The Challenge of Synthesizing Oligomers for Molecular Wires

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
3. Experimental Section
3.1. General
3.2. Synthesis
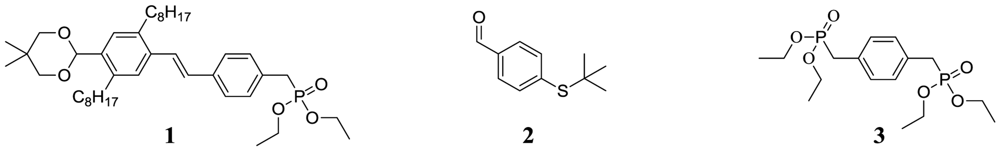
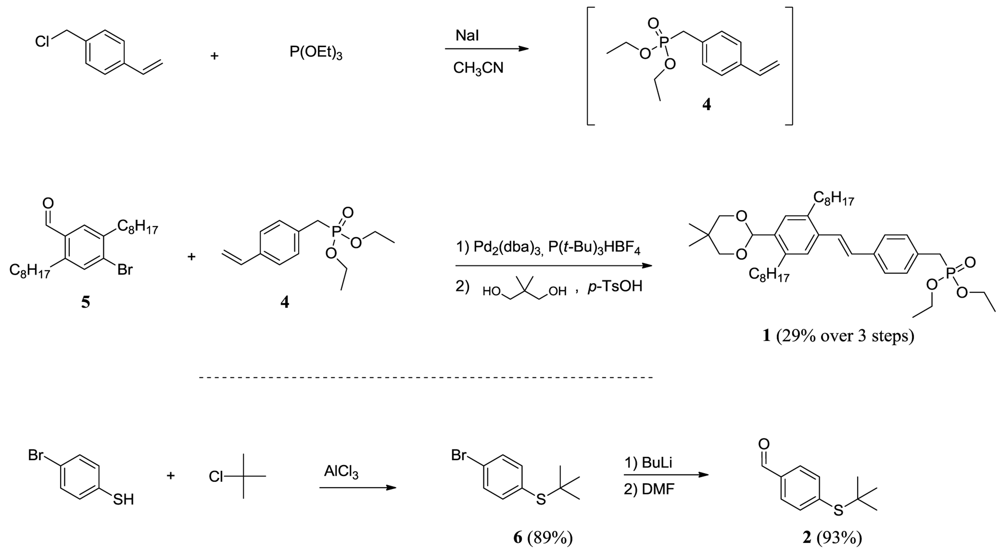
p-xylylene-bis-phosphonic acid tetraethyl ester (3)
4-bromo-2,5-dioctylbenzaldehyde (5)
Diethyl 4-vinylbenzylphosphonate (4)
(E)-diethyl4-(4-(5,5-dimethyl-1,3-dioxan-2-yl)-2,5-dioctylstyryl)benzylphosphonate (1)
(4-bromophenyl)tert-butylthioether (6)
4-(tert-butylthio)benzaldehyde (2)
4-((E)-4-((E)-4-(tert-butylthio)styryl)styryl)-2,5-dioctylbenzaldehyde (7)
4-((E)-4-((E)-4-((E)-4-((E)-4-(tert-butylthio)styryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylbenzaldehyde (8)
4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-(tert-butylthio)styryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylbenzaldehyde (9)
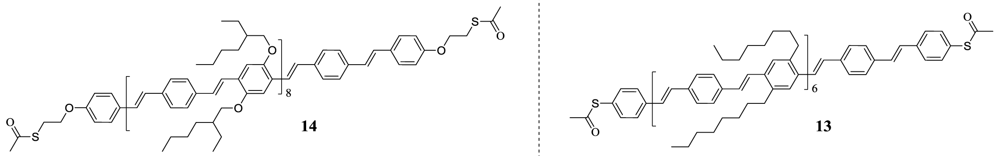
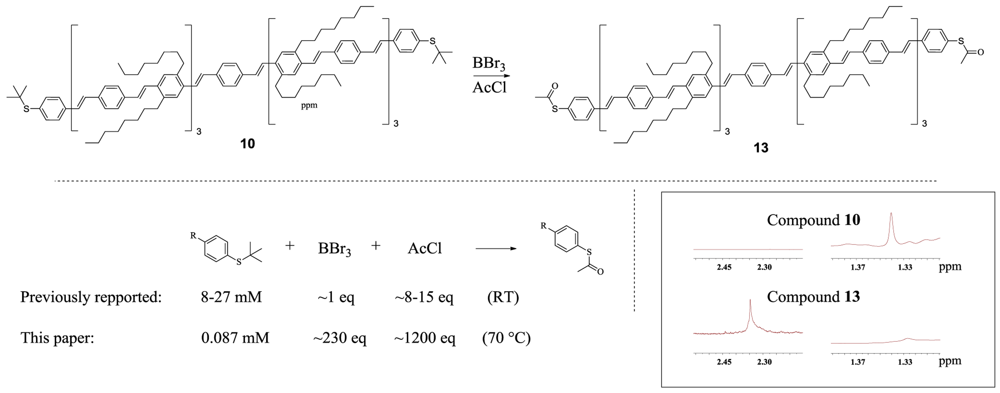
1,4-bis((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-(tert-butylthio)styryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylstyryl)benzene (10)
4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-(tert-butylthio)styryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylbenzaldehyde (11)
1,4-bis((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-(tert-butylthio)styryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylstyryl)benzene (12)
1,4-bis((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-((E)-4-(acetylthio)styryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylstyryl)styryl)-2,5-dioctylstyryl)benzene 13
4. Conclusions
Supplementary Material
polymers-03-00545-s001.pdf




Acknowledgments
Reference
- Ashwell, G.J.; Tyrrell, W.D.; Urasinska, B.; Wang, C.S.; Bryce, M.R. Organic rectifying junctions from an electron-accepting molecular wire and an electron-donating phthalocyanine. Chem. Comm. 2006. [Google Scholar] [CrossRef]
- Ashwell, G.J.; Urasinska, B.; Wang, C.S.; Bryce, M.R.; Grace, I.; Lambert, C.J. Single-molecule electrical studies on a 7 nm long molecular wire. Chem. Comm. 2006. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, B.; Frisbie, CD. Electrical resistance of long conjugated molecular wires. Science 2008, 320, 1482–1486. [Google Scholar]
- Choi, S.H.; Frisbie, CD. Enhanced hopping conductivity in low band gap donor-acceptor molecular wires up to 20 nm in length. J. Am. Chem. Soc. 2010, 132, 16191–16201. [Google Scholar]
- Danilov, A.; Kubatkin, S.; Kafanov, S.; Hedegard, P.; Stuhr-Hansen, N.; Moth-Poulsen, K.; Bjørnholm, T. Electronic transport in single molecule junctions: Control of the molecule-electrode coupling through intramolecular tunneling barriers. Nano Lett. 2008, 8, 1–5. [Google Scholar]
