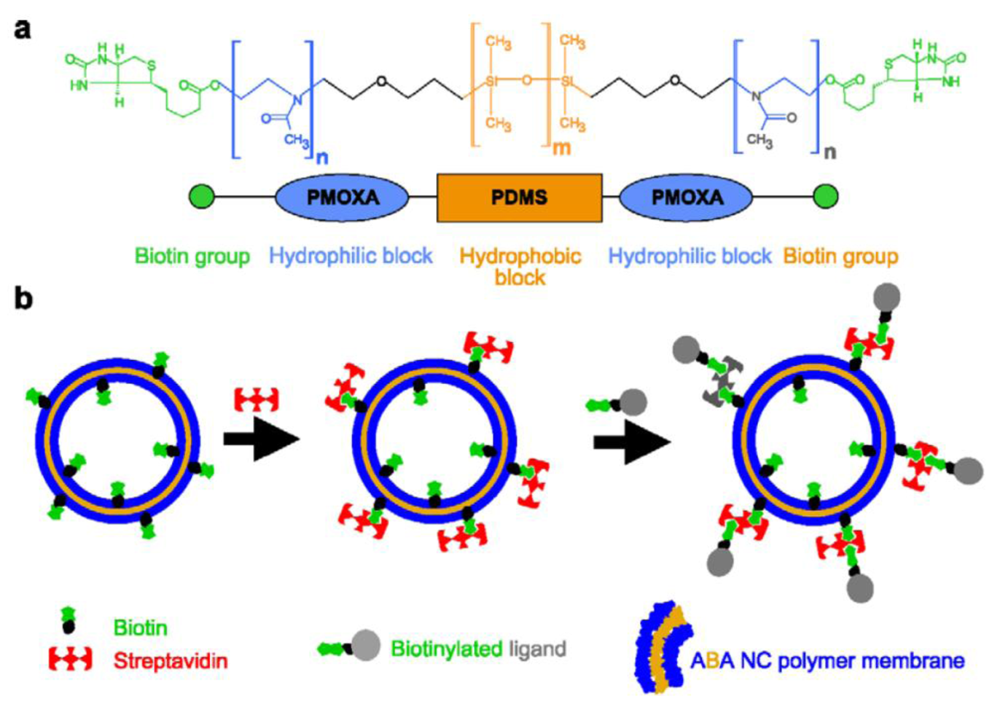
The first method that was used to attach ligands to the surface of preformed polymersomes was the biotin-streptavidin conjugation approach [33], which allows an arbitrary variation of ligands bearing biotin groups [17,20,34,35]. Ligands are bound indirectly by biotin-streptavidin-biotin interactions to the polymersome surface. The modification of block copolymers with biotin has been performed either by N,N'-dicyclohexylcarbodiimide/4-(dimethylamino)pyridine-activated esterification [17,20,34,35] or by preactivation of the terminal hydroxyl functionality by tresyl chloride [44] or 4-fluoro-3-nitrobenzoic acid [36,45] and subsequent reaction with biocytin [33,36]. Both the esterification and the tresylation method resulted in a good yield of biotin modified polymer, which is essential for streptavidin and thus ligand binding after self-assembly of the polymersome.
In order to obtain polymersomes that interact with biotin-modified ligands, biotin-modified block copolymers (Figure 2(a)) were hydrated in aqueous solution to form polymersomes that were then incubated in an excess of streptavidin (Figure 2(b)). The optimal concentrations of polymersomes and streptavidin need to be determined experimentally in order to avoid undesired cross-linking reactions. If the ratio of streptavidin to biotinylated polymersomes is too low, the probability of cross-linking between polymersomes increases. Once the surface of the biotin modified polymersome had been saturated with streptavidin, biotin modified ligands were then attached.
The modification strategy has some possible disadvantages. Although biotin and streptavidin form one of the strongest non-covalent interactions known (with dissociation constant around 10−15 M) [46], it is not a covalent bond. After binding, there is thus the possibility of ligand exchange with other biotinylated molecules in response to a change of ionic strength and/or temperature [47]. Furthermore, streptavidin is a macromolecule of considerable size (52.8 kDa). Therefore, when small ligands are intended for attachment to polymersomes via this strategy, the atom efficiency is not very favorable and steric problems might occur.
The binding of fluorescent dye-modified streptavidin to biotin modified polymersomes, with diameters between 140 and 172 nm, was demonstrated by fluorescence imaging [33], fluorescence correlation spectroscopy (FCS), as well as fluorescence cross-correlation spectroscopy (FCCS) [35]. A model system, investigated by FCS and FCCS and comprising rhodamine-green-biotin modified polymersomes and cyanin5-labeled streptavidin, was utilized in order to determine the number of binding sites and the dissociation constant, KD, of the receptor interaction between biotin-modified polymersomes and streptavidin [35]. An average number of 1921 ± 357 streptavidin-Cy5 per polymersome and an intrinsic KD of (1.7 ± 0.4) × 10−8 M were determined [35]. The latter differs from the value of 10−15 M given in the literature for the biotin-streptavidin interaction, which was explained by entropic and steric effects.
The biotin-streptavidin binding approach was used to modify polymersomes with the oligonucleotide polyguanylic acid (polyG), which specifically targets the macrophage A1 scavenger receptor [20]. Moreover, the decorated polymersome has also proven to be suitable to target tumor cell lines [34]. The receptor-specific binding of these polymersomes in human and transgenic cell lines and in mixed cultures was followed by vesicular uptake. A major improvement in biologic efficacy was obtained for polymersomes (containing pravastatin) compared to free drug, whereas no increased cytotoxic effect was observed in muscle tissue [34].
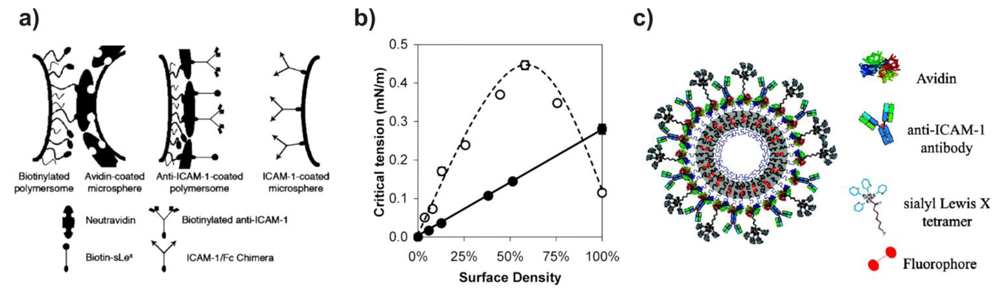
The group of Hammer has conducted extensive work on the surface-modification of polybutadiene-block-poly(ethylene glycol) (PBD-b-PEG) polymersomes via biotin-avidin interactions with the aim of creating and investigating vesicles that stick to biological surfaces under flow conditions, thus mimicking the adhesive properties of leucocites (Figure 3). The first papers in this series investigated the adhesion of biotin-decorated vesicles to avidin-coated surfaces [33,48]. To this end, PBD-b-PEG conjugated to biotin-lysin (biocytin) was synthesized. The functionalized copolymer formed vesicles on its own, or when mixed with unmodified block copolymers. Two different unfunctionalized block copolmers were mixed with the biotinylated block copolymer. One was shorter, the other was the same length as the functionalized polymer. Micropipette aspiration allowed measurement of the critical tension required to peel the membrane away from a bead covered with avidin as a function of biotin surface density and of the length-ratio between unmodified and modified block copolymers. When biocytin was conjugated to a PEG block that was longer than the surrounding unmodified PEG blocks, the critical tension reached a maximum at a content of 55% functionalized blockcopolymer. However, when both copolymers were of the same length, the critical tension was maximal at 10%, after which it increased only marginally. Thus, the biotin end groups need to be accessible at the surface, while they tend to be buried in the hydrophilic polymer layer at higher concentrations.
Biotinylated vesicles were coated with neutravidin, by means of which various biotin-labeled ligands were able to be conjugated to the surface, these include Alexa Fluor 488-biocytin, biotinylated adhesion molecule-1 antibody (anti-ICAM-1) and biotin-sialyl Lewisx (sLex; a selectin ligand) (Figure 3(a)) [36]. The surface density of the antibody was tuned by varying the ratio of anti-ICAM-1 to sLex during the conjugation. The adhesiveness was measured between the decorated vesicles and ICAM-1-coated beads. In contrast to previous studies, the adhesion strength did not depend on the amount of biotinylated block copolymer, but rather increased linearly with the surface-density of the binding ligand, because the flexible polymer chains are buried underneath the coat of neutravidin (Figure 3(b) and Figure 3(c)). Under physiological flow rates, such decorated vesicles do adhere to surfaces coated with inflammatory adhesion molecules P-selectin (to which sLex binds) and ICAM-1 [45,49], as well as to inflamed endothelium [49], indicating possible applications as a targeted drug-delivery system.
Although cell-specific targeting in vitro was shown with polymersomes that were surface-modified by a biotin-streptavidin approach [20,34], such polymersomes would not be of use in vivo studies because streptavidin is known to block essential immune reactions in the human body [50].
Another method of non-covalent but selective attachment of ligands to polymersomes and solid supported membranes was described by Nehring et al. [37,38]. Polymersomes were synthesized based on PBD-b-PEG diblock copolymer comprising carboxylic acid or lysine-nitilotriacetic acid (NTA) functional end groups. No disturbance of self-assembly behavior was observed upon mixing these two polymers. After vesicle formation, the peripheral NTA groups were able to form complexes with Ni2+ or Cu2+ ions. NTA-metal complexes are well known to selectively bind to oligohistidine sequences of proteins. Thus, maltose binding protein labeled with fluoresceine (His10-MBP-FITC) [37], as well as His-tagged enhanced green fluorescent protein (His6-eGFP) [37,38] and red fluorescent protein (His6-RFP) [38], were able to be conjugated to the vesicle surface. After confirmation of the presence of the polymer-metal complex by UV-Vis spectroscopy and electron paramagnetic resonance (EPR), dissociation constants KD of the different His-modified proteins to the Ni2+-NTA functionalized polymersome were determined by FCS. A KD of 7.0 ± 1.2 μM was measured for His10-MBP-FITC, 12.3 ± 1.2 μM for His6-eGFP and 1.99 ± 0.42 μM for His6-RFP [37]. These values are the same order of magnitude as the KD values obtained with Ni2+-NTA-modified liposomes (KD = 4.3 μM) [51] and Cu2+-NTA complexes [52].
