Routes to Nanoparticle-Polymer Superlattices

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
: Nanoparticles can self-assemble into highly ordered two- and three-dimensional superlattices. For many practical applications these assemblies need to be integrated into polymeric matrices to provide stability and function. By appropriate co-assembly of nanoparticles and polymers it has become possible to tailor the nanoparticle superlattice structure via the length and stiffness of the polymer chains. The present article outlines and discusses established routes to nanoparticle-polymer superlattices. Recent progress has been remarkable so that the integration into functional devices has become the next challenge.1. Introduction
Nanoparticles can self-assemble into ordered superlattices leading to the formation of two-dimensional highly ordered monolayers [1] or three-dimensional microcrystals (Figure 1) [2,3]. For many applications the use of bare nanoparticle assemblies is, however, unsuitable. Although free-standing nanoparticle monolayers over dimensions of micrometers have been shown to be stable [4], the stability generally needs to be considerably improved. To provide stability and function, it would be highly desirable to integrate nanoparticle superlattices into polymeric matrices [5]. By appropriate assembly of nanoparticles and polymers, it should additionally be possible to tailor the superlattice structure by the length and stiffness of the polymer chains. Although distance control for nanoparticles with attached low molecular weight ligands is to some degree possible [6], polymers would make a far broader range of distances accessible.
The preparation of such ordered nanoparticle-polymer assemblies, however, represents a considerable challenge. The three most important issues are
to incorporate nanoparticles of sufficient quality and regularity,
to provide compatibility with the polymer matrix,
and to establish a superlattice of high q uality.
2. Routes to Polymer-Nanoparticle Superlattices
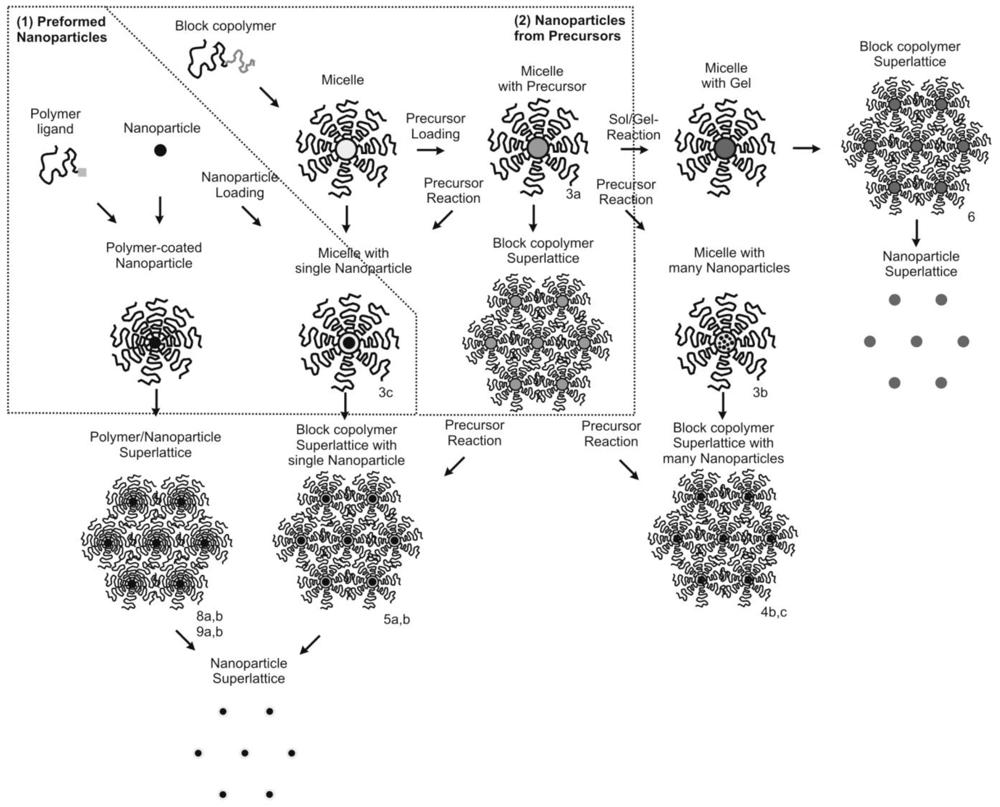
To solve, overcome or circumvent these issues, different routes to polymer nanocomposites have been devised, which are summarized in Figure 2.