- Huang, W.; Masuda, G.; Maeda, S.; Tanaka, H.; Ogawa, T. Molecular junctions composed of oligothiophene dithiol-bridged gold nanoparticles exhibiting photoresponsive properties. Chem. Eur. J. 2006, 12, 607–619. [Google Scholar]
- Kubatkin, S.; Danilov, A.; Hjort, M.; Cornil, J.; Bredas, J.L.; Stuhr-Hansen, N.; Hedegard, P.; Bjørnholm, T. Single-electron transistor of a single organic molecule with access to several redox states. Nature 2003, 425, 698–701. [Google Scholar]
- Li, Z.H.; Pobelov, I.; Han, B.; Wandlowski, T.; Blaszczyk, A.; Mayor, M. Conductance of redox-active single molecular junctions: An electrochemical approach. Nanotechnology 2007, 18, 044018. [Google Scholar]
- Liang, T.T.; Naitoh, Y.; Horikawa, M.; Ishida, T.; Mizutani, W. Fabrication of steady junctions consisting of alpha,omega-bis(thioacetate) oligo(p-phenylene vinylene)s in nanogap electrodes. J. Am. Chem. Soc. 2006, 128, 13720–13726. [Google Scholar]
- Liang, Y.Y.; Wang, H.B.; Yuan, S.W.; Lee, Y.G.; Yu, L.P. Conjugated block copolymers and co-oligomers: From supramolecular assembly to molecular electronics. J. Mater. Chem. 2007, 17, 2183–2194. [Google Scholar]
- Moth-Poulsen, K.; Bjørnholm, T. Molecular electronics with single molecules in solid-state devices. Nat. Nanotechnol. 2009, 4, 551–556. [Google Scholar]
- Osorio, E.A.; O'Neill, K.; Wegewijs, M.; Stuhr-Hansen, N.; Paaske, J.; Bjørnholm, T.; van der Zant, H.S.J. Electronic excitations of a single molecule contacted in a three-terminal configuration. Nano Lett. 2007, 7, 3336–3342. [Google Scholar]
- Seferos, D.S.; Blum, A.S.; Kushmerick, J.G.; Bazan, G.C. Single-molecule charge-transport measurements that reveal technique-dependent perturbations. J. Am. Chem. Soc. 2006, 128, 11260–11267. [Google Scholar]
- van der Zant, H.S.J.; Kervennic, Y.V.; Poot, M.; O'Neill, K.; de Groot, Z.; Thijssen, J.M.; Heersche, H.B.; Stuhr-Hansen, N.; Bjørnholm, T.; Vanmaekelbergh, D.; van Walree, C.A.; Jenneskens, L.W. Molecular three-terminal devices: Fabrication and measurements. Faraday Discuss. 2006, 131, 347–356. [Google Scholar]
- Wang, C.S.; Batsanov, A.S.; Bryce, M.R. Nanoscale aryleneethynylene oligomers incorporating fluorenone units as electron-dopable molecular wires. Faraday Discuss. 2006, 131, 221–234. [Google Scholar]
- Søndergaard, R.; Strobel, S.; Bundgaard, E.; Norrman, K.; Hansen, A.G.; Albert, E.; Csaba, G.; Lugli, P.; Tornow, M.; Krebs, F.C. Conjugated 12 nm long oligomers as molecular wires in nanoelectronics. J. Mater. Chem. 2009, 19, 3899–3908. [Google Scholar]
- Alstrup, J.; Norrman, K.; Jørgensen, M.; Krebs, F.C. Lifetimes of organic photovoltaics: Design and synthesis of single oligomer molecules in order to study chemical degradation mechanisms. Sol. Energy Mater. Sol. Cells 2006, 90, 2777–2792. [Google Scholar]