Solid supported monolayers of the same block copolymers were prepared by Langmuir-Schaefer transfer and incubated with a His-tagged protein, with the aim of investigating protein binding to metal ion-complexing polymer surfaces. Atomic force microscopy (AFM) revealed structural details of the protein-decorated membranes, indicating that polymer monolayers may induce the formation of highly ordered protein arrays. Thus, the immobilization of densely packed proteins to such planar surfaces is a key step for 2D protein crystallization [53].
A further type of non-covalent interaction, the binding of adamantane in the cavity of cyclodextrins, can be exploited to functionalize polymersome surfaces. PS homopolymer terminated with a permethylated β-cyclodextrin (β-CD) group [39], as well as polyether imide (PEI) terminated at both chain ends with β-CDs [40] self-assembled into polymersomes, due to the hydrophilicity of the carbohydrate headgroups and the hydrophobicity of the polymer. The surface of these vesicles is a corona of β-CDs, which allowed the conjugation of adamantane-tagged horse radish peroxidase [39] and adamantane-terminated PEG [40]. Isothermal titration calorimetry (ITC) and static light scattering (SLS) revealed that both the inner and the outer polymersome surfaces were modified with the PEG. The enzyme-coated polymersomes were subjected to multiple washing steps. The amount of enzyme on the vesicles was found to decrease, indicating that the non-covalent interaction between β-CD and adamantane is too weak to withstand the washing procedure [39].
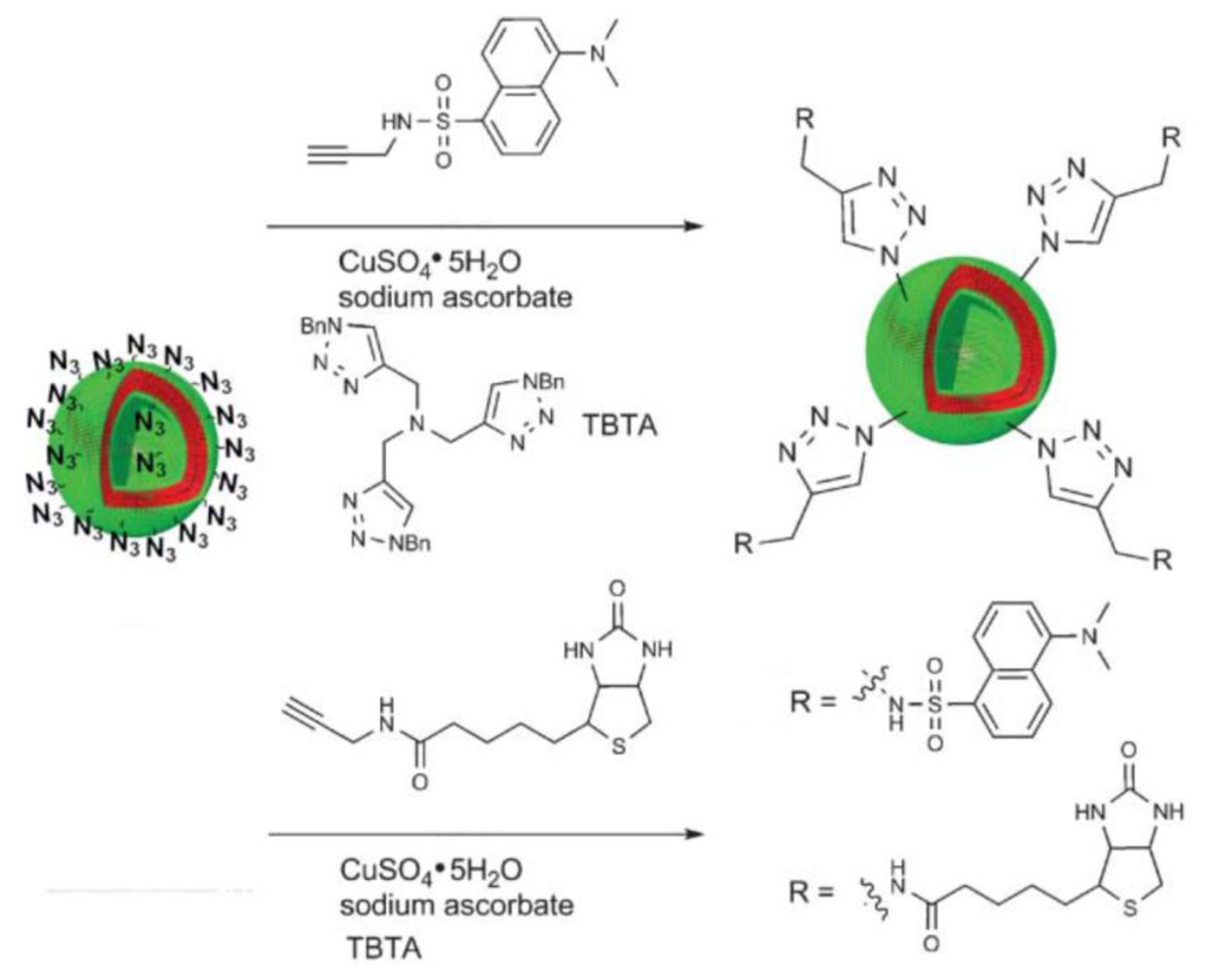
A much more efficient and—in contrast to the biotin-streptavidin, Ni-NTA-His-tag, and β-CD-adamantane interactions—covalent conjugation method is the Huisgen 1,3-dipolar cycloaddition of azides and alkynes, promoted by Sharpless and co-workers as a reaction that fulfills the criteria of click chemistry [54]. This reaction is usually carried out in a variety of organic solvents such as DMF, DMSO, THF, halogenated solvents [55], or in water-alcohol mixtures or pure water [56]. It is driven by heat or a catalytic amount of Cu(I). Since most polymersomes are destabilized in the presence of organic solvents, it is a prerequisite to perform this click chemistry conjugation reaction in aqueous media. Click chemistry with polymersomes in aqueous solution was introduced by van Hest and co-workers using poly(styrene)-block-poly(acrylic acid) (PS-b-PAA) diblock copolymers with terminal azide groups (Figure 4) [32]. Different ligands, such as an alkyne bearing fluorescent dansyl probe, biotin and eGFP, have been conjugated to preformed polymersomes.
The Cu(I) species that is used to catalyze this click reaction is usually formed in situ by mixing sodium ascorbate with a copper sulfate solution. In order to reach high conversion, the reduced Cu(I) species should be kept active for about 24 hours. For this purpose, the Cu(I) was stabilized by tris-(benzyltriazolylmethyl)amine (TBTA) or sodium bathophenantroline disulfonate (BPhT). The degree of functionalization was investigated by measuring the fluorescence intensity of the alkyne bearing fluorescent dansyl probe “clicked” to azide functionalized polymersomes. In order to find optimal reaction conditions and to improve the degree of functionalization of the polymersome surface, different parameters, such as the Cu(I) stabilizing ligands TBTA and BPhT, the copper concentration, the reaction temperature, and the pH of the reaction buffer, were varied. Degrees of functionalization between 24% and 26% were found under all conditions tested, which did not indicate a significant influence by the changed parameters. However, the relatively low degree of functionalization might be explained by functional group availability on the polymersome surface, with the majority of functional groups being buried in the hydrophilic layer of the block copolymer membrane. In addition to azide-functional polymersomes, van Hest and co-workers also introduced polymersomes based on alkyne-functionalized PS-b-PEG mixed with a high ratio of poly(styrene)-block-poly(l-isocyanoalanine(2-thiophen-3-yl-ethyl)amide) (PS-b-PIAT) [29].
An approach used to increase the number of surface-accessible functional groups on polymersomes was shown by Gillies and co-workers. By click chemistry, they attached dendritic and nondendritic displays of mannose, bearing an alkyne functionality, to azide functional polymersomes and compared their binding to the mannose binding protein, concanavalin A, by using a hemagglutination assay [31]. As shown in Figure 5, membranes functionalized with dendritic displays are expected to show a higher availability of the mannose ligands than the nondendritic ones. In fact, this hypothesis was verified by experiments that showed that the binding of polymersomes functionalized with dendritic mannose displays was increased by 1–2 orders of magnitude relative to the nondendritic polymersomes.
Polymersome-enzyme conjugates made by azide-alkyne click chemistry have been applied as nanoreactors for enzymatic cascade reactions [26]. Glucose oxidase (GOx) was encapsulated in the interior of these polymersomes and Candida antarctica lipase B (CalB) was incorporated into the vesicle bilayer membranes. Horseradish peroxidase (HRP) was reacted with imidazole-1-sulfonyl azide hydrochloride to introduce azide functionalities to this enzyme [57,58]. The HRP was then conjugated to the outer surface of the polymersomes via click-chemistry. The resulting construct had the three different enzymes partitioned in three different, well-defined local environments and was able to catalyze a three-step enzymatic reaction cascade, thus mimicking the confinement of individual reaction steps in compartments of living cells. Monoacetylated glucose was deprotected by CalB while diffusing from the bulk solution through the membrane into the cavity of the polymersome. In the cavity, the glucose was oxidized by GOx to gluconolactone and hydrogen peroxide. This small molecule was able to diffuse out of the nanoreactor through the membrane and was utilized by HRP to convert 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) to ABTS•+. In order to determine the enzyme encapsulation and conjugation efficiencies, the enzymes were decorated with a ruthenium complex and the biohybrid polymersomes were analyzed by inductively coupled plasma mass spectrometry. More than 90% (a remarkably high percentage) of available alkyne functionalities on the polymersome surface were thus found to be occupied by HRP enzymes. This example again shows the high efficiency of the azide-alkyne click reaction in aqueous media.
Reactions well-known in the field of protein conjugation chemistry, such as the reactions of maleimide functions with thiol groups [59,60], or N-hydroxy succinimidyl esters with the amine groups of lysins [61], can be applied to modify polymersomes bearing suitable functional groups on their surfaces. An example is the conjugation of the monoclonal antibody OX26 to poly(caprolactone)-block-poly(ethylene glycol) (PCL-b-PEG) polymersomes by reacting cysteine residues of the antibody with maleimido end groups of the PEG block present on the polymersome surface [41]. These decorated polymersomes were investigated as possible vessels for the delivery of peptides to the brain, as the antibody binds to a receptor that can initiate transcytosis of particles across the blood-brain barrier. A quantity of 34 OX26 antibodies on the surface of a polymersome was found to result in the greatest blood-brain barrier permeability.