Two major routes may be distinguished:
- (1)
The incorporation of preformed nanoparticles into polymeric matrices.
This route has the advantage that the best synthesis routes to high quality nanoparticles can be employed. Since methods such as hot-injection techniques have become available, the preparation of nanoparticles with narrow size distribution and high crystallinity has become straightforward, even on larger scales. These synthetic procedures allow one to prepare nanoparticles with different chemical compositions (metals, chalcogenides) and properties (catalytic, conducting, semi-conducting, fluorescent, magnetic). A major difficulty when incorporating these nanoparticles into homopolymer and block copolymer matrices is the incompatibility of the nanoparticles with the polymer matrix. In mixtures with polymers, the nanoparticles will generally aggregate into large clusters, which often deteriorate the properties of the nanoparticles and the nanocomposite. They prohibit the formation of ordered nanoparticle lattices, even if the size distribution of the primary nanoparticles or the block copolymer microdomains is sufficiently narrow.
- (2)
The use of precursors to synthesize nanoparticles within polymer microdomains
The advantage of this method is that low molecular weight precursors can readily be solubilized into polymer microdomains, which can self-assemble into ordered lattices. The structure of these superlattices can be well tailored via the lengths of the polymer blocks. A major issue is how to control the size of the nanoparticles during growth in the polymer matrix. Also for this route, there is the issue of the incompatibility of synthesized nanoparticles with the polymer matrix [7,8].
For both approaches (1) and (2), the problem of incompatibility with the polymer matrix needs to be overcome. This is a major challenge that currently limits the success of nanocomposites in general. For the preformed nanoparticles (route 1) compatibility can be achieved thermodynamically by proper compatibilization of the nanoparticle surface which is possible by coating with appropriate compatibilizers (stearic acid, copolymers, etc.) before mixing with the polymer matrix. When nanoparticles are synthesized from polymer-miscible precursors inside the polymer matrix (route 2), they are usually kinetically entrapped. For any kinetic entrapment without thermodynamic compatibilization, long term stability is an issue. Eventually, the nanoparticles will phase separate, which leads to agglomeration in homopolymer matrices or relocalization and aggregation of the nanoparticles to the interface of the block copolymer microdomains, where their surface energy is minimized.
3. Nanoparticle Arrays from Precursors
The kinetic entrapment of nanoparticles by reaction of polymer-compatible precursors inside block copolymer microdomains is a strategy that simultaneously takes advantage of the capability of block copolymers to assemble into well-ordered superlattices while providing compatibility during, and possibly after, the entrapment of the nanoparticles. The synthesis of nanoparticles within polymer microdomains has been termed the “microreactor” concept, because the chemical reaction from the precursor to the nanoparticle takes place within the microdomains, which themselves can also confine the size of the formed nanoparticles. For this, the microdomains are loaded with a suitable precursor, which can be chemically transformed into the desired nanoparticles either by addition of further reagents or simply by heating. Miscibility or compatibility of precursor and polymer can be provided by the use of secondary interactions (H-bond, coordinative, acid-base) which can be mediated by functional groups in the matrix polymer. Acid-base interactions with precursors such as gold acid (HAuCl4) with basic groups such as polyvinylpyridines, or coordinative interactions between transition metal complexes with amine-groups in polymers, or just the miscibility of hydrophobic precursors in hydrophobic polymers enable to load sufficient precursor material into the polymeric matrix. The precursors are then converted into the nanoparticles by redox-reactions or acid-base reactions [9-11]. Since the addition of reagents into polymeric matrices involves slow diffusion processes, often gaseous reagents or simply decomposition by heating are used. The microreactor domains can either be block copolymer micelles that are dispersed in solvents, which can later be assembled into superlattices, or they can be microdomains within ordered block copolymer phases (see Figure 2).