- Hagemann, O.; Jørgensen, M.; Krebs, F.C. Synthesis of an all-in-one molecule (for organic solar cells). J. Org. Chem. 2006, 71, 5546–5559. [Google Scholar]
- Jørgensen, M.; Krebs, F.C. Stepwise and directional synthesis of end-functionalized single-oligomer OPVs and their application in organic solar cells. J. Org. Chem. 2004, 69, 6688–6696. [Google Scholar]
- Jørgensen, M.; Krebs, F.C. Stepwise unidirectional synthesis of oligo phenylene vinylenes with a series of monomers. Use in plastic solar cells. J. Org. Chem. 2005, 70, 6004–6017. [Google Scholar]
- Peeters, E.; van Hal, P.A.; Knol, J.; Brabec, C.J.; Sariciftci, N.S.; Hummelen, J.C.; Janssen, R.A.J. Synthesis, photophysical properties, and photovoltaic devices of oligo(p-phenylene vinylene)-fullerene dyads. J. Phys. Chem. B 2000, 104, 10174–10190. [Google Scholar]
- Sugiono, E.; Metzroth, T.; Detert, H. Practical synthesis of vinyl-substituted p-phenylenevinylene oligomers and their triethoxysilyl derivatives. Adv. Synth. Catal. 2001, 343, 351–359. [Google Scholar]
- Wong, M.S.; Li, Z.H. Multifunctional properties of monodisperse end-functionalized oligophenylenevinylenes. Pure Appl. Chem. 2004, 76, 1409–1419. [Google Scholar]
- Detert, H.; Sadovski, O. Triarylamines connected via phenylenevinylene segments. Synth. Met. 2003, 138, 185–188. [Google Scholar]
- Giacalone, F.; Segura, J.L.; Martin, N.; Ramey, J.; Guldi, D.M. Probing molecular wires: Synthesis, structural, and electronic study of donor-acceptor assemblies exhibiting long-range electron transfer. Chem. Eur. J. 2005, 11, 4819–4834. [Google Scholar]
- Stuhr-Hansen, N.; Christensen, J.B.; Harrit, N.; Bjørnholm, T. Novel synthesis of protected thiol end-capped stilbenes and oligo(phenylenevinylene)s (OPVs). J. Org. Chem. 2003, 68, 1275–1282. [Google Scholar]
- Krebs, F.C.; Jørgensen, M. High carrier mobility in a series of new semiconducting PPV-type polymers. Macromolecules 2003, 36, 4374–4384. [Google Scholar]
- Pedersen, D.S.; Rosenbohm, C. Dry column vacuum chromatography. Synthesis-Stuttgart 2001. [Google Scholar] [CrossRef]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Søndergaard, R.; Krebs, F.C. The Challenge of Synthesizing Oligomers for Molecular Wires. Polymers 2011, 3, 545-557. https://doi.org/10.3390/polym3010545
Søndergaard R, Krebs FC. The Challenge of Synthesizing Oligomers for Molecular Wires. Polymers. 2011; 3(1):545-557. https://doi.org/10.3390/polym3010545
Chicago/Turabian StyleSøndergaard, Roar, and Frederik C. Krebs. 2011. "The Challenge of Synthesizing Oligomers for Molecular Wires" Polymers 3, no. 1: 545-557. https://doi.org/10.3390/polym3010545
APA StyleSøndergaard, R., & Krebs, F. C. (2011). The Challenge of Synthesizing Oligomers for Molecular Wires. Polymers, 3(1), 545-557. https://doi.org/10.3390/polym3010545