Another example of a mild conjugation reaction known in biochemistry is the addition of thiols to vinyl groups. This reaction was exploited to link cysteine-containing peptides to vesicles with a reactive surface [42]. Poly(ethylene oxide)-block-poly(γ-methyl-ε-caprolactone) (PEG-b-PMCL) was functionalized at its hydrophilic terminus with a vinyl sulfone group. These block copolymers formed vesicles in aqueous solution that displayed vinyl sulfone groups on their surface. Short targeting peptides containing thiol groups did bind to the vesicles, while similar peptides lacking cysteine did not adhere. As this work was only recently published, future experiments are needed to show the efficiency of these peptide-labeled polymersomes in targeted drug-delivery.
The attachment of succinimidyl ester-activated Alexa Fluor 633 to polymersomes consisting of PDMS-b-PMOXA diblock copolymers comprising hydroxyl and amino end groups has been investigated very recently by Egli et al. [43]. The number of fluorophores per vesicle was controlled by adjusting the ratio of amine-terminated- to hydroxyl-terminated block copolymers and was measured by FCS. The fluorescence intensity for polymersomes consisting of 0.3 to 10 mol% AB-NH was determined and plotted, resulting in a linear function that quantitatively describes the number of fluorophores per polymersome. At higher ratios of AB-NH, from 10- to 100 mol%, saturation of the fluorophore attachment to the polymersome was observed, that can be explained by steric hindrance to further dye conjugation.
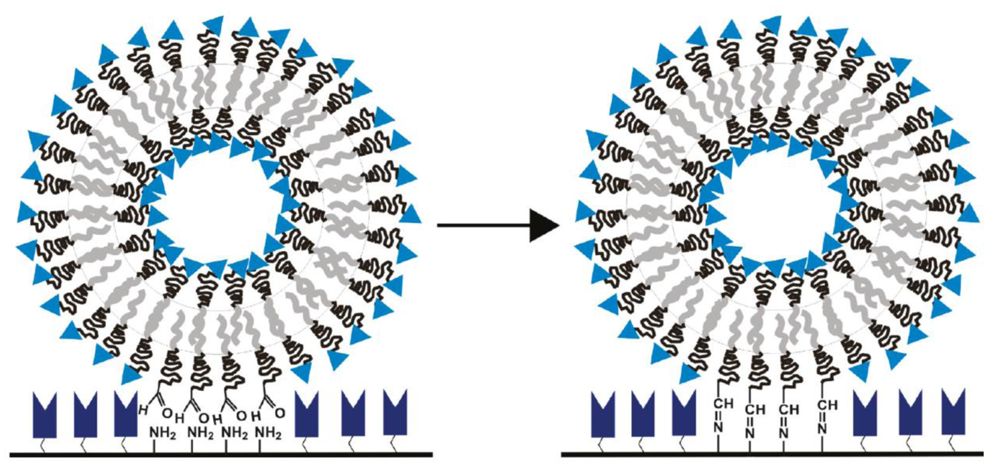
Foster and co-workers reported the immobilization of polymersomes composed of PCL-b-PEG, poly(lactide)-block-poly(ethylene glycol) (PLA-b-PEG), and poly(isoprene)-block-poly(ethylene glycol) (PI-b-PEG), each polymer with aldehyde end groups, to aminated glass surfaces, resulting in a covalent imine bond (Figure 6) [19]. Depending on the different bilayer stiffnesses, the three different species of polymersomes showed different footprint areas upon immobilization.
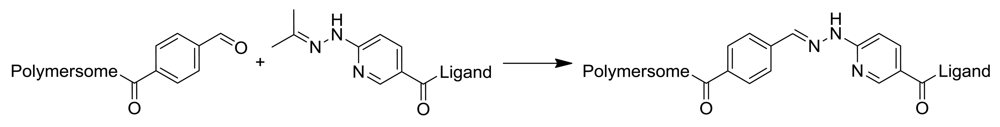
An extension of this concept would involve the conjugation of proteins to the aldehyde decorated polymersome surface, as proteins generally display a multitude of amine groups on their surfaces. However, this approach is unfavorable because cross-linking of polymersomes by proteins could occur, which would lead to large aggregates. Moreover, this approach would not be suitable for the formation of permanent covalent bonds in aqueous environments, since aliphatic imines are known to be unstable and to hydrolyze into their original amine and aldehyde functionalities. In contrast, aromatic imines based on a bis-arylhydrazone bond show no hydrolysis and are stable in aqueous solution in a pH range from 2–10 and up to 95 °C [62], due to their large delocalized electron system. Conjugation chemistry that forms such a bis-aryl hydrazone bond is based on two complementary hetero-bifunctional linkers, comprising an aromatic hydrazine (protected as acetone hydrazone to increase its shelf-life) and an aromatic aldehyde (Figure 7). Using two complementary hetero-bifunctional linkers confers the further advantage that cross-linking cannot occur. Such a system was used to specifically and quantitatively conjugate oligonucleotides to antibodies [63] and to bind proteins to other proteins [64] in aqueous solution. Because this conjugation reaction is carried out under mild conditions in aqueous buffer, without any additional catalyzing agents (e.g., in contrast to the copper catalyzed azide-alkyne click reaction), the resulting conjugates are applicable for pharmaceutical and therapeutic purposes. Recently, Egli et al. used this conjugation strategy to form covalent and stable polymersome-protein (enhanced yellow fluorescent protein, eYFP) and polymersome-antibody conjugates [43]. Polymersomes consisting of PDMS-b-PMOXA diblock copolymers comprising either hydroxyl or amino end groups were functionalized with an excess of succinimidyl ester-activated 4-formyl benzoic acid (S-4FB). In parallel, eYFP and antibodies were modified with succinimidyl ester-activated 6-hydrazinonicotinic acid (S-HyNic) in order to introduce the aromatic acetone hydrazone functionality. The functionalization of polymersomes and ligands with 4FB and HyNic was quantified by colorimetric reactions using 2-hydrazinopyridine (specific absorption of the formed linker at 350 nm) and 4-nitrobenzaldehyde (specific absorption of the formed linker at 390 nm), respectively. Then, solutions of both conjugation partners were mixed and incubated. The successful attachment of approximately 5 eYFP molecules per polymersome was determined by the time-dependent measurement of intensity fluctuations by FCS. Polymersome-antibody conjugates prepared by this strategy were applied to target biotinylated surfaces (using anti-biotin IgG) and Her2 receptors expressed by breast cancer cells SKBR3 (using the antibody trastuzumab). Polymersomes without antibodies attached to their surfaces showed no toxicity and only low inhibition of proliferation, whereas polymersome-trastuzumab conjungates showed specific attachment and clear reduction in proliferation of the cells.
In this section we have reviewed a variety of synthetic routes to modify polymersome surfaces with moieties such as biomolecules. These methods differ by their binding specificities and efficiencies, by their biocompatibilities, as well as by their binding strengths. Non-covalent (e.g., biotin-streptavidin) and covalent binding strategies (e.g., Cu-catalyzed alkyne-azide click chemistry or the reaction of aromatic acetone hydrazones with aromatic aldehydes) are possible. An advantage of non-covalent binding may be that a higher density of functional ligands can be achieved, due to rearrangement of ligands and functional anchor groups. Benefits of covalent binding strategies include improved site-specificity and reproducibility, as well as increased binding stability. The copper catalyzed azide-alkyne click chemistry has proven itself to be applicable to the conjugation of different species such as proteins, enzymes or dendrons to the polymersome surface. The use of copper as a catalyst, as well as the redox conditions while reducing Cu(II) to Cu(I), are unfavorable for biological and pharmaceutical applications. In contrast, the formation of a stable bis-arylhydrazone bond between 4FB modified polymersomes and HyNic modified ligands, such as proteins and antibodies, features the same advantages as the click chemistry, but does not require additional metal-based catalyst species.
Specific reactions to form bonds between block copolymer membranes and biological ligands, as well as the stability, and biocompatibility of the resulting bonds and linkers, are not the only points of interest when surface modified polymersomes are produced. Further investigations on the control of binding site availability, as well as membrane stability following a high degree of modification, will have to be carried out in the future.
2.2. Formation of Polymersomes from End-Functionalized Block Copolymers
In the previous chapter we showed that the surface of an existing block copolymer vesicle can be functionalized by targeting functional groups on the surface with reactive linkers or conjugation reagents that bind a ligand. A different strategy, however, involves functionalization of block copolymers at their hydrophilic chain end(s) with ligands such as carbohydrates, peptides and proteins before self-assembly into vesicles (Figure 1(b)). The advantages of this strategy are that the functionalized copolymer can be purified and characterized as a non-aggregated species and that it can be mixed with the non-functionalized copolymer prior to vesicle formation, thus allowing better control of the surface-density of exposed ligands simply by adjusting the ratio of functionalized to non-functionalized block copolymer. Furthermore, vesicles with a highly functionalized corona are obtained when the ligand-bearing copolymer is allowed to self-assemble on its own. However, the assembly behavior of the block copolymers might be affected by the end-functionalization, so that their ability to form vesicles has to be confirmed by experimental data.
Carbohydrates play an important role in cell-cell recognition and signal transduction in living organisms. Thus, decorating polymer vesicles with sugars can lead to bioactive nanostructures. Lee and co-workers synthesized rod-coil polymer amphiphiles terminated by a carbohydrate, tetra(p-phenylene)-block-PEG12-α-D-mannopyranoside. This glycopolymer formed small vesicles with a diameter of 40 nm, in which the amphiphilic bilayer membrane was covered with mannose residues [65,66]. Indeed, the surface can be regarded as supramolecular, multivalent ligand, as is shown by a hemagglutination inhibition assay with lectin concanavalin A (Con A), which recognizes R-D-mannopyranoside. The vesicle showed 800-fold higher inhibitory potency compared to free methyl mannose [66]. Moreover, the mannose-covered vesicles specifically bind to the pili of the ORN 178 E. coli strain, as imaged by transmission electron microscopy (TEM) (Figure 8) [65]. Bacterial pili are proteinacious fibers that bind to mannose and protrude from the surface of the cell. In control experiments, galactose-terminated amphiphilic polymers were synthesized, but the corresponding self-assembled nanostructures did not bind to the pili [66]. In addition, an E.coli strain that lacks the selective binding protein for mannose was tested. With these bacteria, no adhesion of the vesicles to the pili was observed. These experiments clearly show the significance of the carbohydrate end group in the interaction of polymersome and its surroundings.
Yonese et al. studied the aggregation behavior of lactose-terminated PEG-b-poly(γ-methyl L-glutamate) in water [67]. Dynamic light scattering (DLS) indicated the formation of large aggregates, presumed to be vesicles, with a hydrodynamic radius of about 250 nm. Key to the aggregation behavior might be the association of α-helical poly(γ -methyl L-glutamate) segments, promoting the formation of planar bilayers, which then close into vesicles. The lactose units were recognized by the lectin RCA120, indicating that they are located on the surface of the glycopolymer aggregate.