The direct integration into preformed block copolymer lattices has been first explored by the group of Cohen, where precursors have been loaded into spherical, cylindrical or lamellar microdomains of block copolymers [12,13]. After reaction into the desired nanoparticles, the nanoparticles were shown to be selectively located in the original microdomains, but not in the matrix. However, no nanoparticle superlattices could be established because the nanoparticle growth is difficult to control leading to a large number of small, polydisperse nanoparticles. This is similarly the case for the synthesis of nanoparticles inside block copolymer micelles.
The number and size of the nanoparticles depends on the relative rates of nucleation and growth. The time for the formation of a critical nucleus depends on the precursor concentration and the interfacial energy of the nucleus in the polymer matrix. The growth of the critical nucleus in polymeric matrices is diffusion limited by the transport of precursor molecules through the polymer matrix. If the critical nucleation time, i.e., the time for the formation for the first critical nucleus, is much smaller than the diffusion time to transverse the microdomain, then throughout the microdomain nuclei are formed, resulting in a large number of small nanoparticles per microdomain (see Figure 3).
Although by variation of reaction conditions the mean diameter of the nanoparticles within the polymer microdomain can be varied within a certain range by using different amounts of precursor, the polydispersity is generally large. While this approach is not useful for the preparation of nanoparticle superlattices, it is, however, suitable for the preparation of catalytically active nanoparticles, where it is the high specific surface area, the small size of the nanoparticles together with the stabilization in the polymer microdomain that provides high efficiency and the possibility for catalyst recycling in catalytic reactions [15].
The volume fraction of nanoparticles that can be generated within a microdomain is usually limited by the solubility of precursors. If the amount of nanoparticles in the microdomains is sufficiently large, nanoparticles can be made to fuse inside the polymer microdomains. This can be achieved by chemical vapor deposition of metals onto microphase separated block copolymer films [16]. Since growth is limited by the microdomain size, this can lead to the formation of wires as shown in Figure 4.
In the best case that the critical nucleation time is much larger than the diffusion time, only one nucleus is formed, which then grows until all precursor material within the micellar core has been taken up. If during this time no further nucleus is formed, only one nanoparticle per microdomain is generated. To achieve this goal, i.e., to grow nanoparticle single crystals within a microdomain, requires optimization of precursor concentration, precursor mobility (by addition of solvents or variation of temperature) and interfacial energy (by the presence of stabilizing ligands). Although difficult, this has been achieved for gold nanoparticles in polyvinylpyridine-microdomains of polystyrene-polyvinylpyridine block copolymers (see Figure 5) on surfaces and in bulk. It was shown that a control of the nanoparticle size and distance was independently possible via the amount of precursor in the micellar core and the block length of the block copolymers forming the micelles.
For sol/gel-reactions where the gel is homogeneously formed within the microdomain, proper conditions for the formation of one gel particle per microdomain are easier to find. This extends the use of this concept to prepare nanoparticles of a large class of inorganic oxides such as silica, titania and vanadia via sol/gel-routes (see Figure 6).
4. Nanoparticle Arrays from Preformed Nanoparticles
The direct integration of preformed nanoparticles into polymeric microdomains offers the advantage of using high quality nanoparticles. These nanoparticles can be selectively loaded into microdomains of ordered block copolymers [18,19]. For the formation of regular superlattices the challenge is to load exactly one nanoparticle per microdomain, which is only possible by optimizing loading conditions (see Figure 3(c)). To avoid this problem, preformed nanoparticles can be directly coated with polymer chains, which make them structurally similar to block copolymer micelles, where the nanoparticle represents the micellar core. These could then be similarly assembled into superlattices. In thin films, preformed nanoparticles and block copolymers can exhibit interesting co-assembly behavior, where domain size and orientation can be tailored [20].
For preformed nanoparticles to be miscible with the polymer matrix, the nanoparticle surface has to be compatibilized. Compatibility of nanoparticles with polymers is often achieved by coating the nanoparticles with low molecular weight hydrophobic molecules that have end groups that bind to the nanoparticle surface. Frequent examples are stearic acid that is used for oxidic (basic) nanoparticle surfaces such as ZnO or CaCO3. Also reactions of oxidic nanoparticles with silylation reagents have been used, e.g., for the compatibilization of silica nanoparticles in polymer membranes [21]. If possible, the nanoparticles are covered with a layer of a matrix-miscible polymer, ideally with the matrix polymer itself.
Generally, simple coating of the nanoparticle surface with a compatibilizing layer is still insufficient to provide complete miscibility, because of the loss of conformational entropy of the matrix polymers close to the nanoparticle surface [22]. Due to the large specific surface area of nanoparticles, this purely entropic effect alone leads to immiscibility and aggregation, a phenomenon that in colloidal solutions is known as depletion flocculation. In order to provide sufficient conformational freedom for the matrix polymers close to the nanoparticles, the polymer layer covering the nanoparticles should have a low segment density at its periphery to provide sufficient conformational freedom for the matrix polymers. This type of layer structure is ideally realized in spherical polymer brushes [23].
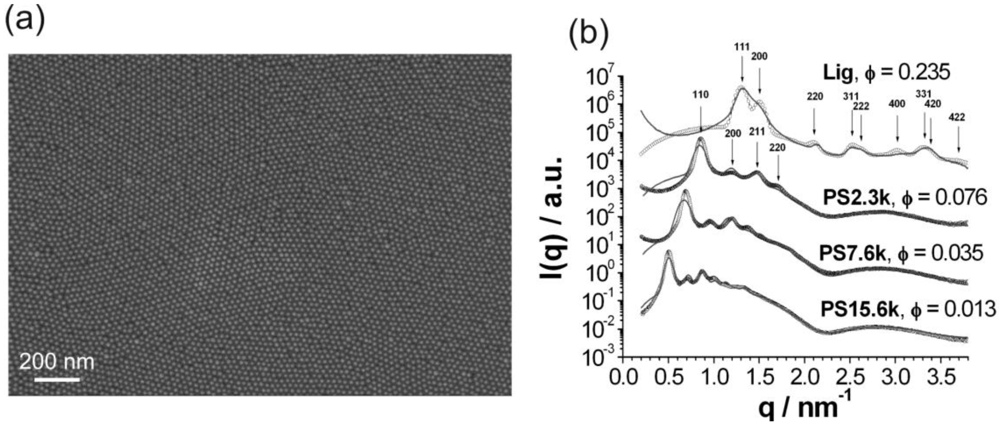
This requires the polymers to be bound to the surface of the nanoparticle with only one chain-end. This is chemically possible by grafting-to or grafting from techniques, where the grafting-from technique offers the advantage of high surface coverage at the cost of more synthetic effort. This has been realized for silica-polystyrene nanoparticles [24]. A more versatile method uses polymer chains containing a ligand end-group that binds to the nanoparticle surface. The attachment of brush-like polymer layers onto nanoparticles has recently been demonstrated for polyethylene glycol and polystyrene by using a ligand exchange procedure [25]. The attachment of polymer chains leads to an increased separation of nanoparticles as shown in Figure 7.
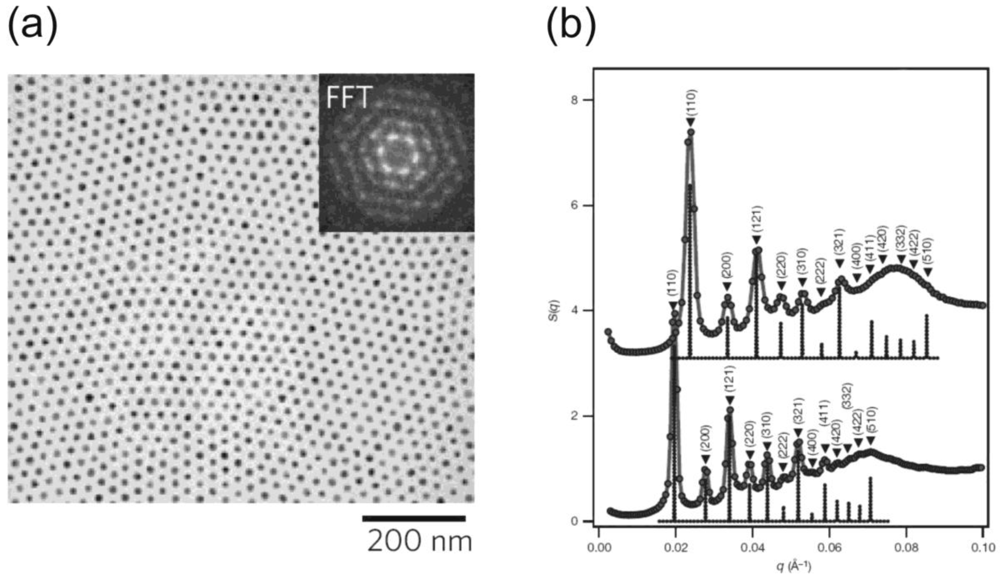
The attachment of a brush-like polymer layer provides complete miscibility of nanoparticles and polymer matrix over the entire range of nanoparticle volume fractions without aggregation [26]. The route is successful for many chemically different nanoparticles such as metals, oxides or selenides, with a variety of different nanoparticle properties (magnetic, catalytic, semiconducting). It so far provides a very versatile way to two- and three-dimensional nanoparticle superlattices in polymeric matrices. Figure 8 shows this for the two-dimensional case of a magnetite nanoparticle monolayer in a polystyrene matrix, and for the three-dimensional case of a CdSe-superlattice in polystyrene. In both cases, the distance between the nanoparticles can be controlled via the length of the attached polymer chains.
5. DNA-Nanoparticle Arrays