Spherical and worm-like polymersomes decorated with folate as a targeting ligand were investigated as nanocarrier system for a combined targeted delivery of an anticancer drug and a magnetic resonance imaging contrast agent [68,69]. To this end, heterobifunctional asymmetric triblock copolymers folate-PEG5000-b-poly(glutamate hydrazone doxorubicin)-b-PEG2000-acrylate, and FA-PEG114-b-PLA-b-PEG46-acrylate were synthesized and allowed to self-assemble into vesicles. The long PEG segments were mostly segregated in the outer hydrophilic leaflet of the membrane, whereas the shorter PEG block was mostly present in the inner hydrophilic layer. Thus, these vesicles presented folate on their surface and could be stabilized by crosslinking the polymers in the inner leaflet via the acrylate functional groups to enhance circulation time. These polymersomes showed higher cellular uptake by HeLa cells than folate-free polymersomes, due to the folate receptor-mediated endocytosis process. Accordingly, doxorubicin loaded polymersomes exhibited higher cytotoxicity than the folate free controls.
Polymersome surfaces can also be functionalized with more sophisticated biological ligands by the strategy of synthesizing end group-functionalized block copolymers and allowing them to self-assemble into polymersomes. Very recently, the group of van Hest expanded their work on functionalized polymersomes by conjugating the cell penetrating peptide Tat to PS-b-PEG-oxanorbornadien block copolymers via a tandem cycloaddition/retro Diels-Alder “click” reaction [70]. These polymers were admixed with 90 wt% PS-b-PIAT to form vesicles that presented Tat on their surfaces. The vesicles were able to encapsulate guest proteins such as green fluorescent protein and HRP, and traffic their cargo into cells with high phagocytic activity (HeLa, Jurkat, and HEK 293). It was clearly shown that the cell penetrating peptide was responsible for the cellular uptake.
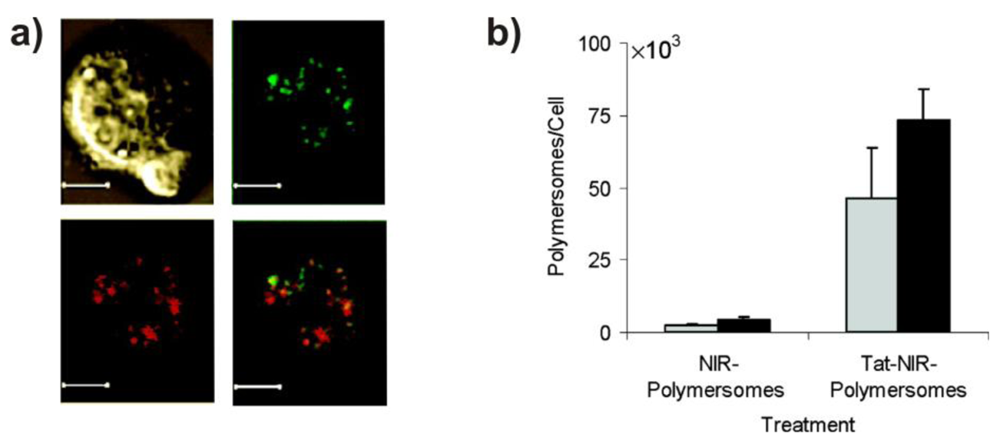
In another example, Hammer and co-workers reported PBD-b-PEG block copolymers that were functionalized with Tat or its fluorescently-labeled version, FITC-Tat, by activating the hydroxyl end group of the block copolymers with succinimidyl carbonate, followed by conjugation with the peptide [71]. When 10 mol% of these functionalized block copolymers were mixed with the non-functionalized PBD-b-PEG, vesicles formed despite the presence of the rather large functional end groups (1,760, or 2,260 Da, respectively). These polymersomes enabled intracellular delivery of a hydrophobic, near-infrared emissive fluorophore embedded in the polymersome membrane (Figure 9). A future application of such a system might be the tracking of dendritic cells by NIR fluorescence based imaging.
The biocompatibility of PBD-b-PEG makes this block copolymer a popular choice for cell targeting studies and potential biomedical applications [71]. Kokkoli and co-workers prepared PBD-b-PEG-based polymersomes that were decorated with a targeting peptide that mimics the cell adhesion site in fibronectin [72]. The peptide was conjugated at its N-terminus with a block copolymer bearing an N-hydroxysuccinimide end group. These functionalized polymers were mixed in 1:1 and 1:99 ratios with non-functionalized PBD-b-PEG and formed vesicles using the film rehydration method. The vesicles were found to be internalized within prostate cancer cells that expressed integrins on their surfaces and were able to deliver a model protein therapeutic (tumor necrosis factor-α) to the cells, dramatically enhancing its cytotoxic potential.
Using the same type of polymer and a similar strategy, Ugaz and coworkers incorporated a small fluorescent label on the surface of vesicles [73]. The coumarins were used to probe the accessibility of functional end groups on the surface of polymersomes. To this end, coumarin-functionalized PBD-b-PEG block copolymers were synthesized by oxidation of the PEG`s hydroxy endgroup into carboxylic acid, followed by N-hydroxysuccinimidyl-ester mediated amidation with 7-amino-4-(trifluoromethyl) coumarin. 10 wt% of coumarin-functionalized block copolymers were admixed with two different non-functionalized block copolymers, one in which the hydrophilic block was shorter than the PEG block of the functionalized block copolymer, and one that had the same length as its functionalized counterpart. The fluorescence quenching of coumarin by iodide ions was greatly reduced on the surface of the vesicles compared to free fluorophores and was smallest when the fluorophore-bearing PEG blocks were the same length as the non-functionalized hydrophilic blocks, due to retarded ion mobility in the presence of PEG.
PBD-b-PEG was also functionalized with another fluorophore, fluorescein, by reacting the terminal hydroxyl group of the PEG chain with fluorescein-5-carbonyl azide [74]. These block copolymers assembled into fluorescent polymersomes and were used to visualize phase separation in mixed polymersomes made of PBD-b-PEG and PBD-b-PAA.
A modular way to tailor the surface properties of polymersomes was recently introduced by Kros and co-workers [75]. They synthesized polypeptides that consisted of a hydrophobic poly(γ-benzyl L-glutamate) (PBLG) block and a hydrophilic coiled-coil forming peptide as the hydrophilic block. The latter binds noncovalently with peptides of complementary amino acid sequence, thus allowing the attachment of ligands to the peptidic block copolymers via the formation of coiled coils prior to vesicle formation. In this first study the proof-of-concept was shown using a complementary peptide and a peptide-PEG conjugate, but it can be easily envisioned that this concept would be useful to attach biological active ligands, thus creating vesicles with a functional corona.
2.3. Formation of Polymersomes from Polymers Comprising Biofunctional Hydrophilic Blocks or Side Groups
Amphiphilic block copolymers can be designed to comprise biomolecules-containing hydrophilic blocks. These blocks can be linear carbohydrates, glycopolymers, that is, synthetic polymers with carbohydrate side groups, peptides and proteins, or oligonucleotides [76,77]. Furthermore, some of these biomolecules can be attached as side chain groups to a homopolymer backbone, resulting in amphiphilic polymers as well. Many of the biomolecules exhibit biological activity, for example, they are recognized as binding sites or signaling moieties by enzymes, proteins and cells. Some of the hybrid block copolymers self-assemble in diluted aqueous solutions into vesicles (Figure 1(c)). The resulting membranes display the hydrophilic blocks towards the water-filled lumen of the vesicles and towards the bulk solution. Therefore, polymersomes with a highly functionalized inner and outer surface are obtained. Moreover, the biomolecules can also be present in deeper layers of the hydrophilic leaflet, which can result in enhanced interaction with, for example, cells, compared to end-functionalized block copolymers discussed above.
As carbohydrates exhibit specific interactions with, or recognition of cell membranes or lectins [78], polymersomes formed from glycopolymers have been extensively investigated. Schlaad et al. prepared a series of well-defined glycopolymers by radical photoaddition of 1-thio-β-D-glucopyranose (Glc) onto PBD homopolymers [79,80] and block copolymers, PBD-b-PS and PBD-b-PEG [81,82]. Glucosylated PBD and PBD-b-PEG exhibited hydrophilic weight fractions whydro > 0.5 and can directly be dispersed in water. The glucosylated PBD-b-PS (whydro = 0.17) had to be dispersed in tetrahydrofuran first, followed by a slow exchange of the solvent against water. PBD-Glc formed large, unilamellar vesicles having hydrodynamic diameters of about 500–600 nm and a membrane less than 4 nm thick, as indicated by light and X-ray scattering analysis and TEM (Figure 10(a)) [79]. The membrane appears to be a bilayer structure. The thickness of the membrane, which is similar to that of a liposome, is not affected by the length of the polymer backbone, suggesting that chains are oriented parallel to the interface. Another consequence of the parallel orientation of chains in the membrane is that vesicles are formed despite the high hydrophilic weight fraction; phospholipids are usually less hydrophilic, and block copolymers of such composition would assemble into spherical or worm-like micelles. The glucosylated PBD-b-PS was found to self-assemble into vesicles in a tetrahydrofuran solution as well as in water (Figure 10(b)) [81]. The apparent hydrodynamic radius of these vesicles in water was about 120 nm. They have a polystyrene bilayered wall, measuring about 20 nm across (TEM), and a Glc corona. Amphiphilic block copolymer samples of glucosylated PBD-b-PEG spontaneously formed very large vesicles of >500 nm in diameter [82]. The existence of unilamellar vesicles was confirmed by TEM (Figure 10(c)) and scattering analyses. Chains consisting of one hydrophobic and two hydrophilic chains can only be packed in a monolayer. These vesicles should therefore have an asymmetric membrane with glucose outside and PEG inside, as evidenced by 2D-NOESY-NMR and surface-enhanced Raman spectroscopy.
To date, these glycopolymer vesicles have not been investigated in terms of their drug release behavior or specific interactions with biological cell membranes or lectins.
The same group synthesized glucopolyoxazoline homopolymers by cationic polymerization and thiol-ene “click” chemistry, which in pure water spontaneously assembled into spherical vesicles and hollow nanotubes [83]. However, aggregates were not observed in HEPES buffered saline solution. Adding Glc-binding lectin Con A to an aqueous glucopolyoxazoline solution led to an increase in the optical density (turbidity), indicating the formation of large aggregates due to multivalent interactions between protein and polymer (“cluster glycoside effect” [84]).