A sophisticated method to tailor nanoparticle self-assembly into superlattices is based on the work of Mirkin [27] and Alivisatos [28] who recognized the potential to control nanoparticle assembly via the attachment of DNA-strands. While this method is mainly limited to gold nanoparticles, where a stable attachment to the DNA-strands is achieved via a DNA-SH-Au link, it illustrates the potential and the quality of nanoparticle superlattices that can be assembled with polymers. Figure 9 shows examples for the two-dimensional case of a gold nanoparticle/DNA monolayer and the three-dimensional assembly of gold nanoparticle/DNA, where the distance between the nanoparticles is controlled via the number of DNA base pairs. By selective assembly of gold nanoparticles on specific sites on the DNA-strands, more complex “nanogrids” can be assembled [29,30].
6. Conclusion and Outlook
Extensive research on the assembly of nanoparticle/polymer composites has shown that the assembly of nanoparticles into superlattices in polymeric matrices is a particularly demanding challenge. It was found that the most critical issue was to prepare and conjugate nanoparticles of sufficient quality with a polymer layer that provided good miscibility with the polymer matrix. Various routes to solve this issue have been developed, of which at least two, the micellar precursor route, and the attached polymer brush route, have successfully led to the formation of nanoparticle superlattices in polymer matrices. These routes enable control of the interparticle distance via the length of the attached polymer chain on the 2–50 nm length scale. This is a feature that allows one to address critical design concepts in magnetic storage and hybrid solar cell applications, where the interparticle distance has to be carefully adapted to the length scale of magnetic dipolar interactions or the excition diffusion length. The possibility to translate structure into function is thus a currently exciting research area where progress will have profound influences for the use of polymer nanocomposites in functional devices.
Current challenges include assembling nanoparticles in a regular fashion at particular positions in two-phase nanostructures. This could be within the interface to provide electronic excitation and charge separation for the use in hybrid solar cells (Figure 10(a)), or the inclusion of two or more chemically different nanoparticles for directed energy transfer in light harvesting assemblies, the emission of white light in organic light emitting diodes, or the development of multiple contrast agents for medical diagnostics (Figure 10(b)).










Acknowledgments
The author would like to thank Frank Caruso for his hospitality during the sabbatical at the University of Melbourne, where the article has been written.
References
- Bigioni, T.P.; Lin, X.-M.; Nguyen, T.T.; Corwin, E.I.; Witten, T.A.; Jaeger, H.M. Kinetically driven self-assembly of highly ordered nanoparticles monolayers. Nat. Mater. 2006, 5, 265–270. [Google Scholar]
- Murray, C.B.; Kagan, C.R.; Bawendi, M.G. Self-organization of CdSe nanocrystallines into 3-dimensional quantum-dot superlattices. Science 2008, 270, 1335–1338. [Google Scholar]
- Shevchenko, E.V.; Talapin, D.; Kornowski, A.; Wiekhorst, F.; Kötzler, J.; Haase, M.; Rogach, A.L.; Weller, H. Colloidal crystals of monodisperse FePt nanoparticles grown by a three-layer technique of controlled oversaturation. Adv. Mater. 2002, 14, 287–290. [Google Scholar]
- Mueggenburg, K.E.; Lin, X.-M.; Goldsmith, R.H.; Jaeger, H.M. Elastic membranes of close-packed nanoparticle arrays. Nat. Mater. 2007, 6, 656–660. [Google Scholar]
- Haryono, A.; Binder, W. Controlled arrangement of nanoparticle arrays in block copolymer domains. Small 2006, 2, 600–611. [Google Scholar]
- Martin, J.E.; Wilcoxon, J.P.; Odinek, J.; Provencio, P. Control of the interparticle spacing in gold nanoparticle superlattices. J. Phys. Chem. B 2000, 104, 9475–9486. [Google Scholar]
- Förster, S.; Antonietti, M. Amphiphilic block copolymers in structure-controlled nanomaterials hybrids. Adv. Mater. 1998, 10, 195–217. [Google Scholar]
- Förster, S.; Plantenberg, T. From self-organizing polymers to nanohybrid and biomateirals. Angew. Chem. Int. Ed. 2002, 41, 688–714. [Google Scholar]
- Antonietti, M.; Förster, S.; Hartmann, J.; Oestreich, S. Novel amphiphilic block copolymers by polymer analogous reactions and their use for solubilization of metal salts and metal colloids. Macromolecules 1996, 29, 3800–3806. [Google Scholar]
- Spatz, J.P.; Herzog, T.; Mössmer, S.; Ziemann, P.; Möller, M. Inorganic-polymer micellar hybrid systems—A tool for nanolithography. Adv. Mater. 1999, 11, 149–153. [Google Scholar]
- Spatz, J.P. Nano- and micropatterning by organic-inorganic templating of hierarchical self-assembled structures. Angew. Chem. Int. Ed. 2002, 114, 3359–3362. [Google Scholar]
- Clay, R.T.; Cohen, R.E. Synthesis of metal nanoclustes within microphase-separated diblock copolymers: Sodium carboxylate vs. carboxylic acid functionlization. Supramol. Sci. 1998, 5, 41–48. [Google Scholar]
- Abes, J.I.; Cohen, R.E.; Ross, C.A. Selective growth of cobalt nanoclusters in domains of block copolymer films. Chem. Mater. 2003, 12, 1125–1131. [Google Scholar]
- Förster, S.; Konrad, M. From self-organizing polymers to nano- and biomaterials. J. Mater. Chem. 2003, 13, 2671. [Google Scholar]
- Klingelhöfer, S.; Heitz, W.; Greiner, A.; Oestreich, S.; Förster, S.; Antonietti, M. Preparation of palladium colloids in block copolymer micelles and their use for the catalysis of the Heck reaction. J. Am. Chem. Soc. 1997, 119, 10116–10120. [Google Scholar]
- Lopez, W.A.; Jaeger, H.M. Hierarchical self-assembly of metal nanostructures on diblock copolymer scaffolds. Nature 2001, 414, 735–738. [Google Scholar]
- Frömsdorf, A.; Kornowski, A.; Pütter, S.; Lee, L.T. Highly ordered nanostructured surfaces obtained by silica filled diblock copolymer micelles as templates. Small 2007, 5, 880–889. [Google Scholar]
- Fogg, D.E.; Radzilowski, L.H.; Blanski, R.; Schrock, R.R.; Thomas, E.L.; Bawendi, M.G. Fabrication of quantum dot-polymer composites: Semiconductor nanoclusters in dual-function polymer matrices with electron-transporting and cluster-passivating properties. Macromolecules 1997, 30, 8433–8439. [Google Scholar]