Amphiphilic block copolymers that include sugar moieties in one of the blocks are a further example of vesicle-forming glycopolymers. Li et al. synthesized a series of amphiphilic block copolymers consisting of hydrophobic PS and hydrophilic poly(2-(β-D-glucopyranosyloxy)ethyl acrylate) (PGEA) blocks using atom transfer radical polymerization (ATRP) [85,86]. Copolymers were first dissolved in a common solvent (N,N-dimethylformamide, tetrahydrofuran, dioxane, and their mixtures), followed by the addition of water to induce self-assembly. The observed morphologies included micelles, vesicles, hollow tubules, porous spheres, and large compound vesicles, depending on the composition of the copolymer and the solvent mixture. Vesicles usually exhibited polydisperse size distributions, diameters ranging from 70 to 350 nm, and the membranes measured about 20–25 nm across. The structures are envisioned to find applications as models for cell-specific drug delivery systems; to date, experimental studies have not been reported.
Dong et al. investigated a series of biodegradable and biomimetic star-shaped block copolymers based on PCL and either poly(lactobionamidoethyl methacrylate) (PLAMA) [87] or poly(gluconamidoethyl methacrylate) (PGAMA) [88], synthesized via ATRP (Figure 11). Biohybrid amphiphiles with hydrophobic PCL mole fractions of 62% and 76%, respectively, were found to self-assemble into vesicles measuring about 150–250 nm, eventually 750 nm, in diameter in dilute aqueous solution. The PCL-PLAMA and PCL-PGAMA copolymers showed specific recognition for RCA120 lectin and Con A, respectively.
Pasparakis and Alexander synthesized block copolymers based on hydrophilic poly(2-glucosyl-oxyethyl methacrylate) (PGEMA) and poly(diethyleneglycol methacrylate) (PDEGMA) by ATRP and reversible addition-fragmentation chain-transfer (RAFT) polymerization [89]. In dilute aqueous solution, the polymers assembled into vesicles with mean diameters of approximately 250 nm (polymer containing 15 wt% Glc) and 500 nm (34 wt% Glc) at 20 °C, as indicated by dynamic light scattering and scanning electron microscopy. At 37 °C, above the lower critical solution temperature (LCST) of PDEGMA, the size of the vesicles decreased to 180 nm and 300 nm, respectively, which was attributed to the increased hydrophobicity and collapse of the PDEGMA blocks. It was demonstrated that the glycopolymer vesicles were able to accommodate the protein lecitin at their surfaces. Further studies involved carbohydrate interaction of the vesicles with a mutant E. coli strain that is both green fluorescent and expresses the fimH protein, which has binding specificity for glucose and mannose. The smaller vesicles formed large aggregates with the bacteria, the clusters comprising about 100–150 bacteria and 60–90 vesicles; the larger vesicles were bound to E. coli but did not form clusters. Hence, the size of vesicles controls the vesicle-cell interactions from bulk aggregation to individual associations. Furthermore, it was demonstrated that a molecular “information” load, that is a fluorescent dye, could be specifically transferred from the vesicle membrane to the cytoplasm of the bacteria (Figure 12). The vesicles can thus be considered to represent primitive mimics of natural cells with their associated glycocalix, with potential applications in cell sensing, therapeutics, and synthetic biology.
Hydrophilic polysaccharides can be converted to amphiphilic polymers by conjugation of hydrophobic side chains to the sugar moieties, as described, for example, by Mély and Babak et al. [90] and Li et al. [91]. In neutral water N,N-dialkyl chitosans (alkyl = octyl, decyl, and dodecyl) formed stable bilayered vesicles having hydrodynamic diameters in the range of 100–200 nm as determined by dynamic light scattering [91]. The size of vesicles was found to increase with the increasing molecular weight of the hydrophilic chitosan backbone and/or the increasing length of the hydrophobic alkyl side chains. This effect was attributed to a more compact structure of the membrane. The drug (vitamin B12) release behavior exhibited by N,N-dialkyl chitosan vesicles are characterized by biphasic drug release kinetics, including an initial burst phase and a following plateau phase. Studies on specific interactions of the polysaccharide surface have not been reported.
Mishra and Lecommandoux et al. synthesized poly(γ-benzyl L-glutamate)-block-hyaluronan (PBLG-b-HYA) by “click” coupling of PBLG-azide (Mn ∼ 5 kDa) and HYA-alkyne (Mw ∼ 5 kDa), which self-assembled into polymersomes in dilute aqueous solution [22]. The intracellular delivery of doxorubicin loaded polymersomes (PolyDOX) was investigated in high (MCF-7) and low (U87) CD44 expressing cancer cell models. HYA is a major ligand for CD44 and can be used to target breast cancer cells having over-expressed CD44 glycoproteins. In fact, flow cytometry data suggested successful uptake of PolyDOX in cells and a higher accumulation in MCF-7 than U87 cells. PolyDOX significantly enhanced reactive oxygen species level in both cells. PolyDOX suppressed growth of breast tumor on female Sprague-Dawley rats as compared to phosphate buffer saline pH 7.4 (PBS) control group and exhibited lesser cardiotoxicity.
Polypeptides and their larger equivalents—fully folded proteins or enzymes—are macromolecules that expose hydrophilic functional groups towards an aqueous medium. When these biomolecules are linked to hydrophobic polymers, amphiphilic molecules that can assemble into vesicular structures are obtained. Block copolymers comprising a polypeptide block, and the polymersomes that result, have been extensively reviewed elsewhere [77,92,93]. Simple polypeptides (glutamate, lysine, etc.) can be considered as polyelectrolytes without special biological or other specific functions. Functionality is absent in the corona of such vesicles and therefore they are beyond the scope of this review.
Functional enzymes can form the surfaces of vesicles if the protein is incorporated in giant amphiphiles as macromolecular hydrophilic head groups of hydrophobic polymers. Thus, these vesicles possess a functional surface. Nolte and co-workers introduced a route to giant amphiphiles by cofactor reconstitution (Figure 13(a)). A synthetic polymer, PS-b-PEG, was first functionalized with a heme cofactor, protoporphyrin IX, and subsequently reconstituted with an apoprotein, myoglobin (Mb) or HRP [94,95]. The PS-b-PEG precursor formed small spherical micelles in water; the solution was prepared by injection of the polymer in THF into water. After reconstitution with the hydrophilic apo-HRP, the triblock biohybrid sample PEG-b-PS-b-HRP assembled into submicron vesicles with broad size distribution (Figure 13(b)). The hydrophobic membrane is built by the polystyrene middle block and is stabilized by PEG chains and protein. Whether the membrane has a mono- or a bilayered structure, and whether the PEG and HRP are segregated or mixed in the corona, is not yet known. The enzyme-polymer hybrid vesicles were further investigated according to their enzymatic activity [95,96]. The enzymatic activity decreased for HRP and the stability of the oxy complex was reduced for Mb. This may be explained by the introduction of a polystyrene chain, which seems to result in a somewhat modified binding of the heme in the reconstituted protein. Several factors could be responsible for this behavior, such as an unfavorable interaction of the protein with the PS chain or a disturbance of the three-dimensional structure of the protein, due to the aggregation of the hybrid molecules. In addition, both proteins have their substrate access channels located near the site of attachment of the polystyrene chains, which means that these channels might be partly shielded from the aqueous solution. However, the residual activity also might have arisen from smaller (micellar) aggregates having a more dynamic structure than the larger vesicles.
Another route to giant amphiphiles is to graft a hydrophobic polymer chain from a protein. Such amphiphiles were realized by Le Droumaguet and Velonia [97]. They initiated the ATRP of styrene with an ATRP-initiator conjugated to serum albumins and reduced human calcitonin. The resulting amphiphiles spontaneously formed vesicles with diameters between 20 and 100 nm. The giant amphiphiles were able to encapsulate active enzymes, so that nanoreactors were formed. However, a specific interaction of the protein corona with other biomolecules was not investigated.
Vesicles with an oligonucleotide corona have been produced by the self-assembly of block copolymers with a hydrophobic PBD block and a hydrophilic oligonucleotide block [98,99]. The diblock copolymers were synthesized by linking an amine-terminated PBD to a carboxy-modified, 12-mer oligonucleotide sequence on the solid support on which the oligonucleotide had been synthesized. After cleavage from the solid support and purification, the block copolymer formed vesicles in dilute aqueous solution. These vesicles were stabilized further by photo-crosslinking the PBD blocks [99]. The oligonucleotide shell of such vesicles was able to bind to complementary oligonucleotide sequences on a glass substrate, thus allowing immobilization of the vesicles. Furthermore, the oligonucleotide sequences on the substrate and/or on the vesicles was recognized by microorganisms, as shown by enhanced expression of bacterial organelles of adhesion (curli) in E. coli cells during biofilm growth [99]. This is the first example of oligonucleotide-coated vesicles being recognized by microorganisms and cells. Such vesicles potentially influence cell-signaling, internalization and bacterial adhesion. The surface of vesicles might be further modified with ligands through the binding of a complementary oligonucleotide sequence.
2.4. Reconstitution of Membrane Proteins in Polymersomes
A very specific way to introduce recognition sites on vesicle surfaces is the reconstitution of membrane proteins in amphiphilic block copolymer membranes during the self-assembly process. Meier and co-workers pioneered the field of membrane-protein equipped polymersomes. They created polymer vesicles with defined, tunable permeability that resulted from the presence of channel proteins and porins in the membrane [5,10,13,23,100,101]. However, channel proteins provide not only pores, but can also act as binding sites for viral phages through their surface-exposed regions. Indeed, LamB channel proteins reconstituted in polymersomes from PMOXA-b-PDMS-b-PMOXA triblock copolymers were recognized by bacteriophage λ [7]. The phage underwent binding to the poylmersome and injected its DNA into the vesicle (Figure 14). This work clearly showed that membrane proteins incorporated in polymersomal membranes can provide specific binding sites on the polymersome surface. However, the concept has not yet been applied to other examples.
3. Conclusions
Cells and cellular compartments interact with their environment through a multitude of functional moieties that include antigens and receptors present on their surfaces. These functional ligands are often anchored within the lipid bilayer membrane that forms the boundary shell of cells and organelles.
In the search for synthetic analogs of vesicles, organelles, and even cells, structures formed by amphiphilic block copolymers have attained a great deal of attention and research activity. Amphiphilic block copolymers can self-assemble into membranes comprising a central hydrophobic layer between two hydrophilic leaflets. This structural similarity to lipid-based membranes makes them mimics of natural membranes. Moreover, block copolymer membranes tend to form vesicles, i.e., closed nano-to micro-sized compartments that engulf a central pool of water. Such polymersomes are more stable than their lipid-based counterparts and thus show promising applications in biomedical settings such as drug-delivery. However, the surface of a conventional polymer vesicle does not display any functional moieties that could orchestrate interaction with the environment. Therefore, a variety of methods have been developed to functionalize the surface of polymersomes with active ligands, binding sites, or signaling residues. These methods can either target fully assembled polymer vesicles by non-covalent or covalent conjugation, or the polymers that form vesicles can be designed to encompass functional groups prior to vesicle formation. Both strategies have proven successful in yielding highly functionalized polymersomes. Thus, a new generation of multifunctional polymer vesicles is emerging, capable of being designed to specifically bind, adhere to, or target surfaces, cell lines, or biomolecules while still retaining the ability to encapsulate therapeutic and diagnostic cargo, or active (bio)catalysts.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymersome functional group | Ligand / substrate functional group | Resulting bond | Block copolymer | Ligand / substrate |
|---|---|---|---|---|
| Alkyne | Azide | 1,4-disubstituted 1,2,3-triazole | PS40-b-PIAT50, or mixture of PS40-b-PIAT50 with PS40-b-PEG68 [26,29] | Horseradish peroxidase [26] and an azidofunctionalized Candida antarctica Lipase B [29] |
| Azide | Alkyne | 1,4-disubstituted 1,2,3-triazole | PBD-b-PEG [30,31], PS-b-PAA [32] | Mannose functional polyester dendron [30,31], dansyl probe [32], biotin [32], eGFP [32] |
| Biotin | Streptavidin | Biotin-Streptavidin interaction | PBD-b-PEG and PEG-b-PEE [33],PMOXA-b-PDMS-b-PMOXA [17,20,34,35] | Superavidin-coated microspheres [33], antibody [36], polyguanylic acid [20,34], streptavidin-modified glass surface [17], fluorescently labeled avidin [33,35] |
| Ni2+-nitrilotriacetic acid (Ni2+-NTA) | Oligohistidine sequence | Complexation | PDB-b-PEG [37,38] | Fluorescein labeled maltose binding protein [37],eGFP [37,38], RFP [38] |
| Cyclodextrin (CD) | Adamantane | Hydrophobic interactions | PS-CD [39], CD-PEI-CD [40] | HRP [39], PEG [40] |
| N-hydroxysuccinimidyl ester | Thiol | C-S bond | PCL-b-PEG [41] | Antibody OX26 [41] |
| Vinyl sulfone | Thiol | C-S bond | PEG-b-PMCL [42] | Cysteine-RGD peptide [42] |
| Amine | Succinimidyl ester | Amide | PDMS-b-PMOXA [43] | Alexa Fluor 633 succinimidyl ester [43] |
| Aldehyde | Amine | Imine | PCL-b-PEG, PLA-b-PEG and PI-b-PEG [19] | Aminated glass surface |
| 4-Formyl benzoic amide | 6-Hydrazinonicotinic | amide Bis-arylhydrazone | PDMS-b-PMOXA [43] | eYFP, anti-biotin-IgG, trastuzumab [43] |
Enhanced green fluorescent protein (eGFP), enhanced yellow fluorescent protein (eYFP), horseradish peroxidase (HRP), immunoglobulin G (IgG), poly(butadiene) (PBD), poly(caprolactone) (PCL), poly(dimethylsiloxane) (PDMS), poly(ethylethylene) (PEE), poly(ethylene glycol) (PEG), poly(ether imide) (PEI), poly(l-isocyanoalanine(2-thiophen-3-yl-ethyl)amide) (PIAT), poly(isoprene) (PI), poly(lactide) (PLA), poly(γ-methyl-ε-caprolactone) (PMCL), poly(2-methyl-2-oxazoline) (PMOXA), polystyrene (PS), red fluorescent protein (RFP).
Acknowledgments
This work was financially supported by the European Science Foundation through their collaborative research project Biofunctional Self-Organized Nanostructures of ionic/non-ionic amphiphilic copolymers, biopolymers-biomacromoleculres and nanoparticles (BIOSONS), by the Swiss National Science Foundation, and the NCCR Nanosciences. Nico Bruns gratefully acknowledges the Marie Curie Actions of the European Commission for an Intra-European Fellowship. We thank Mark Inglin for editing the manuscript.
References
- Singer, S.J.; Nicolson, G.L. Fluid mosaic model of the structure of cell membranes. Science 1972, 175, 720–731. [Google Scholar]
- Kita-Tokarczyk, K.; Grumelard, J.; Haefele, T.; Meier, W. Block copolymer vesicles-using concepts from polymer chemistry to mimic biomembranes. Polymer 2005, 46, 3540–3563. [Google Scholar]
- Antonietti, M.; Förster, S. Vesicles and liposomes: A self-assembly principle beyond lipids. Adv. Mater. 2003, 15, 1323–1333. [Google Scholar]
- Discher, D.E.; Eisenberg, A. Materials science: Soft surfaces: Polymer vesicles. Science 2002, 297, 967–973. [Google Scholar]
- Meier, W.; Nardin, C.; Winterhalter, M. Reconstitution of channel proteins in (polymerized) ABA triblock copolymer membranes. Angew. Chem. Int. Ed. 2000, 39, 4599–4602. [Google Scholar]
- Luo, L.; Eisenberg, A. Thermodynamic size control of block copolymer vesicles in solution. Langmuir 2001, 17, 6804–6811. [Google Scholar]
- Graff, A.; Sauer, M.; Van Gelder, P.; Meier, W. Virus-assisted loading of polymer nanocontainer. Proc. Natl. Acad. Sci. USA 2002, 99, 5064–5068. [Google Scholar]
- Ahmed, F.; Pakunlu, R.I.; Brannan, A.; Bates, F.; Minko, T.; Discher, D.E. Biodegradable polymersomes loaded with both paclitaxel and doxorubicin permeate and shrink tumors, inducing apoptosis in proportion to accumulated drug. J. Control. Release 2006, 116, 150–158. [Google Scholar]
- Meng, F.; Zhong, Z.; Feijen, J. Stimuli-responsive polymersomes for programmed drug delivery. Biomacromolecules 2009, 10, 197–209. [Google Scholar]
- Onaca, O.; Enea, R.; Hughes, D.W.; Meier, W. Stimuli-responsive polymersomes as nanocarriers for drug and gene delivery. Macromol. Biosci. 2009, 9, 129–139. [Google Scholar]
- Li, M.-H.; Keller, P. Stimuli-responsive polymer vesicles. Soft Matter 2009, 5, 927–937. [Google Scholar]
- Du, J.; O'Reilly, R.K. Advances and challenges in smart and functional polymer vesicles. Soft Matter 2009, 5, 3544–3561. [Google Scholar]
- Renggli, K.; Baumann, P.; Langowska, K.; Onaca, O.; Bruns, N.; Meier, W. Selective and responsive nanoreactors. Adv. Funct. Mater. 2010. in press. [Google Scholar]
- Boerner, H.G.; Schlaad, H. Bioinspired functional block copolymers. Soft Matter 2007, 3, 394–408. [Google Scholar]
- Duncan, R. The dawning era of polymer therapeutics. Nat. Rev. Drug Discovery 2003, 2, 347–360. [Google Scholar]
- McCarron, P.A.; Marouf, W.M.; Donnelly, R.F.; Scott, C. Enhanced surface attachment of protein-type targeting ligands to poly(lactide-co-glycolide) nanoparticles using variable expression of polymeric acid functionality. J. Biomed. Mater. Res. A 2008, 87A, 873–884. [Google Scholar]
- Grzelakowski, M.; Onaca, O.; Rigler, P.; Kumar, M.; Meier, W. Immobilized Protein-polymer nanoreactors. Small 2009, 5, 2545–2548. [Google Scholar]
- Li, F.; Ketelaar, T.; Cohen Stuart, M.A.; Sudhoelter, E.J.R.; Leermakers, F.A.M.; Marcelis, A.T.M. Gentle immobilization of nonionic polymersomes on solid substrates. Langmuir 2008, 24, 76–82. [Google Scholar]
- Domes, S.; Filiz, V.; Nitsche, J.; Fromsdorf, A.; Forster, S. Covalent attachment of polymersomes to surfaces. Langmuir 2010, 26, 6927–6931. [Google Scholar]
- Broz, P.; Benito, S.M.; Saw, C.; Burger, P.; Heider, H.; Pfisterer, M.; Marsch, S.; Meier, W.; Hunziker, P. Cell targeting by a generic receptor-targeted polymer nanocontainer platform. J. Control. Release 2005, 102, 475–488. [Google Scholar]
- Uchegbu, I.F. Pharmaceutical nanotechnology: Polymeric vesicles for drug and gene delivery. Expert Opin. Drug Delivery 2006, 3, 629–640. [Google Scholar]
- Upadhyay, K.K.; Bhatt, A.N.; Mishra, A.K.; Dwarakanath, B.S.; Jain, S.; Schatz, C.; Le Meins, J.-F.; Farooque, A.; Chandraiah, G.; Jain, A.K.; Misra, A.; Lecommandoux, S. The intracellular drug delivery and anti tumor activity of doxorubicin loaded poly(gamma -benzyl -glutamate)-b-hyaluronan polymersomes. Biomaterials 2010, 31, 2882–2892. [Google Scholar]
- Nardin, C.; Thoeni, S.; Widmer, J.; Winterhalter, M.; Meier, W. Nanoreactors based on (polymerized) ABA-triblock copolymer vesicles. Chem. Commun. 2000, 36, 1433–1434. [Google Scholar]