- Yeh, S.-W.; Wu, T.-L.; Wei, K.-H. Spatial position control of pre-synthesized CdS-nanoclusters using a self-assembled diblock copolymer template. Nanotechnology 2005, 16, 683–687. [Google Scholar]
- Lin, Y.; Böker, A.; He, J.; Sill, K.; Xiang, H.; Abetz, C.; Li, X.; Wank, J.; Emrick, T.; Long, S.; Wang, O.; Balasz, A.; Russell, T.P. Self-directed self-assembly of nanoparticle/copolymer mixtures. Nature 2004, 434, 55–59. [Google Scholar]
- Merkel, T.C.; Freeman, B.D.; Spontak, R.J.; He, Z.; Pinnau, I.; Meakin, P.; Hill, A.J. Ultrapermeable, reverse-selective nanocomposite membranes. Science 2002, 296, 519–522. [Google Scholar]
- De Gennes, P.G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1979. [Google Scholar]
- Milner, S.T. Polymer brushes. Science 1991, 251, 905–914. [Google Scholar]
- Pyun, J.; Matyjaszewski, C. Synthesis of nanocomposites organic/inorganic hybrid materials using controlled/ “living”polymerization. Chem. Mater. 2001, 13, 3436–3448. [Google Scholar]
- Nikolic, M.A.; Alexandrovic, V.; Krack, M.; Kornowski, A.; Förster, S.; Weller, H. Taylor-made ligands for biocompatible nanoparticles. Angew. Chem. Int. Ed. 2006, 45, 6577–6580. [Google Scholar]
- Fischer, S.; Salcher, A.; Kornowski, A.; Weller, H.; Förster, S. Completely miscible nanocomposites. Angew. Chem. Int. Ed. 2011. in press. [Google Scholar]
- Mirkin, C.A.; Letsinger, R.L.; Mucic, R.C.; Storhoff, J.J. A DNA-based method for rationally assembling nanoparticles into macroscopic materials. Nature 1996, 382, 609–611. [Google Scholar]
- Alivisatos, A.P.; Johnsson, K.P.; Peng, X.; Wilson, T.E.; Loweth, C.J.; Bruchez, M.P., Jr.; Schultz, P.G. Organization of ‘nanocrystal molecules’ using DNA. Nature 1996, 382, 609–611. [Google Scholar]
- Zhang, J.P.; Liu, Y.; Ke, Y.G.; Yan, H. Periodic square-like gold nanoparticle arrays templated by self-assembled 2D DNA nanogrids on a surface. Nano Lett. 2006, 6, 248–251. [Google Scholar]
- Zheng, J.W.; Constantinou, P.E.; Micheel, C.; Alivisatos, A.P.; Kiehl, R.A.; Seeman, N.C. Two-dimensional nanoparticle arrays show the organizational power of robust DNA motifs. Nano Lett. 2006, 6, 1502–1504. [Google Scholar]
- Cheng, W.; Campolongo, M.J.; Cha, J.J.; Tan, S.J.; Umbach, C.C.; Muller, D.A.; Luo, D. Free-standing nanoparticle superlattice sheets controlled by DNA. Nat. Mater. 2009, 8, 519. [Google Scholar]
- Nykypanchuk, D.; Maye, M.M.; van der Lelie, D.; Gang, O. DNA-guided crystallization of colloidal nanoparticles. Nature 2008, 451, 549–552. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mehdizadeh Taheri, S.; Fischer, S.; Förster, S. Routes to Nanoparticle-Polymer Superlattices. Polymers 2011, 3, 662-673. https://doi.org/10.3390/polym3020662
Mehdizadeh Taheri S, Fischer S, Förster S. Routes to Nanoparticle-Polymer Superlattices. Polymers. 2011; 3(2):662-673. https://doi.org/10.3390/polym3020662
Chicago/Turabian StyleMehdizadeh Taheri, Sara, Steffen Fischer, and Stephan Förster. 2011. "Routes to Nanoparticle-Polymer Superlattices" Polymers 3, no. 2: 662-673. https://doi.org/10.3390/polym3020662
APA StyleMehdizadeh Taheri, S., Fischer, S., & Förster, S. (2011). Routes to Nanoparticle-Polymer Superlattices. Polymers, 3(2), 662-673. https://doi.org/10.3390/polym3020662