- Axthelm, F.; Casse, O.; Koppenol, W.H.; Nauser, T.; Meier, W.; Palivan, C.G. Antioxidant nanoreactor based on superoxide dismutase encapsulated in superoxide-permeable vesicles. J. Phys. Chem. B 2008, 112, 8211–8217. [Google Scholar]
- De Vocht, C.; Ranquin, A.; Willaert, R.; Van Ginderachter, J.A.; Vanhaecke, T.; Rogiers, V.; Versees, W.; van Gelder, P.; Steyaert, J. Assessment of stability, toxicity and immunogenicity of new polymeric nanoreactors for use in enzyme replacement therapy of mitochondrial neurogastrointestinal encephalomyopathy. J. Control. Release 2009, 137, 246–254. [Google Scholar]
- van Dongen, S.F.M.; Nallani, M.; Cornelissen, J.J.L.M.; Nolte, R.J.M.; van Hest, J.C.M. A three-enzyme cascade reaction through positional assembly of enzymes in a polymersome nanoreactor. Chem.-Eur. J. 2009, 15, 1107–1114. [Google Scholar]
- Onaca, O.; Hughes, D.W.; Balasubramanian, V.; Grzelakowski, M.; Meier, W.; Palivan, C.G. SOD Antioxidant nanoreactors: Influence of Block copolymer composition on the nanoreactor efficiency. Macromol. Biosci. 2010, 10, 531–538. [Google Scholar]
- Broz, P.; Driamov, S.; Ziegler, J.; Ben-Haim, N.; Marsch, S.; Meier, W.; Hunziker, P. Toward intelligent nanosize bioreactors: A ph-switchable, channel-equipped, functional polymer nanocontainer. Nano Lett. 2006, 6, 2349–2353. [Google Scholar]
- van Dongen, S.F.M.; Nallani, M.; Schoffelen, S.; Cornelissen, J.J.L.M.; Nolte, R.J.M.; van Hest, J.C.M. A block copolymer for functionalisation of polymersome surfaces. Macromol. Rapid Commun. 2008, 29, 321–325. [Google Scholar]
- Li, B.; Martin, A.L.; Gillies, E.R. Multivalent polymer vesicles via surface functionalization. Chem. Commun. 2007, 43, 5217–5219. [Google Scholar]
- Martin, A.L.; Li, B.; Gillies, E.R. Surface functionalization of nanomaterials with dendritic groups: Toward enhanced binding to biological targets. J. Am. Chem. Soc. 2009, 131, 734–741. [Google Scholar]
- Opsteen, J.A.; Brinkhuis, R.P.; Teeuwen, R.L.M.; Loewik, D.W.P.M.; van Hest, J.C.M. “Clickable” polymersomes. Chem. Commun. 2007, 43, 3136–3138. [Google Scholar]
- Lin, J.J.; Silas, J.A.; Bermudez, H.; Milam, V.T.; Bates, F.S.; Hammer, D.A. The effect of polymer chain length and surface density on the adhesiveness of functionalized polymersomes. Langmuir 2004, 20, 5493–5500. [Google Scholar]
- Broz, P.; Ben-Haim, N.; Grzelakowski, M.; Marsch, S.; Meier, W.; Hunziker, P. Inhibition of macrophage phagocytotic activity by a receptor-targeted polymer vesicle-based drug delivery formulation of pravastatin. J. Cardiovasc. Pharmacol. 2008, 51, 246–252. [Google Scholar]
- Rigler, P.; Meier, W. Encapsulation of fluorescent molecules by functionalized polymeric nanocontainers: Investigation by confocal fluorescence imaging and fluorescence correlation spectroscopy. J. Am. Chem. Soc. 2006, 128, 367–373. [Google Scholar]
- Lin, J.J.; Ghoroghchian, P.P.; Zhang, Y.; Hammer, D.A. Adhesion of antibody-functionalized polymersomes. Langmuir 2006, 22, 3975–3979. [Google Scholar]
- Nehring, R.; Palivan, C.G.; Casse, O.; Tanner, P.; Tuxen, J.; Meier, W. Amphiphilic diblock copolymers for molecular recognition: Metal-nitrilotriacetic acid functionalized vesicles. Langmuir 2009, 25, 1122–1130. [Google Scholar]
- Nehring, R.; Palivan, C.G.; Moreno-Flores, S.; Mantion, A.; Tanner, P.; Toca-Herrera, J.L.; Thuenemann, A.; Meier, W. Protein decorated membranes by specific molecular interactions. Soft Matter 2010, 6, 2815–2824. [Google Scholar]
- Felici, M.; Marza-Perez, M.; Hatzakis, N.S.; Nolte, R.J.M.; Feiters, M.C. P-Cyclodextrin-appended giant amphiphile: Aggregation to vesicle polymersomes and immobilisation of enzymes. Chem. Eur. J. 2008, 14, 9914–9920. [Google Scholar]
- Guo, M.; Jiang, M.; Zhang, G. Surface modification of polymeric vesicles via host-guest inclusion complexation. Langmuir 2008, 24, 10583–10586. [Google Scholar]
- Pang, Z.; Lu, W.; Gao, H.; Hu, K.; Chen, J.; Zhang, C.; Gao, X.; Jiang, X.; Zhu, C. Preparation and brain delivery property of biodegradable polymersomes conjugated with OX26. J. Control. Release 2008, 128, 120–127. [Google Scholar]
- Petersen, M.A.; Yin, L.; Kokkoli, E.; Hillmyer, M.A. Synthesis and characterization of reactive PEO-PMCL polymersomes. Polym. Chem. 2010, 1, 1281–1290. [Google Scholar]
- Egli, S.; Nussbaumer, M.G.; Balasubramanian, V.; Chami, M.; Bruns, N.; Palivan, C.G.; Meier, W. Biocompatible functionalization of polymersome surfaces: New approach for surface immobilization and cell targeting using polymersomes. J. Am. Chem. Soc. 2010. submitted. [Google Scholar]
- Nilsson, K.; Mosbach, K. Immobilization of ligands with organic sulfonyl chlorides. Methods Enzymol. 1984, 104, 56–69. [Google Scholar]
- Hammer, D.A.; Robbins, G.P.; Haun, J.B.; Lin, J.J.; Qi, W.; Smith, L.A.; Peter Ghoroghchian, P.; Therien, M.J.; Bates, F.S. Leuko-polymersomes. Faraday Discuss. 2008, 139, 129–141. [Google Scholar]
- Weber, P.C.; Ohlendorf, D.H.; Wendoloski, J.J.; Salemme, F.R. Structural origins of high-affinity biotin binding to streptavidin. Science 1989, 243, 85–88. [Google Scholar]
- Holmberg, A.; Blomstergren, A.; Nord, O.; Lukacs, M.; Lundeberg, J.; Uhlen, M. The biotin-streptavidin interaction can be reversibly broken using water at elevated temperatures. Electrophoresis 2005, 26, 501–510. [Google Scholar]
- Lin, J.J.; Bates, F.S.; Hammer, D.A.; Silas, J.A. Adhesion of polymer vesicles. Phys. Rev. Lett. 2005, 95, 026101. [Google Scholar]
- Robbins, G.P.; Saunders, R.L.; Haun, J.B.; Rawson, J.; Therien, M.J.; Hammer, D.A. Tunable Leuko-polymersomes That Adhere Specifically to Inflammatory Markers. Langmuir 2010, 26, 14089–14096. [Google Scholar]
- Alon, R.; Hershkoviz, R.; Bayer, E.A.; Wilchek, M.; Lider, O. Streptavidin blocks immune reactions mediated by fibronectin-VLA-5 recognition through an Arg-Gly-Asp mimicking site. Eur. J. Immunol. 1993, 23, 893–898. [Google Scholar]
- Dorn, I.T.; Neumaier, K.R.; Tampe, R. Molecular recognition of histidine-tagged molecules by metal-chelating lipids monitored by fluorescence energy transfer and correlation spectroscopy. J. Am. Chem. Soc. 1998, 120, 2753–2763. [Google Scholar]
- Rahimi, Y.; Shrestha, S.; Banerjee, T.; Deo, S.K. Copper sensing based on the far-red fluorescent protein, HcRed, from Heteractis crispa. Anal. Biochem. 2007, 370, 60–67. [Google Scholar]
- Kornberg, R.D.; Darst, S.A. Two-dimensional crystals of proteins on lipid layers. Curr. Opin. Struct. Biol. 1991, 1, 642–646. [Google Scholar]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar]
- Binder, W.H.; Sachsenhofer, R. ‘Click’ chemistry in polymer and materials science. Macromol. Rapid Commun. 2007, 28, 15–54. [Google Scholar]
- Evans, C.E.; Lovell, P.A. Click chemistry as a route to surface functionalization of polymer particles dispersed in aqueous media. Chem. Commun. 2009, 45, 2305–2307. [Google Scholar]
- Goddard-Borger, E.D.; Stick, R.V. An efficient, inexpensive, and shelf-stable diazotransfer reagent: Imidazole-1-sulfonyl Azide hydrochloride. Org. Lett. 2007, 9, 3797–3800. [Google Scholar]
- van Dongen, S.F.M.; Teeuwen, R.L.M.; Nallani, M.; van Berkel, S.S.; Cornelissen, J.J.L.M.; Nolte, R.J.M.; van Hest, J.C.M. Single-step azide introduction in proteins via an aqueous diazo transfer. Bioconjugate Chem. 2009, 20, 20–23. [Google Scholar]
- Ghosh, S.S.; Kao, P.M.; McCue, A.W.; Chappelle, H.L. Use of maleimide-thiol coupling chemistry for efficient syntheses of oligonucleotide-enzyme conjugate hybridization probes. Bioconjugate Chem. 1990, 1, 71–76. [Google Scholar]
- Velonia, K.; Rowan, A.E.; Nolte, R.J.M. Lipase polystyrene giant amphiphiles. J. Am. Chem. Soc. 2002, 124, 4224–4225. [Google Scholar]
- Xu, H.; Kaar, J.L.; Russell, A.J.; Wagner, W.R. Characterizing the modification of surface proteins with poly(ethylene glycol) to interrupt platelet adhesion. Biomaterials 2006, 27, 3125–3135. [Google Scholar]
- Hermanson, G.T. Bioconjugate Techniques, 2nd ed.; Academic Press: London, UK, 2008. [Google Scholar]
- Kozlov, I.A.; Melnyk, P.C.; Stromsborg, K.E.; Chee, M.S.; Barker, D.L.; Zhao, C. Efficient strategies for the conjugation of oligonucleotides to antibodies enabling highly sensitive protein detection. Biopolymers 2004, 73, 621–630. [Google Scholar]
- Bruns, N.; Pustelny, K.; Bergeron, L.M.; Whitehead, T.A.; Clark, D.S. Mechanical nanosensor based on fret within a thermosome: Damage-reporting polymeric materials. Angew. Chem., Int. Ed. 2009, 48, 5666–5669. [Google Scholar]
- Kim, B.-S.; Yang, W.-Y.; Ryu, J.-H.; Yoo, Y.-S.; Lee, M. Carbohydrate-coated nanocapsules from amphiphilic rod-coil molecule: Binding to bacterial type 1 pili. Chem. Commun. 2005, 41, 2035–2037. [Google Scholar]
- Kim, B.-S.; Hong, D.-J.; Bae, J.; Lee, M. Controlled self-assembly of carbohydrate conjugate rod-coil amphiphiles for supramolecular multivalent ligands. J. Am. Chem. Soc. 2005, 127, 16333–16337. [Google Scholar]
- Toyotama, A.; Kugimiya, S.-I.; Yamanaka, J.; Yonese, M. Preparation a novel aggregate like sugar-ball micelle composed of poly(methylglutamate) and poly(ethyleneglycol) modified by lactose and its molecular recognition by lectin. Chem. Pharm. Bull. 2001, 49, 169–172. [Google Scholar]
- Yang, X.; Grailer, J.J.; Rowland, I.J.; Javadi, A.; Hurley, S.A.; Matson, V.Z.; Steeber, D.A.; Gong, S. Multifunctional stable and ph-responsive polymer vesicles formed by heterofunctional triblock copolymer for targeted anticancer drug delivery and ultrasensitive MR imaging. ACS Nano 2010, 4, 6805–6817. [Google Scholar]
- Yang, X.; Grailer, J.J.; Rowland, I.J.; Javadi, A.; Hurley, S.A.; Steeber, D.A.; Gong, S. Multifunctional SPIO/DOX-loaded wormlike polymer vesicles for cancer therapy and MR imaging. Biomaterials 2010, 31, 9065–9073. [Google Scholar]
- van Dongen, S.F.M.; Verdurmen, W.P.R.; Peters, R.J.R.W.; Nolte, R.J.M.; Brock, R.; van Hest, J.C.M. Cellular integration of an enzyme-loaded polymersome nanoreactor. Angew. Chem. Int. Ed. 2010, 49, 7213–7216. [Google Scholar]
- Christian, N.A.; Milone, M.C.; Ranka, S.S.; Li, G.; Frail, P.R.; Davis, K.P.; Bates, F.S.; Therien, M.J.; Ghoroghchian, P.P.; June, C.H.; Hammer, D.A. Tat-functionalized near-infrared emissive polymersomes for dendritic cell labeling. Bioconjugate Chem. 2007, 18, 31–40. [Google Scholar]
- Demirgoz, D.; Pangburn, T.O.; Davis, K.P.; Lee, S.; Bates, F.S.; Kokkoli, E. PR_b-targeted delivery of tumor necrosis factor-α by polymersomes for the treatment of prostate cancer. Soft Matter 2009, 5, 2011–2019. [Google Scholar]
- Chang, Y.-W.; Silas, J.A.; Ugaz, V.M. A direct probe of the interplay between bilayer morphology and surface reactivity in polymersomes. Langmuir 2010, 26, 12132–12139. [Google Scholar]
- Rajagopal, K.; Christian, D.A.; Harada, T.; Tian, A.; Discher, D.E. Polymersomes and wormlike micelles made fluorescent by direct modifications of block copolymer amphiphiles. Int. J. Polym. Sci. 2010. [Google Scholar] [CrossRef]
- Marsden, H.R.; Handgraaf, J.-W.; Nudelman, F.; Sommerdijk, N.A.J.M.; Kros, A. Uniting polypeptides with sequence-designed peptides: Synthesis and assembly of poly(γ-benzyl l-glutamate)-b-coiled-coil peptide copolymers. J. Am. Chem. Soc. 2010, 132, 2370–2377. [Google Scholar]
- Schatz, C.; Lecommandoux, S. Polysaccharide-containing block copolymers: Synthesis, properties and applications of an emerging family of glycoconjugates. Macromol. Rapid Commun. 2010, 31, 1664–1684. [Google Scholar]
- van Dongen, S.F.M.; de Hoog, H.P.M.; Peters, R.; Nallani, M.; Nolte, R.J.M.; van Hest, J.C.M. Biohybrid polymer capsules. Chem. Rev. 2009, 109, 6212–6274. [Google Scholar]
- Wu, A.; Lisowska, E.; Duk, M.; Yang, Z. Lectins as tools in glycoconjugate research. Glycoconjugate J. 2009, 26, 899–913. [Google Scholar]
- Hordyjewicz-Baran, Z.; You, L.; Smarsly, B.; Sigel, R.; Schlaad, H. Bioinspired polymer vesicles based on hydrophilically modified polybutadienes. Macromolecules 2007, 40, 3901–3903. [Google Scholar]
- ten Brummelhuis, N.; Diehl, C.; Schlaad, H. Thiol-ene modification of 1,2-polybutadiene using UV light or sunlight. Macromolecules 2008, 41, 9946–9947. [Google Scholar]
- You, L.; Schlaad, H. An easy way to sugar-containing polymer vesicles or glycosomes. J. Am. Chem. Soc. 2006, 128, 13336–13337. [Google Scholar]
- Schlaad, H.; You, L.; Sigel, R.; Smarsly, B.; Heydenreich, M.; Mantion, A.; Mašić, A. Glycopolymer vesicles with an asymmetric membrane. Chem. Commun. 2009, 45, 1478–1480. [Google Scholar]
- Gress, A.; Smarsly, B.; Schlaad, H. Formation of glycopolyamide nanofibers. Macromol. Rapid Commun. 2008, 29, 304–308. [Google Scholar]
- Lundquist, J.J.; Toone, E.J. The cluster glycoside effect. Chem. Rev. 2002, 102, 555–578. [Google Scholar]
- Li, Z.-C.; Liang, Y.-Z.; Li, F.-M. Multiple morphologies of aggregates from block copolymers containing glycopolymer segments. Chem. Commun. 1999, 35, 1557–1558. [Google Scholar]
- Liang, Y.-Z.; Li, Z.-C.; Li, F.-M. Multiple morphologies of molecular assemblies formed by polystyrene-block-poly[2-(β-D-glucopyranosyloxy)ethyl acrylate] in water. New J. Chem. 2000, 24, 323–328. [Google Scholar]
- Zhou, W.; Dai, X.-H.; Dong, C.-M. Biodegradable and biomimetic poly(ε-caprolactone)/ poly(lactobionamidoethyl methacrylate) biohybrids: Synthesis, lactose-installed nanoparticles and recognition properties. Macromol. Biosci. 2008, 8, 268–278. [Google Scholar]
- Dai, X.-H.; Dong, C.-M. Synthesis, self-assembly and recognition properties of biomimetic star-shaped poly(ε-caprolactone)-b-glycopolymer block copolymers. J. Polym. Sci. A Polym. Chem. 2008, 46, 817–829. [Google Scholar]
- Pasparakis, G.; Alexander, C. Sweet talking double hydrophilic block copolymer vesicles. Angew. Chem. Int. Ed. 2008, 47, 4847–4850. [Google Scholar]
- Ercelen, S.; Zhang, X.; Duportail, G.; Grandfils, C.; Desbrières, J.; Karaeva, S.; Tikhonov, V.; Mély, Y.; Babak, V. Physicochemical properties of low molecular weight alkylated chitosans: A new class of potential nonviral vectors for gene delivery. Colloids Surf. B 2006, 51, 140–148. [Google Scholar]
- Li, M.; Su, S.; Xin, M.; Liao, Y. Relationship between N,N-dialkyl chitosan monolayer and corresponding vesicle. J. Colloid Interf. Sci. 2007, 311, 285–288. [Google Scholar]
- Bertin, A.; Hermes, F.; Schlaad, H. Biohybrid and peptide-based polymer vesicles. Adv. Polym. Sci. 2010, 224, 167–195. [Google Scholar]
- Carlsen, A.; Lecommandoux, S. Self-assembly of polypeptide-based block copolymer amphiphiles. Curr. Opin. Colloid Interface Sci. 2009, 14, 329–339. [Google Scholar]
- Reynhout, I.C.; Cornelissen, J.J.L.M.; Nolte, R.J.M. Self-assembled architectures from biohybrid triblock copolymers. J. Am. Chem. Soc. 2007, 129, 2327–2332. [Google Scholar]
- Boerakker, M.J.; Botterhuis, N.E.; Bomans, P.H. H.; Frederik, P.M.; Meijer, E.M.; Nolte, R.J.M.; Sommerdijk, N.A.J.M. Aggregation behavior of giant amphiphiles prepared by cofactor reconstitution. Chem.-Eur. J. 2006, 12, 6071–6080. [Google Scholar]
- Boerakker, M.J.; Hannink, J.M.; Bomans, P.H. H.; Frederik, P.M.; Nolte, R.J.M.; Meijer, E.M.; Sommerdijk, N.A.J.M. Giant Amphiphiles by cofactor reconstitution. Angew. Chem. Int. Ed. 2002, 41, 4239–4241. [Google Scholar]
- Le Droumaguet, B.; Velonia, K. In situ ATRP-Mediated hierarchical formation of giant amphiphile bionanoreactors. Angew. Chem., Int. Ed. 2008, 47, 6263–6266. [Google Scholar]
- Teixeira, F.; Rigler, P.; Vebert-Nardin, C. Nucleo-copolymers: Oligonucleotide-based amphiphilic diblock copolymers. Chem. Commun. 2007, 43, 1130–1132. [Google Scholar]
- Cottenye, N.; Teixeira, F.; Ponche, A.; Reiter, G.; Anselme, K.; Meier, W.; Ploux, L.; Vebert-Nardin, C. Oligonucleotide Nanostructured surfaces: Effect on escherichia coli curli expression. Macromol. Biosci. 2008, 8, 1161–1172. [Google Scholar]
- Nardin, C.; Widmer, J.; Winterhalter, M.; Meier, W. Amphiphilic block copolymer nanocontainers as bioreactors. Eur. Phys. J. E Soft Matter Biol. Phys. 2001, 4, 403–410. [Google Scholar]
- Kumar, M.; Grzelakowski, M.; Zilles, J.; Clark, M.; Meier, W. Highly permeable polymeric membranes based on the incorporation of the functional water channel protein Aquaporin Z. Proc. Natl. Acad. Sci. USA 2007, 104, 20719–20724. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Egli, S.; Schlaad, H.; Bruns, N.; Meier, W. Functionalization of Block Copolymer Vesicle Surfaces. Polymers 2011, 3, 252-280. https://doi.org/10.3390/polym3010252
Egli S, Schlaad H, Bruns N, Meier W. Functionalization of Block Copolymer Vesicle Surfaces. Polymers. 2011; 3(1):252-280. https://doi.org/10.3390/polym3010252
Chicago/Turabian StyleEgli, Stefan, Helmut Schlaad, Nico Bruns, and Wolfgang Meier. 2011. "Functionalization of Block Copolymer Vesicle Surfaces" Polymers 3, no. 1: 252-280. https://doi.org/10.3390/polym3010252
APA StyleEgli, S., Schlaad, H., Bruns, N., & Meier, W. (2011). Functionalization of Block Copolymer Vesicle Surfaces. Polymers, 3(1), 252-280. https://doi.org/10.3390/polym3010252